
From Kinases to Diseases: Investigating the Role of AMPK in Human Pathologies

Instituto de Investigación Sanitaria del Principado de Asturias (ISPA), 33011 Oviedo, Spain
*
Author to whom correspondence should be addressed.
Kinases Phosphatases 2023, 1(3), 181-205; https://doi.org/10.3390/kinasesphosphatases1030012
Submission received: 18 June 2023
/
Revised: 7 July 2023
/
Accepted: 10 July 2023
/
Published: 1 August 2023
Abstract
:Adenosine Monophosphate-Activated Protein Kinase (AMPK) is the major conserved regulator of cellular metabolism in eukaryotic cells, from yeast to mammals. Given its pivotal role, it is not surprising that alterations in its function may contribute to the pathogenesis of numerous human diseases. Indeed, AMPK has become a promising therapeutic target for several pathologies. In this context, significant efforts have been dedicated to discovering new pharmacological agents capable of activating AMPK based on next-generation sequencing (NGS) technology and personalized medicine. Thanks to computational methodologies and high-throughput screening, the identification of small molecules and compounds with the potential to directly activate AMPK or modulate its intricate signaling network has become viable. However, the most widely used drug to activate AMPK in human patients is still metformin, which has shown promising results in the treatment of various diseases, such as type II diabetes, atherosclerosis, Alzheimer’s disease, Huntington’s disease, and several types of cancer. In this review, we present a comprehensive analysis of the involvement of AMPK in human pathology, emphasizing its significant potential as a therapeutic target.
1. Introduction
The substantial increase in human life expectancy in recent decades can be primarily attributed to remarkable advancements in the field of biomedicine. The accessibility and cost-effectiveness of next-generation sequencing (NGS), in conjunction with the development of personalized medicine and the discovery of novel pharmaceutical compounds, have served as pivotal factors in elevating both the quality of life and overall longevity of individuals [1]. Moreover, these advancements have facilitated a profound understanding of the molecular mechanisms involved in the development of several diseases, thereby presenting prospective therapeutic targets within the genetic landscape. In this context, from a cellular perspective, eukaryotic cells possess an extraordinary capacity to adapt to unfavorable alterations in the cellular environment, thus ensuring the preservation of homeostatic conditions [2].
Specifically, cells have developed molecular sensors that detect disruptions in cellular homeostasis. One of these fundamental sensors is Adenosine Monophosphate-Activated Protein Kinase (AMPK) which acts as a central hub, connecting diverse cellular functions and processes to energy availability [3]. From a molecular point of view, AMPK is a conserved protein kinase found in both unicellular organisms like baker’s yeast and more complex multicellular eukaryotes such as mammals. In this regard, it was described as the primary energy sensor in yeast with the identification of its yeast ortholog, SNF1 (Sucrose Non-fermenting 1), in 1981 [4].
Due to its role in the energy balance at cellular and organismal levels, it is not surprising that alterations in the AMPK system hold significant implications for human health, contributing to the development of several diseases, including atherosclerosis, diabetes, cancer, inflammatory alterations, neurodegenerative disorders, or viral infections, among others [5,6,7,8].
In this review, we describe the pivotal role of AMPK as the primary energy-sensing mechanism in eukaryotic cells, orchestrating the intricate regulation of diverse cellular processes, such as autophagy, lipid metabolism, mitochondrial biogenesis, and glucose metabolism, among others. Additionally, we will explore in depth the complex mechanisms by which disruptions in AMPK function can actively contribute to the initiation or progression of diverse human pathologies. Moreover, we will thoroughly analyze the potential of AMPK as a promising therapeutic target for combating these pathologies from molecular and integrative perspectives.
