
Six Undescribed Capnosane-Type Macrocyclic Diterpenoids from South China Sea Soft Coral Sarcophyton crassocaule: Structural Determination and Biological Evaluation
 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Structure Elucidation of New Compounds 1–6
2.2. In Vitro Biological Assay
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Animal Materials
3.3. Extraction and Isolation
3.4. X-ray Crystallographic Analysis
3.5. Computational Details
3.6. Cytotoxic Detection
3.7. Anti-Inflammatory Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, B.; Zhou, X.F.; Lin, X.P.; Liu, J.; Peng, Y.; Yang, X.W.; Liu, Y.H. Cembrane diterpenes chemistry and biological properties. Curr. Org. Chem. 2012, 16, 1512–1539. [Google Scholar] [CrossRef]
- Dauben, W.G.; Thiessen, W.E.; Resnick, P.R. Cembrene, a fourteen-membered ring diterpene hydrocarbon. J. Org. Chem. 1965, 30, 1693–1698. [Google Scholar] [CrossRef]
- Du, Y.Q.; Chen, J.; Wu, M.J.; Zhang, H.Y.; Liang, L.F.; Guo, Y.W. Uncommon capnosane diterpenes with neuroprotective potential from South China Sea soft coral Sarcophyton boettgeri. Mar. Drugs 2022, 20, 602. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Nakano, E. Stereochemical course of the transannular cyclization, in chloroform, of epoxycembranoids derived from the geometrical isomers of (14S)-14-hydroxy-l,3,7,11-cembratetraenes. J. Org. Chem. 1990, 55, 1947–1951. [Google Scholar] [CrossRef]
- Li, Y.; Pattenden, G. Perspectives on the structural and biosynthetic interrelationships between oxygenated furanocembranoids and their polycyclic congeners found in corals. Nat. Prod. Rep. 2011, 28, 1269–1310. [Google Scholar] [CrossRef]
- Liu, Z.; Cheng, W.; Liu, D.; van Ofwegen, L.; Proksch, P.; Lin, W.H. Capnosane-type cembranoids from the soft coral Sarcophyton trocheliophorum with antibacterial effects. Tetrahedron 2014, 70, 8703–8713. [Google Scholar] [CrossRef]
