

Short-Chain Fatty Acids Suppress mTOR Signaling in Colon Cancer Cells via Long Non-Coding RNA RMST

Abstract
:1. Introduction
2. Results
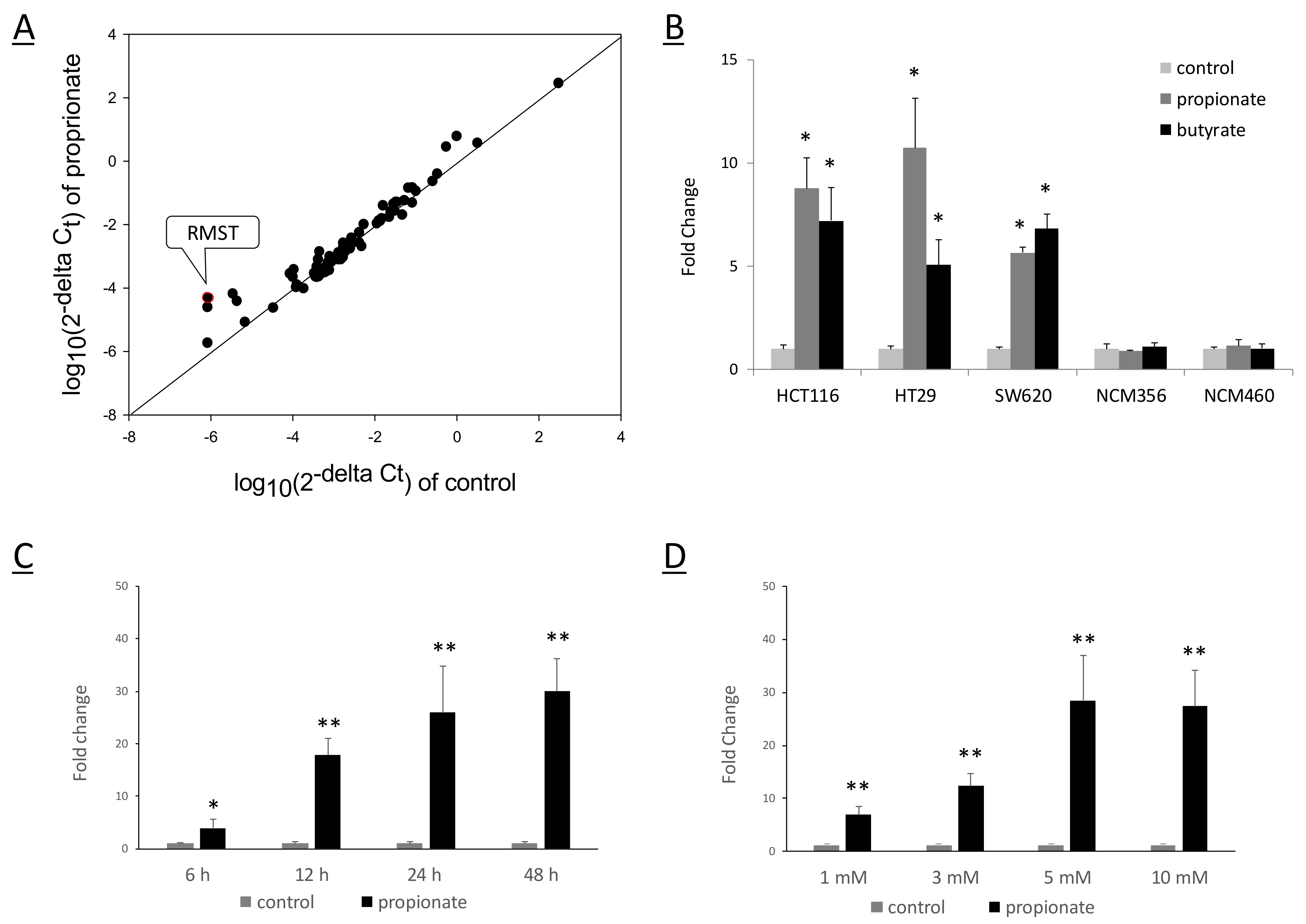
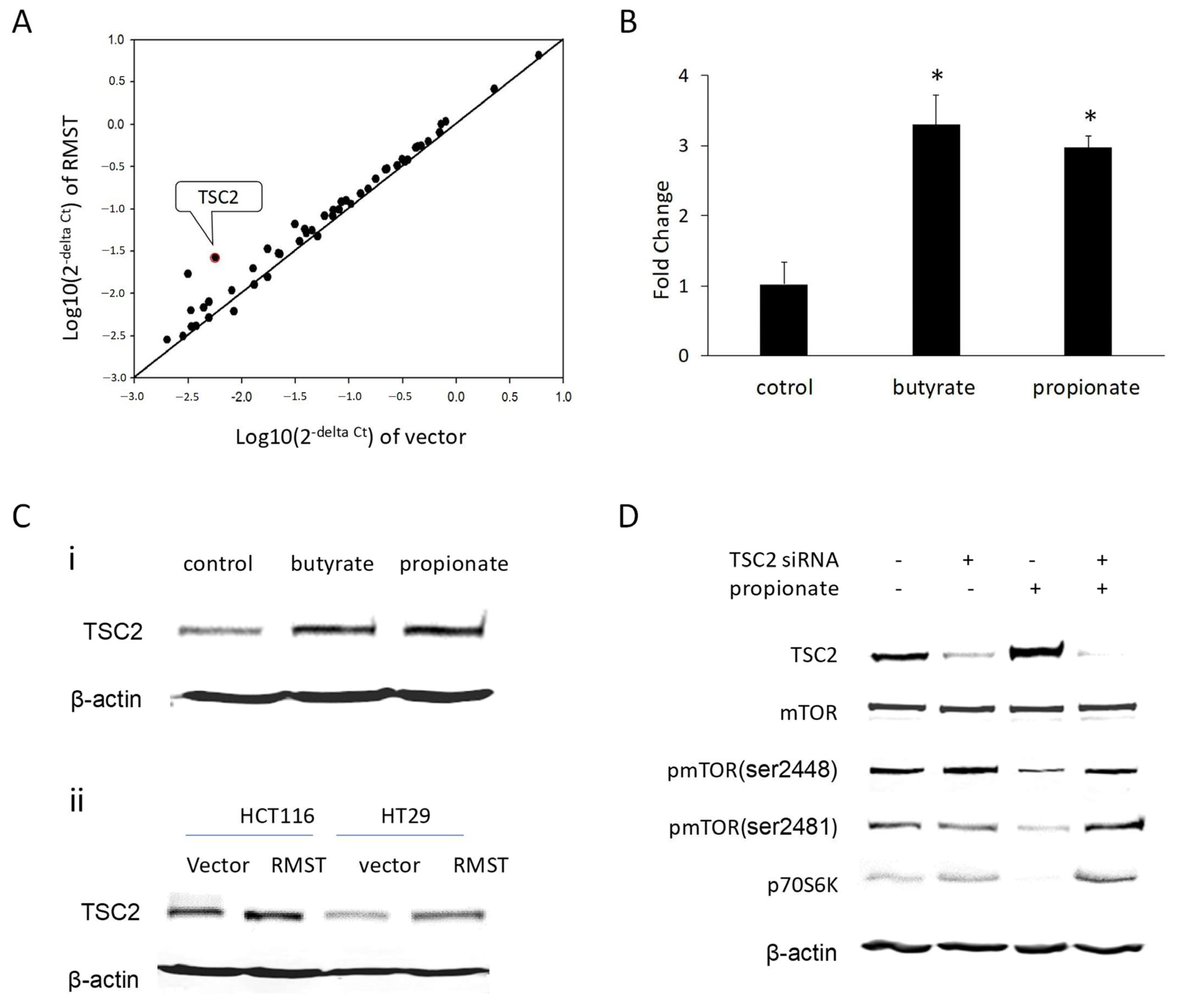
2.1. SCFAs Induced RMST Expression in Colon Cancer Cells
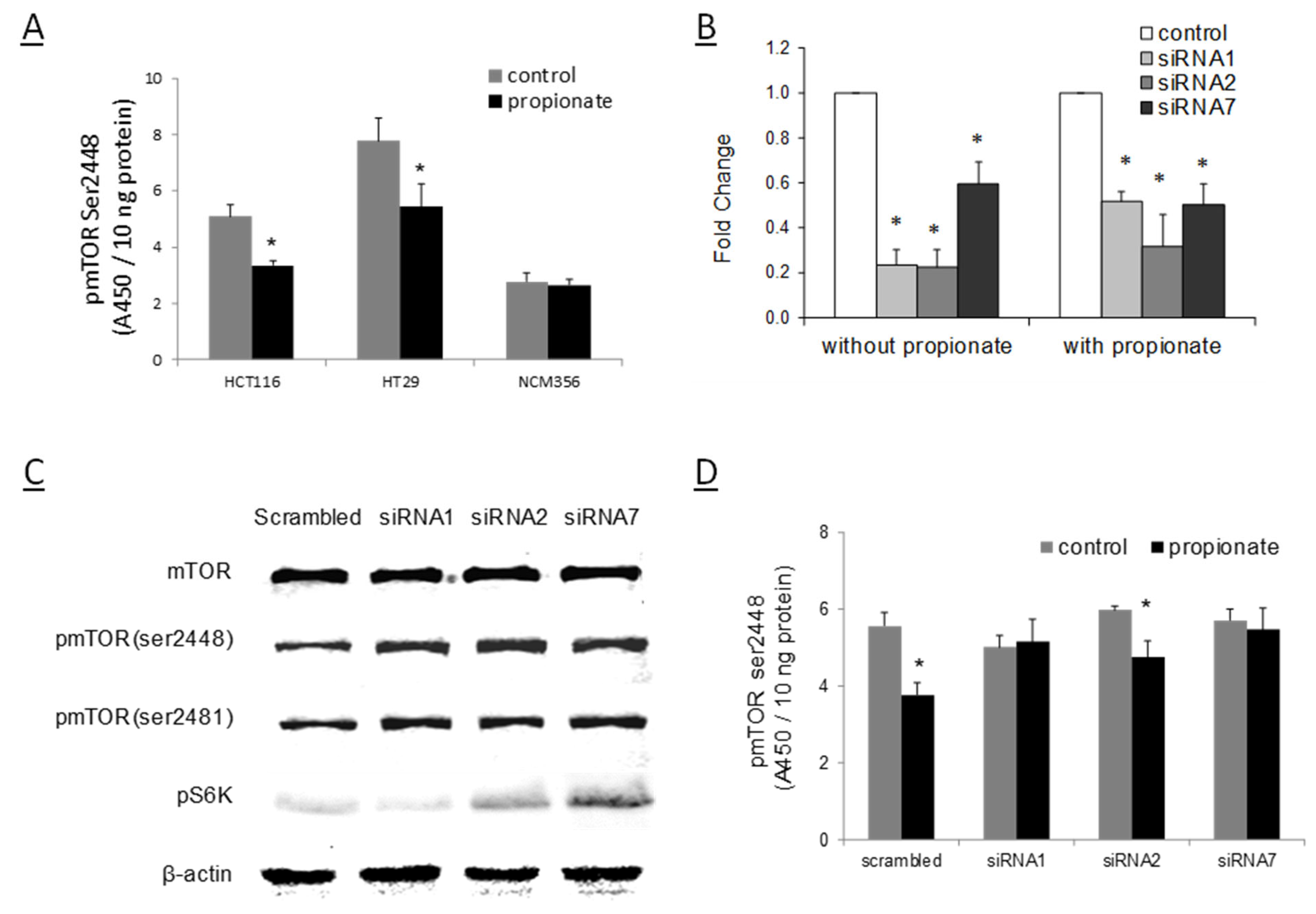
2.2. RMST Is Required for SCFA to Reduce mTOR Activation
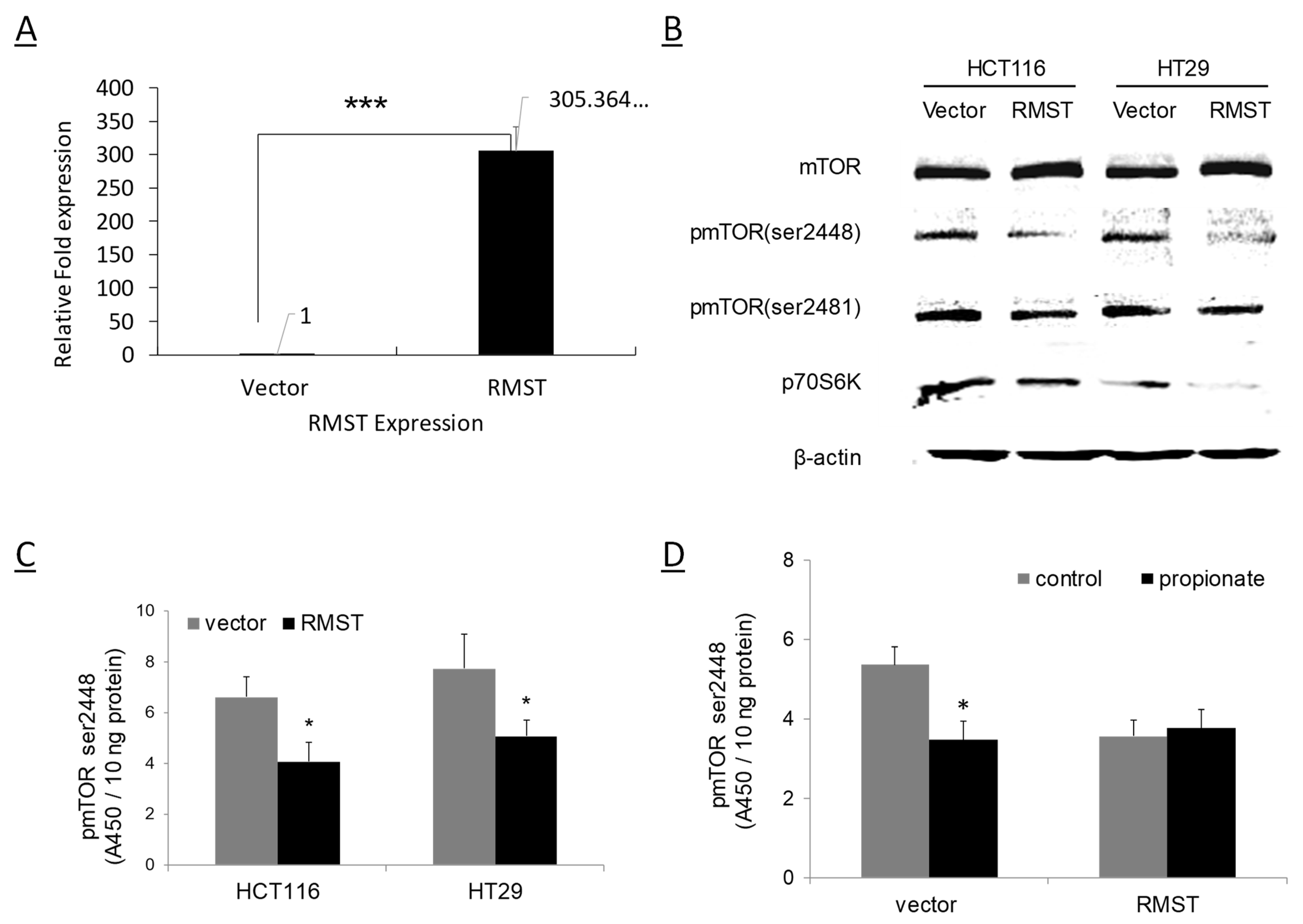
2.3. RMST Suppresses mTOR Activation
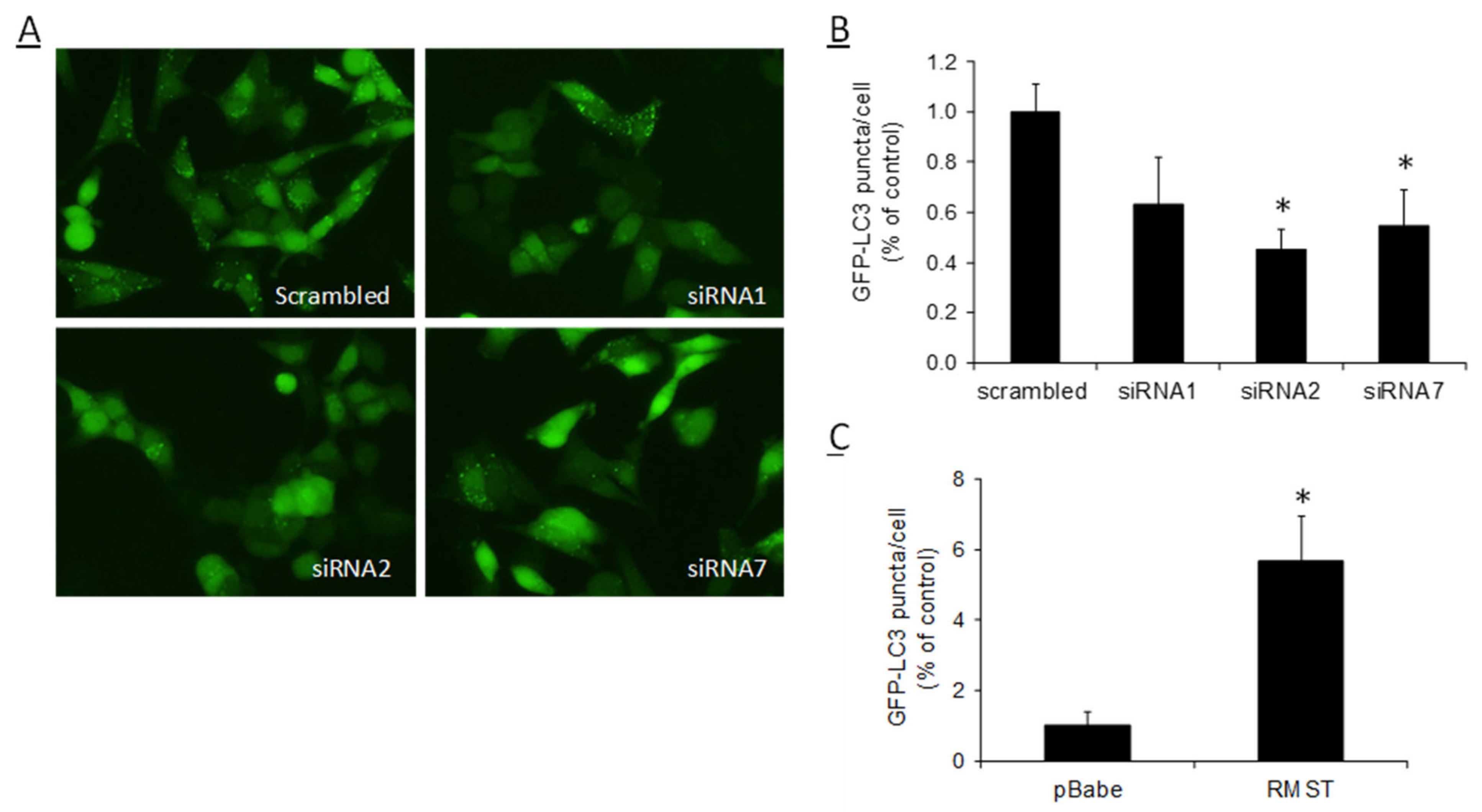
2.4. RMST Modulates SCFAs-Induced Autophagy
2.5. TSC2 Is Involved in the Suppression of mTOR Signaling by SCFA/RMST
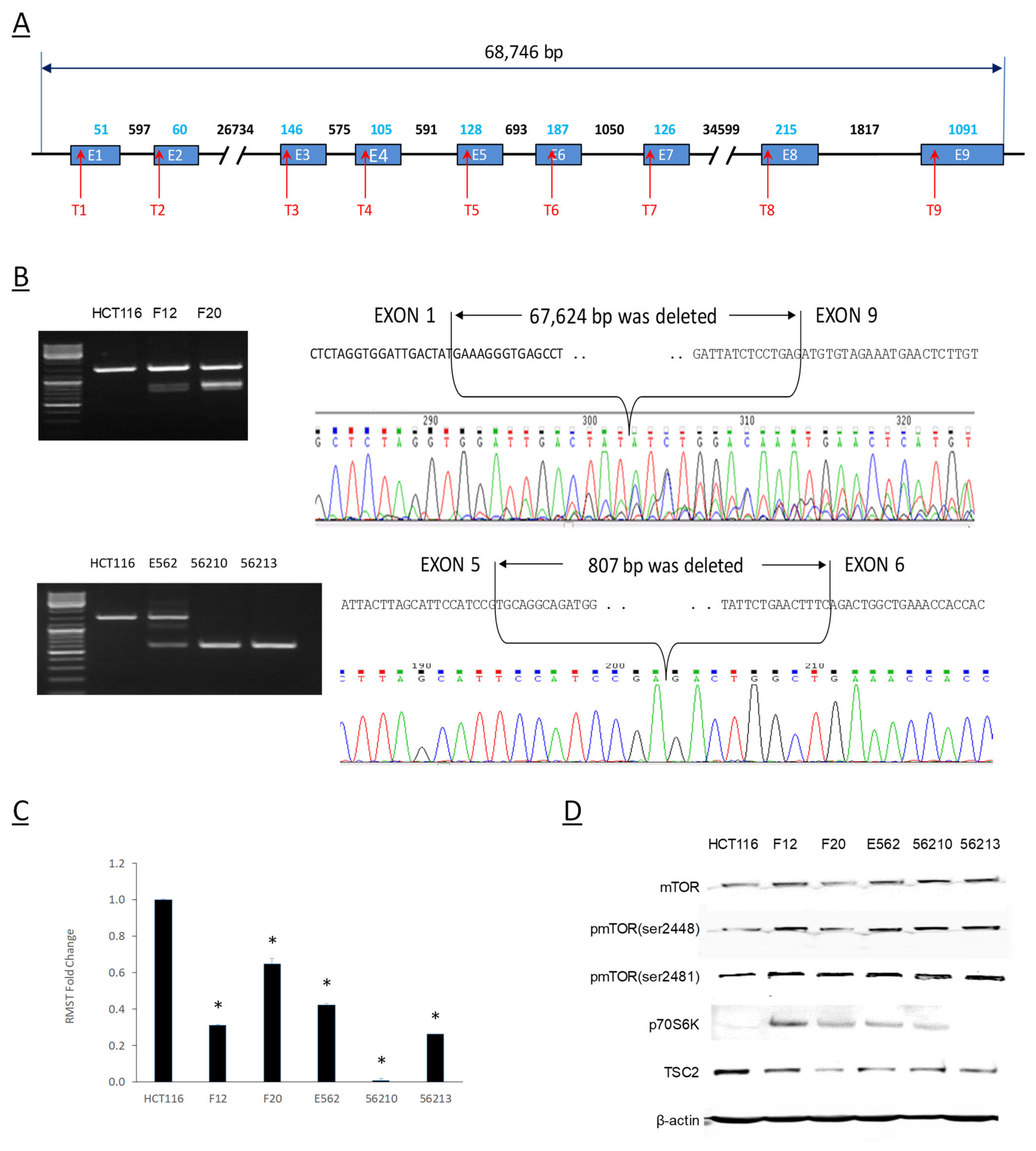
2.6. Genomic Editing of RMST Gene Affects mTOR Activities and TSC2 Expression
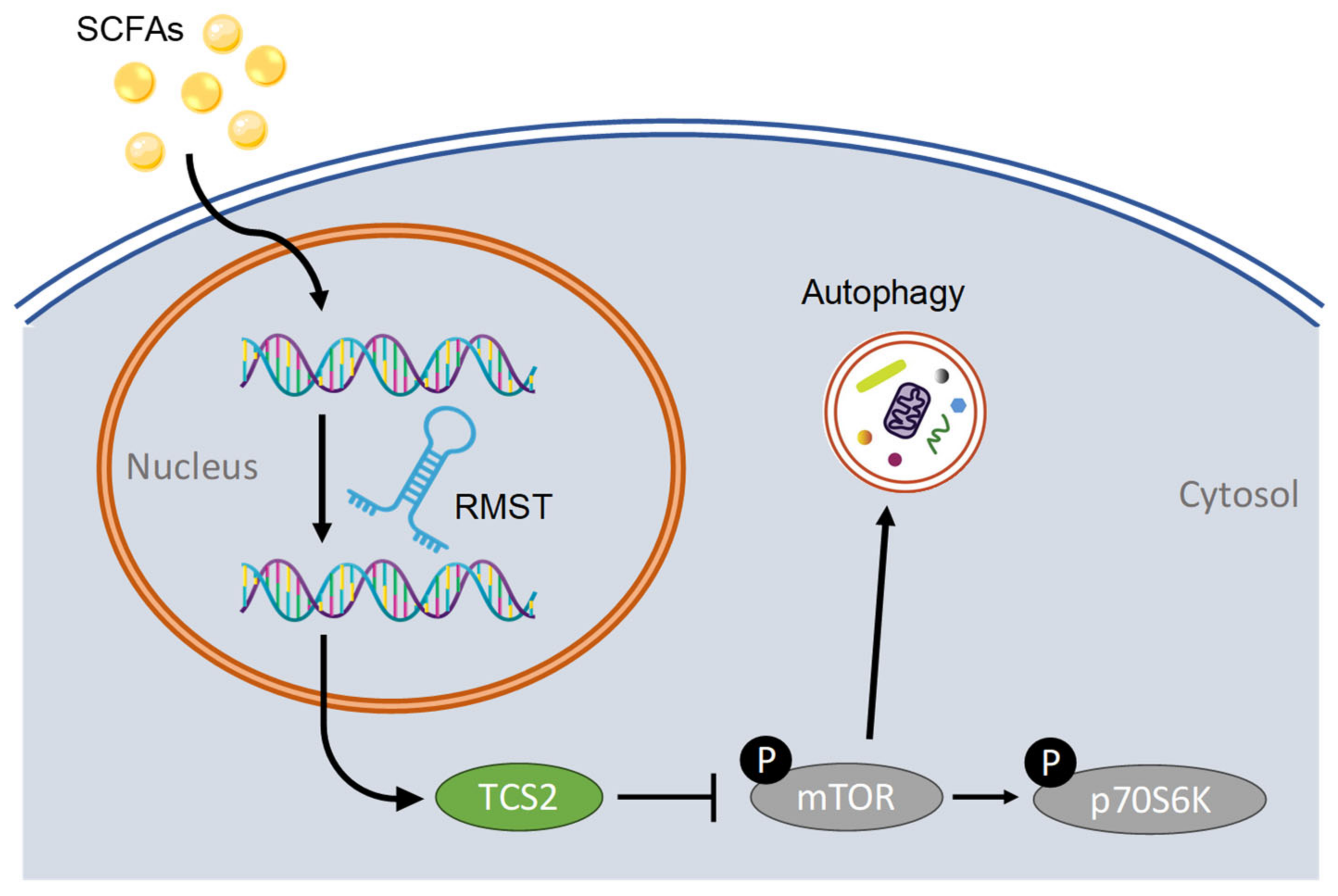
3. Discussion
4. Materials and Methods
4.1. Constructs and Gene Cloning
4.2. Cell Culture and Stable Transfection
4.3. siRNA Transfections
4.4. RNA Isolation and Real-Time RT-PCR
4.5. RMST Genomic Deletion with CRISPR/CAS9 System
4.6. Western Blot
4.7. ELISA for mTOR pser2448
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Topping, D.L.; Clifton, P.M. Short-chain fatty acids and human colonic function: Roles of resistant starch and nonstarch polysaccharides. Physiol. Rev. 2001, 81, 1031–1064. [Google Scholar] [CrossRef]