2. Structural Configuration and Mechanism of Activation
Given its function as the main energy sensor responsible for maintaining cellular homeostasis, it is unsurprising that AMPK has been extensively studied and characterized from an evolutionary perspective. In this regard, AMPK represents a highly conserved serine/threonine protein kinase that belongs to the AMPK-related kinase family, comprising thirteen kinases found in the human genome [9]. Within mammalian cells, AMPK takes the form of a heterotrimeric complex, consisting of a catalytic α subunit along with regulatory β and γ subunits. Each subunit is encoded by distinct genes, called PRKAA (5′-AMP-activated protein kinase catalytic subunit alpha) for the α subunit, PRKAB (5′-AMP-activated protein kinase subunit beta) for the β subunit, and PRKAG (5′-AMP-activated protein kinase subunit gamma) for the γ subunit [10].
Specifically, in humans, AMPK subunits are encoded by seven genes: PRKAA1 and PRKAA2 for the two isoforms of the α subunit (α1 and α2), PRKAB1 and PRKAB2 for the two isoforms of the β subunit (β1 and β2), and PRKAG1, PRKAG2, and PRKAG3 for the three isoforms of the γ subunit (γ1, γ2, and γ3), respectively. A functional AMPK complex comprises one α subunit, one β subunit, and one γ subunit, forming a total of twelve distinct combinations [11]. These conformations are associated with specific tissues, cell types, or subcellular locations, and they arise due to the ability to form different combinations of α, β, and γ subunits (Figure 1).
At the molecular level, the α-subunit of AMPK harbors a serine/threonine kinase domain at the N-terminal region where a residue Thr172 is situated. In fact, this residue presents a fundamental role in the enzymatic activity and regulation of AMPK. Phosphorylation of the conserved residue Thr172 by multiple upstream kinases represents the principal mechanism for the short-term regulation of AMPK activity. These kinases phosphorylate Thr172, leading to the modulation of cellular processes involved in energy homeostasis. By undergoing phosphorylation, Thr172 plays a crucial role in regulating the catalytic activity and overall functionality of AMPK in response to intracellular signals and energy status changes [12]. The unique characteristics of the Thr172 residue in AMPK highlight the existence of an intricate network of kinases responsible for regulating AMPK activity. This network encompasses multiple signaling pathways that can impact the phosphorylation status of Thr172 [9]. On the one hand, LKB1 (Liver kinase B1) has been identified as a kinase capable of phosphorylating Thr172 in response to a diverse range of signals [13]. On the other hand, another kinase known as CAMKK2 (Calcium/calmodulin-dependent protein kinase kinase 2) has been shown to phosphorylate Thr172 independently of LKB1, particularly in response to changes in calcium levels [14]. In addition, MAPKKK family member TAK1/MAP3K7 (Transforming growth factor beta-activated kinase 1)/(Mitogen-activated protein kinase 7) could phosphorylate AMPK in the position Thr172 too. The presence of different kinases, such as LKB1, CAMKK2, or TAK1/MAP3K7, underscores the complexity and versatility of the regulatory mechanisms involved in modulating AMPK activity [15]. The diverse signaling pathways associated with these kinases allow for the integration of various cellular signals and environmental signals, enabling AMPK to respond and adapt to different physiological and metabolic conditions.
In the context of AMPK regulation, the γ-subunit of AMPK functions as a sensor that enables the protein to respond to changes in AMP/ATP or ADP/ATP levels. In this sense, AMP binding followed by conformational changes allows the phosphorylation of Thr172. Specifically, the modulation of AMPK activity may occur through three distinct mechanisms. For example, AMP can inhibit the dephosphorylation of Thr172 by shielding it from the activity of phosphatases. Additionally, AMP promotes the phosphorylation of Thr172 by LKB1 and AMP also modulates AMPK through allosteric activation [16,17,18]. In fact, AMP can inhibit the dephosphorylation of Thr172 by shielding it from the activity of phosphatases. Additionally, AMP can also enhance AMPK activity once Thr172 is phosphorylated, exerting an allosteric effect. Therefore, the multifaceted mechanisms of AMP regulation, including phosphorylation, allosteric modulation, and the sensing role of the γ-subunit, collectively illustrate the intricate interplay between AMP, phosphorylation, and allosteric modulation. These mechanisms work in synergy to precisely adjust AMPK activity, allowing it to effectively respond to changes in cellular energy status, in which the phosphorylation status of Thr172 has an essential function as a critical switch in the activation of AMPK [18].