- Chen, W.T.; Yao, L.G.; Li, X.W.; Guo, Y.W. Sarcophytrols A-C, new capnosane diterpenoids from the South China Sea soft coral Sarcophyton trocheliophorum. Tetrahedron Lett. 2015, 56, 1348–1352. [Google Scholar] [CrossRef]
- Liang, L.F.; Kurtán, T.; Mándi, A.; Gao, L.X.; Li, J.; Zhang, W.; Guo, Y.W. Sarsolenane and capnosane diterpenes from the Hainan soft coral Sarcophyton trocheliophorum marenzeller as PTP1B inhibitors. Eur. J. Org. Chem. 2014, 2014, 1841–1847. [Google Scholar] [CrossRef]
- Song, Y.T.; Yu, D.D.; Su, M.Z.; Luo, H.; Cao, J.G.; Liang, L.F.; Yang, F.; Guo, Y.W. Structurally diverse diterpenes from the South China Sea soft coral Sarcophyton trocheliophorum. Mar. Drugs 2023, 21, 69. [Google Scholar] [CrossRef]
- Tseng, W.R.; Ahmed, A.F.; Huang, C.Y.; Tsai, Y.Y.; Tai, C.J.; Orfali, R.S.; Hwang, T.L.; Wang, Y.H.; Dai, C.F.; Sheu, J.H. Bioactive capnosanes and cembranes from the soft coral Klyxum flaccidum. Mar. Drugs 2019, 17, 461. [Google Scholar] [CrossRef]
- Lai, D.; Geng, Z.F.; Deng, Z.W.; van Ofwegen, L.; Proksch, P.; Lin, W.H. Cembranoids from the soft coral Sinularia rigida with antifouling activities. J. Agric. Food Chem. 2013, 61, 4585–4592. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Zhu, Z.D.; Gu, Y.C.; Li, J.; Zhu, W.L.; Guo, Y.W. Further new diterpenoids as PTP1B inhibitors from the Xisha soft coral Sinularia polydactyla. Mar. Drugs 2018, 16, 103. [Google Scholar] [CrossRef] [PubMed]
- Bu, Q.; Yang, M.; Yan, X.Y.; Li, S.W.; Ge, Z.Y.; Zhang, L.; Yao, L.G.; Guo, Y.W.; Liang, L.F. Mililatensols A-C, new records of sarsolenane and capnosane diterpenes from soft coral Sarcophyton mililatensis. Mar. Drugs 2022, 20, 566. [Google Scholar] [CrossRef]
- Shen, S.; Zhu, H.J.; Chen, D.W.; Liu, D.; van Ofwegen, L.; Proksch, P.; Lin, W.H. Pavidolides A-E, new cembranoids from the soft coral Sinularia pavida. Tetrahedron Lett. 2012, 53, 5759–5762. [Google Scholar] [CrossRef]