- Hamer, H.M.; Jonkers, D.; Venema, K.; Vanhoutvin, S.; Troost, F.J.; Brummer, R.J. Review article: The role of butyrate on colonic function. Aliment. Pharmacol. Ther. 2008, 27, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.M.; de Souza, R.; Kendall, C.W.; Emam, A.; Jenkins, D.J. Colonic health: Fermentation and short chain fatty acids. J. Clin. Gastroenterol. 2006, 40, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef]
- Fung, K.Y.; Brierley, G.V.; Henderson, S.; Hoffmann, P.; McColl, S.R.; Lockett, T.; Head, R.; Cosgrove, L. Butyrate-induced apoptosis in HCT116 colorectal cancer cells includes induction of a cell stress response. J. Proteome Res. 2011, 10, 1860–1869. [Google Scholar] [CrossRef]
- Clarke, J.M.; Young, G.P.; Topping, D.L.; Bird, A.R.; Cobiac, L.; Scherer, B.L.; Winkler, J.G.; Lockett, T.J. Butyrate delivered by butyrylated starch increases distal colonic epithelial apoptosis in carcinogen-treated rats. Carcinogenesis 2012, 33, 197–202. [Google Scholar] [CrossRef]
- Krishnan, M.; Singh, A.B.; Smith, J.J.; Sharma, A.; Chen, X.; Eschrich, S.; Yeatman, T.J.; Beauchamp, R.D.; Dhawan, P. HDAC inhibitors regulate claudin-1 expression in colon cancer cells through modulation of mRNA stability. Oncogene 2010, 29, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Chen, Y.; Jiang, H.; Nie, D. The role of short-chain fatty acids in orchestrating two types of programmed cell death in colon cancer. Autophagy 2011, 7, 235–237. [Google Scholar] [CrossRef]
- Tang, Y.; Chen, Y.; Jiang, H.; Nie, D. Short-chain fatty acids induced autophagy serves as an adaptive strategy for retarding mitochondria-mediated apoptotic cell death. Cell Death Differ. 2011, 18, 602–618. [Google Scholar] [CrossRef]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Reviews. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, S.; Das, S.; Chakrabarti, J. Long noncoding RNAs: New players in the molecular mechanism for maintenance and differentiation of pluripotent stem cells. Stem Cells Dev. 2013, 22, 2240–2253. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Donaghey, J.; Carey, B.W.; Garber, M.; Grenier, J.K.; Munson, G.; Young, G.; Lucas, A.B.; Ach, R.; Bruhn, L.; et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011, 477, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [PubMed]