Despite the well-established understanding of the mechanisms involved in AMPK activation, there is limited knowledge about the processes that lead to the downregulation of AMPK activity. In this regard, a mechanism involving the phosphorylation of specific sites on AMPKα1 and AMPKα2 has been described, specifically at Ser487 (Ser485 in rodent) and Ser491 (equivalent to rodent Ser491) in humans, which inhibit AMPK activity [19,20,21]. From a molecular perspective, Akt plays a role in this inhibitory phosphorylation process. Specifically, in response to insulin or insulin-like growth factor (IGF-1), Akt phosphorylates Ser487 site on AMPKα1 in several cell types, including heart cells, adipocytes, or tumor cell lines, among others [22]. In fact, the phosphorylation of AMPKα1 Ser487 by Akt has been identified as a mechanism that inhibits the phosphorylation of Thr172, leading to a reduction in AMPK activity.
Additionally, in vitro studies have demonstrated that recombinant PKA (cAMP-dependent protein kinase) can also phosphorylate AMPKα1 Ser487. It has been reported that agents capable of increasing intracellular cAMP levels can stimulate the phosphorylation of AMPKα1 Ser487 in mouse embryonic fibroblasts and insulin-secreting cell lines [23]. Conversely, AMPKα2 Ser491 has been found to be a less favorable substrate for Akt phosphorylation in vitro. However, it has been reported that p70S6 kinase, downstream of Akt, is involved in the leptin-mediated phosphorylation of AMPKα2 Ser491 in the mouse hypothalamus and a neuronal cell line [24]. These findings contribute to understanding the intricate regulatory mechanisms involved in AMPK phosphorylation and highlight the role of various signaling pathways in modulating AMPK activity.
3. Metabolic Regulation
AMPK is responsible for triggering alternative metabolic pathways when there are variations in the main carbon or nitrogen sources, thereby enabling cellular metabolism to adapt to internal environmental fluctuations [25]. In higher eukaryotes, AMPK is activated in response to an increase in the ratios of AMP (adenosine monophosphate) to ATP (adenosine triphosphate) and/or ADP (adenosine diphosphate) to ATP. Once activated, AMPK maintains energy homeostasis through two complementary actions: Inhibiting energy-consuming anabolic processes and promoting energy-generating catabolic processes [26]. Due to its main role in cellular energy balance, AMPK activity is tightly regulated by multiple upstream regulators, ensuring that cellular metabolism is finely tuned to fluctuating parameters within the internal environment and dynamic changes in nutritional and energetic demands [27]. Moreover, AMPK signaling pathways participate in a wide range of physiological processes beyond their primary metabolic functions, encompassing cytoskeleton remodeling, transcriptional control, and the regulation of essential cellular processes, such as autophagy or apoptosis [28] (Figure 2).
3.1. Glucose Metabolism
AMPK activity contributes to ATP production by modulating several catabolic and anabolic pathways involved in glucose metabolism. In this regard, AMPK facilitates glucose uptake through its inhibitory phosphorylation of TBC1D1 (TBC domain family member 1) and TXNIP (thioredoxin-interacting protein). Specifically, TBC1D1 and TXNIP act as negative regulators of the translocation process for glucose transporters GLUT1 and GLUT4 [29,30]. Consequently, increased AMPK activity results in the enhanced presence of GLUT1 and GLUT4 transporters on the cell surface. Additionally, AMPK positively regulates glycolysis by phosphorylating PFKFB3 (6-phosphofructo-2-kinase/fructose-2,6-biphosphatase), promoting glucose metabolism [31]. In fact, AMPK also inhibits glycogen synthesis by phosphorylating several isoforms of GYS (glycogen synthase), preventing glucose conversion into glycogen [32]. However, AMPK is also involved in glycogen supercompensation regulation in skeletal muscle. In this sense, during prolonged physical exercise, sustained AMPK activation stimulates glycogen synthesis, particularly in skeletal muscle. This occurs due to increased glucose uptake, leading to intracellular accumulation of G6P (glucose 6 phosphate), which allosterically activates GYS, overriding the inhibitory effect of AMPK on this enzyme. In addition to its direct effects on specific enzymes, AMPK modulates glucose metabolism transcriptionally. For instance, during fasting or reduced glucose intake, gluconeogenesis is activated to maintain blood glucose levels [33]. Upon re-feeding, an elevation in insulin levels results in the phosphorylation of liver AMPK by LKB1 kinases, leading to transcriptional inhibition of key gluconeogenesis genes. This effect involves AMPK-dependent phosphorylation and nuclear exclusion of CRTC2 (cyclic-AMP-regulated transcriptional co-activator 2) and HDACs (class IIA histone deacetylases), essential for gluconeogenic gene transcription. Thereby, AMPK exerts influence on glucose metabolism through direct regulation of specific proteins and transcriptional control of key genes involved in glucose metabolism [34,35].