- Xi, Z.F.; Bie, W.; Chen, W.; Liu, D.; van Ofwegen, L.; Proksch, P.; Lin, W.H. Sarcophyolides B-E, new cembranoids from the soft coral Sarcophyton elegans. Mar. Drugs 2013, 11, 3186–3196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, X.W.; Yao, L.G.; Wu, B.; Guo, Y.W. Three new capnosane-type diterpenoids from the South China Sea soft coral Lobophytum sp. Fitoterapia 2019, 133, 70–74. [Google Scholar] [CrossRef]
- Zhang, M.; Long, K.H.; Shi, S.H.; Mak, T.C.W. A novel diterpenolide from the soft coral Sarcophyton solidun. J. Nat. Prod. 1992, 55, 1672–1675. [Google Scholar] [CrossRef]
- Bowden, B.F.; Coll, J.C.; Gulbis, J.M.; Mackay, M.F.; Willis, R.H. Studies of Australian soft corals. XXXVIII. Structure determination of several diterpenes derived from a cespitularia species (coelenterata, octocorallia, xeniidae). Aust. J. Chem. 1986, 39, 803–812. [Google Scholar] [CrossRef]
- D’Ambrosio, M.; Guerriero, A.; Pietra, F. Novel cembranolides (coralloidolide D and E) and a 3,7-cyclized cembranolide (coralloidolide C) from the mediterranean coral Alcyonium coralloides. Helv. Chim. Acta. 1989, 72, 1590–1596. [Google Scholar] [CrossRef]
- Zhang, D.; Li, Y.Y.; Li, X.L.; Han, X.; Wang, Z.; Li, G.Q. A new capnosane-type diterpenoid from the South China sea soft coral Lobophytum pauciflorum. Nat. Prod. Res. 2024, 38, 97–102. [Google Scholar] [CrossRef]
- Li, J.; Huan, X.J.; Wu, M.J.; Chen, Z.H.; Chen, B.; Miao, Z.H.; Guo, Y.W.; Li, X.W. Chemical constituents from the South China sea soft coral Sinularia humilis. Nat. Prod. Res. 2022, 36, 3324–3330. [Google Scholar] [CrossRef] [PubMed]
- Elkhawas, Y.A.; Elissawy, A.M.; Elnaggar, M.S.; Mostafa, N.M.; Al-Sayed, E.; Bishr, M.M.; Singab, A.N.B.; Salama, O.M. Chemical diversity in species belonging to soft coral genus Sacrophyton and its impact on biological activity: A review. Mar. Drugs 2020, 18, 41. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, I.G.; Miguel, M.G.; Mnif, W. A brief review on new naturally occurring cembranoid diterpene derivatives from the soft corals of the genera Sarcophyton, Sinularia, and Lobophytum since 2016. Molecules 2019, 24, 781. [Google Scholar] [CrossRef] [PubMed]