- Paralkar, V.R.; Weiss, M.J. Long noncoding RNAs in biology and hematopoiesis. Blood 2013, 121, 4842–4846. [Google Scholar] [CrossRef]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef]
- Chan, A.S.; Thorner, P.S.; Squire, J.A.; Zielenska, M. Identification of a novel gene NCRMS on chromosome 12q21 with differential expression between rhabdomyosarcoma subtypes. Oncogene 2002, 21, 3029–3037. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.Y.; Bogu, G.K.; Soh, B.S.; Stanton, L.W. The long noncoding RNA RMST interacts with SOX2 to regulate neurogenesis. Mol. Cell 2013, 51, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Uhde, C.W.; Vives, J.; Jaeger, I.; Li, M. Rmst is a novel marker for the mouse ventral mesencephalic floor plate and the anterior dorsal midline cells. PLoS ONE 2010, 5, e8641. [Google Scholar] [CrossRef] [PubMed]
- Mohan, N.; Shen, Y.; Dokmanovic, M.; Endo, Y.; Hirsch, D.S.; Wu, W.J. VPS34 regulates TSC1/TSC2 heterodimer to mediate RheB and mTORC1/S6K1 activation and cellular transformation. Oncotarget 2016, 7, 52239–52254. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Lakshmaiah, K.C.; Jacob, L.A.; Aparna, S.; Lokanatha, D.; Saldanha, S.C. Epigenetic therapy of cancer with histone deacetylase inhibitors. J. Cancer Res. Ther. 2014, 10, 469–478. [Google Scholar] [PubMed]
- Tang, Y.; Chen, Y.; Jiang, H.; Robbins, G.T.; Nie, D. G-protein-coupled receptor for short-chain fatty acids suppresses colon cancer. Int. J. Cancer 2011, 128, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Beermann, J.; Piccoli, M.T.; Viereck, J.; Thum, T. Non-coding RNAs in Development and Disease: Background, Mechanisms, and Therapeutic Approaches. Physiol. Rev. 2016, 96, 1297–1325. [Google Scholar] [CrossRef]
- Han, R.; Sun, Q.; Wu, J.; Zheng, P.; Zhao, G. Sodium Butyrate Upregulates miR-203 Expression to Exert Anti-Proliferation Effect on Colorectal Cancer Cells. Cell Physiol Biochem. 2016, 39, 1919–1929. [Google Scholar] [CrossRef]
- Hu, S.; Liu, L.; Chang, E.B.; Wang, J.Y.; Raufman, J.P. Butyrate inhibits pro-proliferative miR-92a by diminishing c-Myc-induced miR-17-92a cluster transcription in human colon cancer cells. Mol. Cancer 2015, 14, 180. [Google Scholar] [CrossRef]