3.2. Lipid Metabolism
As previously mentioned, for glucose metabolism, AMPK has been related to lipid metabolism in a cellular context. Upon activation, AMPK functions to downregulate the activity of key enzymes involved in lipid synthesis and related processes, ensuring their activity is aligned with cellular energy levels. One important aspect of this regulation is the inhibitory phosphorylation of ACC1 (acetyl-CoA carboxylase 1) and ACC2 (acetyl-CoA carboxylase 2), which are key enzymes responsible for the initial step in lipid synthesis. In fact, AMPK exerts its phosphorylation on specific sites such as Ser79 in ACC1 and Ser221 in ACC2 to modulate lipid homeostasis [36].
Furthermore, AMPK can inhibit HMGCR (3-hydroxy-3-methylglutaryl-coenzyme A reductase), a crucial enzyme involved in cholesterol synthesis. On the other hand, AMPK promotes the breakdown of triglycerides into fatty acids by stimulating lipases such as ATGL (Adipocyte Triglyceride Lipase) and HSL (Hormone-Sensitive Lipase). In this regard, in cellular energy stress conditions, AMPK facilitates the import of free fatty acids into mitochondria for β-oxidation, a process that relies on the activity of acyl-transferases from the CPT1 (Carnitine palmitoyl-transferase 1) family [37,38]. AMPK indirectly regulates CPT1 activity by reducing the levels of malonyl-CoA, a potent inhibitor of CPT1, through the inhibition of ACC1 and ACC2 [39].
Despite its direct activity in lipid metabolism regulation, AMPK also controls several transcription factors specifically involved in lipid metabolic processes. In this sense, AMPK-mediated phosphorylation leads to the inhibition of several transcriptional factors, including ChREBP (carbohydrate-responsive element binding protein) [40], SREBP1 (sterol regulatory element binding protein 1), and HNF4α (hepatocyte nuclear factor 4α) [41], among others [42].
3.3. Autophagic Process
Autophagy, a highly conserved catabolic pathway found in all nucleated cells, is critical for maintaining cellular homeostasis. While basal autophagic degradation occurs constitutively, nutrient deprivation and energy scarcity serve as the primary physiological inducers of autophagy. In this context, autophagy is characterized by the formation of double-membrane vesicles known as autophagosomes, which sequester and enclose specific portions of the cytoplasm [43]. This sequestration process serves as a crucial quality control mechanism, ensuring a harmonious balance between macromolecule synthesis and degradation [44]. In mammalian cells, autophagic degradation is primarily regulated by two key kinases: mTOR (mechanistic/mammalian Target of Rapamycin) and AMPK. mTOR exists in two distinct protein complexes, named mTORC1 and mTORC2. While mTORC2, as far as we know, has a limited impact on autophagy dynamics, mTORC1 plays a dominant role as a suppressor of autophagy in mammalian cells [45]. Under conditions of high energy levels, abundant cellular amino acids, or stimulation by growth factors, mTORC1 is activated, leading to a negative effect on autophagic degradation.