- Li, J.F.; Zeng, Y.B.; Li, W.S.; Luo, H.; Zhang, H.Y.; Guo, Y.W. Xishaglaucumins A-J, new cembranoids with anti-inflammatory activities from the South China Sea soft coral Sarcophyton glaucum. Chin. J. Chem. 2022, 40, 79–90. [Google Scholar] [CrossRef]
- Zeng, Y.B.; Wang, Z.; Chang, W.J.; Zhao, W.B.; Wang, H.; Chen, H.Q.; Dai, H.F.; Lv, F. New azaphilones from the marine-derived fungus Penicillium sclerotiorum E23Y-1A with their anti-inflammatory and antitumor activities. Mar. Drugs 2023, 21, 75. [Google Scholar] [CrossRef]
- Chen, H.Q.; Guo, D.S.; Wang, H.; Cai, C.H.; Yuan, J.Z.; Dai, H.F.; Yang, L.; Mei, W.L. Sesquiterpenoids and bibenzyl derivative from Dendrobium hercoglossum. Fitoterapia 2024, 172, 105748. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
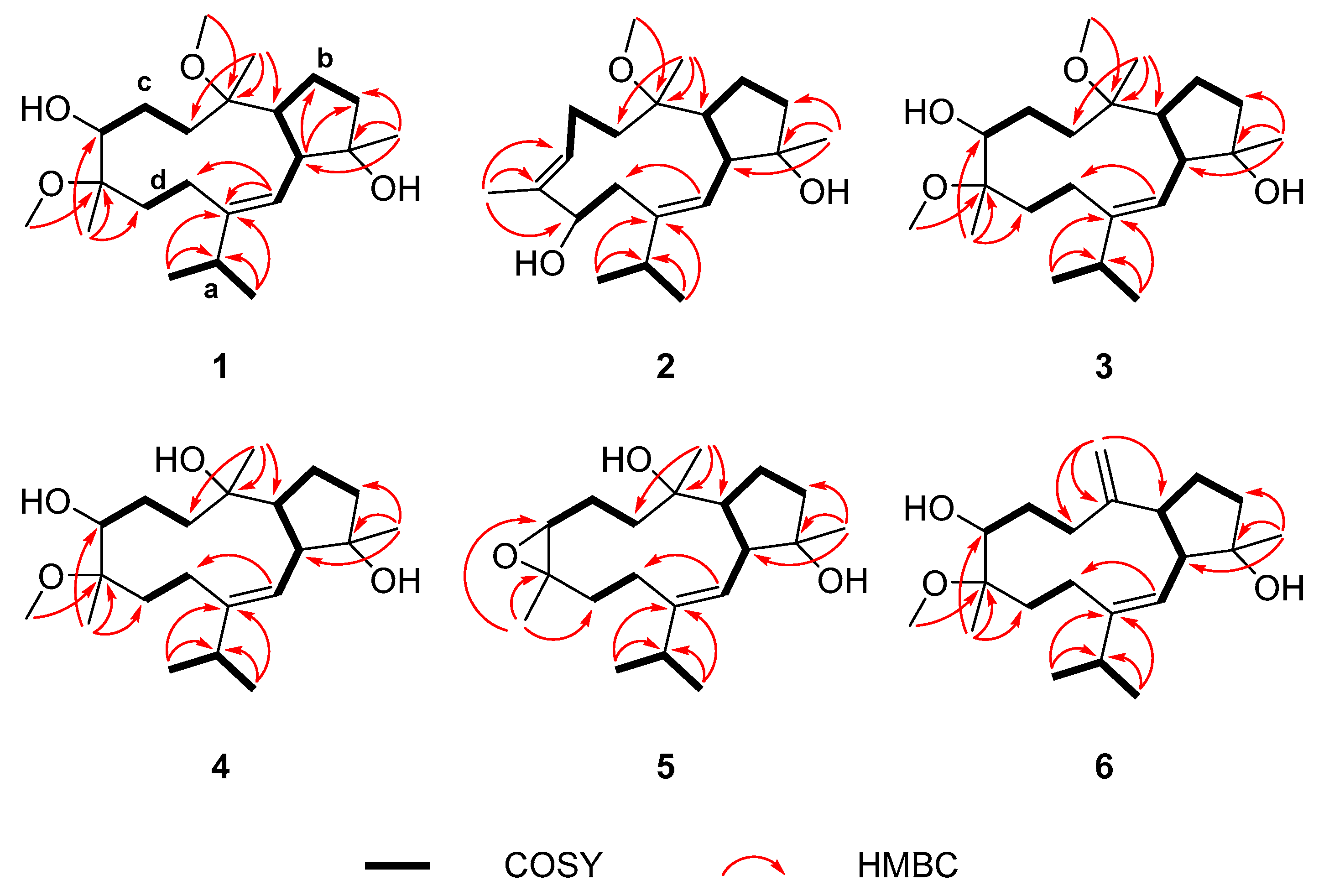{kind=link}
{kind=link}
{kind=link}
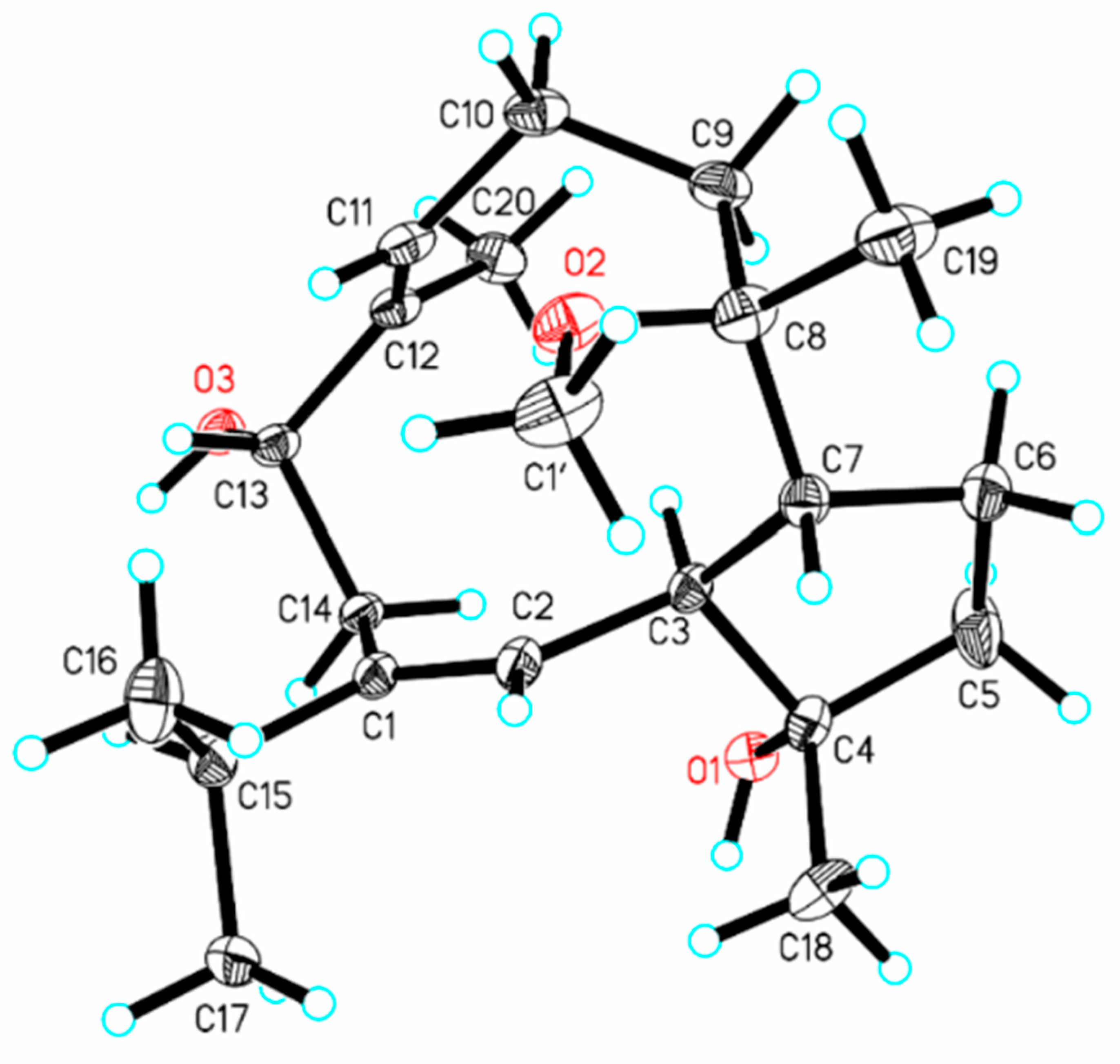{kind=link}
{kind=link}
| No. | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 2 | 5.02 d (11.7) | 5.00 d (9.3) | 5.16 d (10.3) | 5.21 d (10.4) | 5.31 d (12.0) | 4.88 d (10.7) |
| 3 | 2.49 dd (11.7, 5.3) | 2.34 t (9.3) | 2.36 dd (11.5, 10.3) | 2.41 t (10.9) | 2.97 t (11.5) | 2.41 dd (10.7, 5.9) |
| 5 | 1.77 m 1.66 m | 1.58–1.66 m | 1.74 m 1.69 m | 1.74 m | 1.79 m | 1.79 m 1.69 m |
| 6 | 1.86 m 1.75 m | 1.59 m 1.48 m | 1.68 m 1.48 m | 1.72 m 1.50 m | 1.97 m 1.72 m | 1.85–1.95 m |
| 7 | 2.88 m | 2.15 m | 1.97 m | 1.93 m | 2.73 m | 2.17 m |
| 9 | 1.97 m 1.62 m | 2.00 m 1.55 m | 1.74 m 1.58 ddd (15.5, 6.7, 2.2) | 1.69 m | 1.99 m 1.73 m | 2.27 m 2.16 m |
| 10 | 1.56 m 1.32 m | 2.08–2.19 m | 1.90 m 1.30 m | 1.67 m 1.41 m | 2.14 m 1.22 m | 1.58 m 1.45 m |