- Malakar, P.; Shilo, A.; Mogilavsky, A.; Stein, I.; Pikarsky, E.; Nevo, Y.; Benyamini, H.; Elgavish, S.; Zong, X.; Prasanth, K.V.; et al. Long noncoding RNA MALAT1 promotes hepatocellular carcinoma development by SRSF1 up-regulation and mTOR activation. Cancer Res. 2016, 77, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Pawar, K.; Hanisch, C.; Palma Vera, S.E.; Einspanier, R.; Sharbati, S. Down regulated lncRNA MEG3 eliminates mycobacteria in macrophages via autophagy. Sci. Rep. 2016, 6, 19416. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Luo, J. Long Non-Coding RNA (lncRNA) Urothelial Carcinoma-Associated 1 (UCA1) Enhances Tamoxifen Resistance in Breast Cancer Cells via Inhibiting mTOR Signaling Pathway. Med. Sci. Monit. 2016, 22, 3860–3867. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Dibble, C.C.; Matsuzaki, M.; Manning, B.D. The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol. Cell. Biol. 2008, 28, 4104–4115. [Google Scholar] [CrossRef]
- Lee, J.S.; Kim, Y.J.; Kim, C.L.; Lee, G.M. Differential induction of autophagy in caspase-3/7 down-regulating and Bcl-2 overexpressing recombinant CHO cells subjected to sodium butyrate treatment. J. Biotechnol. 