In contrast, AMPK activation has a positive regulatory effect on autophagy, promoting its pro-catabolic functions. The activities of mTORC1 and AMPK are interconnected at the molecular level, with inhibition of mTORC1 being a key mechanism through which AMPK enhances autophagic degradation. In fact, this antagonistic relationship between mTORC1 and AMPK highlights their coordinated control over autophagy regulation [46]. From a molecular point of view, under conditions of abundance in energy, growth factors, or amino acids, mTORC1 inhibits autophagy by phosphorylating ATG13, leading to decreased activity of the ULK1 complex. This reduction in ULK1 activity impairs the formation of autophagosomes, thereby repressing the autophagic process [47]. Furthermore, mTORC1 directly targets ULK1 itself, providing an additional mechanism for inhibiting autophagy. In contrast, AMPK plays an opposing role to mTORC1 by promoting the activity of the ULK1 complex, positively regulating the initial stages of autophagosome formation [48]. In this context, AMPK directly phosphorylates specific sites on ULK1, such as Ser467, Ser555, Thr574, and Ser637. These phosphorylation events enhance the recruitment of autophagy-related proteins (ATG proteins) to membrane domains involved in autophagosome formation [49].
Additionally, AMPK exerts a negative regulation on mTORC1 activity, counteracting its inhibitory effect on ULK1 through two complementary mechanisms. Firstly, AMPK phosphorylates Thr1227 and Ser1345 residues on TSC2 (Tuberous sclerosis complex 2), leading to the assembly of the TSC1/TSC2 heterodimer and subsequent inhibition of mTORC1 activity. Secondly, AMPK directly phosphorylates Ser722 and Ser792 residues on RAPTOR, resulting in the inhibition of mTORC1. In fact, AMPK also plays a role in promoting autophagy by exerting differential effects on various components of autophagy-initiating protein complexes [50,51]. Through specific phosphorylation events, AMPK influences the regulation of autophagy at different levels. In this autophagic regulation context, the activities of AMPK and mTORC1 are antagonistic and function together to integrate autophagy regulation with multiple signaling pathways. Specifically, the ULK1 complex serves as a key checkpoint in the control of autophagy initiation, where AMPK and mTORC1 converge to modulate autophagic activity [52].
AMPK is involved in the regulation of the Class III PI3K complex too. In this regard, ULK1 exerts its pro-autophagic function by phosphorylating various components of this complex, including Beclin1, AMBRA1, and the catalytic subunit VPS34 [53]. Furthermore, AMPK can influence autophagic activity through the specific phosphorylation of ATG9, a transmembrane protein involved in autophagosome biogenesis, by supplying vesicles that contribute to autophagosome elongation. AMPK-mediated phosphorylation of ATG9 at Ser761 enhances the recruitment of ATG9A to LC3-positive autophagosomes, thereby promoting autophagosome formation [54].
Furthermore, the acetylation status of autophagy-associated proteins is regulated by signaling molecules that respond to cellular nutrients and growth factors. In this context, SIRT1 can be activated in an AMPK-dependent manner when glucose availability is limited [55]. In addition, SIRT1 and AMPK present a mutual reinforcement in their activity. For example, SIRT1 plays a role in deacetylating and thereby enhancing the stability and activity of LKB1 [56], whereas AMPK stimulates SIRT1 activity by increasing the expression of nicotinamide phosphoribosyltransferase (NAMPT), a crucial enzyme for the regeneration of NAD+, which serves as a substrate for SIRT1 [57,58]. From an autophagic perspective, SIRT1 and AMPK play a complementary role in promoting autophagy. On the one hand, SIRT1 contributes to this process by deacetylating multiple factors, which, in their deacetylated state, actively participate in the formation of autophagosomes. On the other hand, SIRT1 also facilitates autophagosome/lysosome fusion by deacetylating the FOXO1 transcription factor, which promotes the transcription of the gene encoding Rab7, a crucial G protein involved in this fusion process [59,60].