| 11 | 3.59 t (11.0) | 5.38 t (7.7) | 3.66 t (10.1) | 3.68 t (9.5) | 3.16 dd (10.0, 4.3) | 3.60 t (9.3) |
| 13 | 2.15 m 1.19 m | 3.97 dd (11.0, 2.2) | 2.10 m 1.21 m | 2.14 m 1.24 m | 2.07 m 1.72 m | 2.09 m 1.15 m |
| 14 | 2.09 m 1.97 m | 2.81 t (11.9) 1.98 m | 2.13 m 1.94 m | 2.18 m 2.00 m | 2.60 td (13.3, 6.3) 1.97 m | 1.99 m 1.89 m |
| 15 | 2.23 m | 2.26 sept (6.8) | 2.22 m | 2.28 m | 2.37 sept (6.8) | 2.22 m |
| 16 | 1.08 d (6.8) | 1.08 d (6.8) | 1.10 d (6.8) | 1.11 d (6.8) | 1.14 d (6.8) | 1.06 d (6.8) |
| 17 | 0.99 d (6.8) | 1.07 d (6.8) | 1.02 d (6.8) | 1.05 d (6.8) | 1.05 d (6.8) | 0.98 d (6.8) |
| 18 | 1.10 s | 1.06 s | 1.06 s | 1.08 s | 1.16 s | 1.14 s |
| 19 | 1.07 s | 1.15 s | 1.08 s | 1.16 s | 1.24 s | 5.12 br s 4.89 br s |
| 20 | 1.26 s | 1.69 s | 1.25 s | 1.27 s | 1.17 s | 1.24 s |
| 8-OMe | 3.19 s | 3.12 s | 3.10 s | |||
| 12-OMe | 3.22 s | 3.20 s | 3.21 s | 3.21 s |
| No. | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | 146.7, C | 141.6, C | 146.8, C | 151.0, C | 146.5, C | 146.6, C |
| 2 | 121.0, CH | 127.2, CH | 122.7, CH | 121.4, CH | 120.4, CH | 123.0, CH |
| 3 | 52.0, CH | 50.0, CH | 51.6, CH | 51.7, CH | 50.0, CH | 56.9, CH |
| 4 | 83.2, C | 82.4, C | 81.4, C | 81.6, C | 82.2, C | 82.6, C |
| 5 | 37.5, CH2 | 39.5, CH2 | 37.7, CH2 | 38.4, CH2 | 39.6, CH2 | 40.7, CH2 |
| 6 | 23.1, CH2 | 24.8, CH2 | 24.7, CH2 | 25.0, CH2 | 25.3, CH2 | 31.7, CH2 |
| 7 | 50.0, CH | 51.0, CH | 52.7, CH | 55.7, CH | 48.5, CH | 51.5, CH |
| 8 | 79.3, C | 78.0, C | 80.0, C | 75.8, C | 74.5, C | 154.3, C |
| 9 | 35.7, CH2 | 36.2, CH2 | 30.4, CH2 | 33.4, CH2 | 39.2, CH2 | 34.2, CH2 |
| 10 | 25.1, CH2 | 23.0, CH2 | 24.9, CH2 | 25.6, CH2 | 23.4, CH2 | 28.8, CH2 |
| 11 | 78.7, CH | 127.9, CH | 78.9, CH | 78.4, CH | 61.7, CH | 74.3, CH |
| 12 | 79.4, C | 134.3, C | 79.4, C | 79.3, C | 60.1, C | 79.7, C |
| 13 | 33.2, CH2 | 76.2, CH | 33.7, CH2 | 33.7, CH2 | 33.9, CH2 | 33.5, CH2 |