2012, 161, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, H.; Ochiai, K.; Suzuki, N.; Otsuka, K. Butyrate, a bacterial metabolite, induces apoptosis and autophagic cell death in gingival epithelial cells. J. Periodontal Res. 2010, 45, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef]
- Ng, S.Y.; Johnson, R.; Stanton, L.W. Human long non-coding RNAs promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. EMBO J. 2012, 31, 522–533. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target 1 | TGAAACTTGGAGCAAAGTGG(AGG) |
| Target 2 | GCTGAATATCTTCAGGAAAA(TGG) |
| Target 3 | TTTTTATTTTGCAGATTCTC(AGG) |
| Target 4 | CTATATTTTGTTAGAAAGGA(AGG) |
| Target 5 | ACTTAGCATTCCATCCGTGC(AGG) |
| Target 6 | CTTATTCTGAACTTTCAGAC(TGG) |
| Target 7 | AACCTCTATTGTATTCCAGT(TGG) |
| Target 8 | TGTCTGGCTTTCCTTGCAGT(GGG) |
| Target 9 | (CCT)GCTCGATGTTTGCATTTAAA |
| RMST | ||
| siRNA1 | sense | CGGAGACUUGGUAGUGAAAUUCUTC |
| antisense | GAAGAAUUUCACUACCAAGUCUCCGUG | |
| siRNA2 | sense | GUUUGAUUGACCUUUGUUAGGGUGA |
| antisense | UCACCCUAACAAAGGUCAAUCAAACUU | |
| siRNA3 | sense | CGAAUUAUUUGCAAUGCCAUUUAAT |
| antisense | AUUAAAUGGCAUUGCAAAUAAUUCGUU | |
| Name | Forward Primer | Reverse Primer |
|---|---|---|