| 14 | 21.3, CH2 | 38.3, CH2 | 21.6, CH2 | 21.7, CH2 | 25.0, CH2 | 21.9, CH2 |
| 15 | 32.8, CH | 34.5, CH | 32.9, CH | 33.0, CH | 31.3, CH | 32.1, CH |
| 16 | 21.6, CH3 | 22.2, CH3 | 22.1, CH3 | 22.1, CH3 | 20.2, CH3 | 21.8, CH3 |
| 17 | 23.1, CH3 | 24.3, CH3 | 24.0, CH3 | 23.8, CH3 | 24.4, CH3 | 23.4, CH3 |
| 18 | 26.5, CH3 | 24.9, CH3 | 24.9, CH3 | 24.9, CH3 | 24.3, CH3 | 25.1, CH3 |
| 19 | 19.2, CH3 | 24.9, CH3 | 24.1, CH3 | 30.2, CH3 | 25.9, CH3 | 111.5, CH2 |
| 20 | 19.1, CH3 | 11.7, CH3 | 19.0, CH3 | 19.0, CH3 | 20.9, CH3 | 19.0, CH3 |
| 8-OMe | 48.1, CH3 | 48.8, CH3 | 49.4, CH3 | |||
| 12-OMe | 49.5, CH3 | 49.5, CH3 | 49.5, CH3 | 49.5, CH3 |
| Compounds | IC50 ± SD (μM) | Compounds | IC50 ± SD (μM) |
|---|---|---|---|
| 1 | >100 | 5 | 93.0 ± 3.8 |
| 2 | >100 | 6 | >100 |
| 3 | >100 | 7 | >100 |
| 4 | 76.8 ± 8.0 | Quercetin | 12.7 ± 2.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, H.; Zeng, Y.; Wang, H.; Chang, W.; Chen, H.; Zhou, F.; Dai, H.; Wang, X. Six Undescribed Capnosane-Type Macrocyclic Diterpenoids from South China Sea Soft Coral Sarcophyton crassocaule: Structural Determination and Biological Evaluation. Mar. Drugs 2023, 21, 645. https://doi.org/10.3390/md21120645
Peng H, Zeng Y, Wang H, Chang W, Chen H, Zhou F, Dai H, Wang X. Six Undescribed Capnosane-Type Macrocyclic Diterpenoids from South China Sea Soft Coral Sarcophyton crassocaule: Structural Determination and Biological Evaluation. Marine Drugs. 2023; 21(12):645. https://doi.org/10.3390/md21120645
Chicago/Turabian StylePeng, Hanyang, Yanbo Zeng, Hao Wang, Wenjun Chang, Huiqin Chen, Fengjuan Zhou, Haofu Dai, and Xiachang Wang. 2023. "Six Undescribed Capnosane-Type Macrocyclic Diterpenoids from South China Sea Soft Coral Sarcophyton crassocaule: Structural Determination and Biological Evaluation" Marine Drugs 21, no. 12: 645. https://doi.org/10.3390/md21120645