| qPCR | CCCAGTGCCACCAACTGGGAT | CCAGCTGGCGGGTACGATCAG |
| gRMST1 | GGG AAA CAG AGG GAG CAC | GGT ATG GAA CAA GGC AGC |
| gRMST2 | ATC AAT GCC CCT ACT CAC | ACG CTG CTC TGC ATT TTC |
| gRMST3 | TAG CCC GGC TTT TAG GTC | ACC CGT CAG TTT CCA AGA |
| gRMST4 | AGC TCT GTC GTT CCA CCT | GTC ACT CCT CGG AAA GC |
| gRMST5 | CAG TAG TGC TTG GGA CTC | ACA GCT GAT GTC CCG TTT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Guo, Y.; Fang, X.; Zhang, Y.; Nie, D. Short-Chain Fatty Acids Suppress mTOR Signaling in Colon Cancer Cells via Long Non-Coding RNA RMST. Kinases Phosphatases 2024, 2, 136-150. https://doi.org/10.3390/kinasesphosphatases2020008
Wang J, Guo Y, Fang X, Zhang Y, Nie D. Short-Chain Fatty Acids Suppress mTOR Signaling in Colon Cancer Cells via Long Non-Coding RNA RMST. Kinases and Phosphatases. 2024; 2(2):136-150. https://doi.org/10.3390/kinasesphosphatases2020008
Chicago/Turabian StyleWang, Jiuhui, Yande Guo, Xiangwei Fang, Yuanqin Zhang, and Daotai Nie. 2024. "Short-Chain Fatty Acids Suppress mTOR Signaling in Colon Cancer Cells via Long Non-Coding RNA RMST" Kinases and Phosphatases 2, no. 2: 136-150. https://doi.org/10.3390/kinasesphosphatases2020008