Despite its direct activity in autophagic process regulation, AMPK also regulates different transcription factors involved in autophagy. For example, AMPK and mTOR present an antagonistic role in the context of TFEB/TFE transcription factors, which are responsible for controlling the expression of genes involved in lysosomal biogenesis and autophagy [61]. During high energy conditions, mTOR phosphorylates these transcription factors, inhibiting their function. However, it has been shown that the nuclear translocation of TFEB/TFE is significantly reduced in cells lacking AMPK or treated with AMPK inhibitors. This suggests that AMPK is necessary for promoting the nuclear translocation and activity of TFEB/TFE, thereby facilitating the expression of genes involved in lysosomal function and autophagy [62].
3.4. Mitochondrial Biogenesis
AMPK plays a crucial role in maintaining mitochondrial function, particularly in response to energy imbalances. In this sense, during disruptions in energy levels, AMPK can promote mitochondrial biogenesis, increasing mitochondrial mass and promoting the expression of mitochondrial genes. The main genetic regulator in this process is PGC1-α, a transcription factor that controls the expression of a wide range of mitochondrial genes [63]. In fact, PGC1-α interacts with PPAR-γ and ERRs to regulate mitochondrial gene expression, and its activity is tightly regulated by post-translational modifications. From a tissular perspective, for example, in muscle cells, overexpression of PGC1-α leads to the conversion of type IIb fibers into type I and type II characterized by higher mitochondrial content. In the context of PGC1-α regulation, AMPK contributes by direct phosphorylation of PGC1-α in Thr177 and Ser538 residues [64]. On the other hand, AMPK indirectly influences PGC1-α by phosphorylating HDAC5, SIRT1, and p38 MAPK, which further regulate PGC1-α function [65,66,67,68]. As previously mentioned, AMPK also promotes the activity of TFEB, which, in the context of mitochondrial biogenesis, activates the gene encoding PGC1-α (PPARGC1A) along with other genes involved in autophagy [69].
3.5. Selective Degradation of Mitochondria by Autophagy
In addition to its general regulatory role in autophagic degradation, AMPK also plays a specific role in the regulation of mitophagy, which is the selective removal of damaged mitochondria through the process of autophagy [70]. Mitochondria exist in a dynamic network that undergoes morphological changes through a combination of fission (division) and fusion (merging). Consequently, mitochondria can exhibit various distributions, ranging from a single interconnected network to numerous small, fragmented units. In fact, an increase in mitochondrial fission is necessary to facilitate mitophagy [71]. This process makes mitochondria more susceptible to being engulfed by pre-autophagosomal isolation membranes, thus enabling the occurrence of mitophagy. Furthermore, it has been observed that several mitochondrial stressors cause damage to mitochondria and enhance mitochondrial fission. In this regard, Activation of AMPK plays a crucial role in promoting mitochondrial fission, thereby linking mitochondrial dynamics to mitophagic degradation [72]. From a molecular point of view, AMPK phosphorylates and activates a protein called mitochondrial fission factor (MFF). MFF is located in the outer membrane of mitochondria and acts as a recruiter for cytoplasmic dynamin 1-like protein (DRP1), guiding it to the outer membrane of the mitochondria. This specific recruitment of DRP1 to the mitochondrial outer membrane increases mitochondrial fission, leading to the formation of fragmented mitochondria that are then targeted for mitophagy [73].
On the other hand, SIRT1 also plays a role in promoting mitophagy by increasing the expression levels of Parkin and Pink1, proteins that tag dysfunctional mitochondria for autophagic degradation [74]. Moreover, SIRT1 and AMPK not only collaborate to promote mitophagy but also work together to stimulate mitochondrial biogenesis by activating post-translational modifications of PPAR and PGC-1α [75,76]. In this regard, it has been demonstrated that the phosphorylation of ULK1 on Ser555 by AMPK is also crucial for the induction of mitophagy [77].
By coordinating these actions, AMPK enables the formation of autophagosomes and regulates mitochondrial size, thus facilitating efficient autophagic degradation of mitochondria. In fact, this integrated regulation ensures the effective removal of dysfunctional mitochondria and maintenance of cellular homeostasis.
4. Role of AMPK in Human Pathologies
Due to its pivotal role as a metabolic sensor for maintaining homeostasis, any perturbation in the function of AMPK can result in genetic dysregulation that impacts diverse processes, including autophagy, glucose metabolism, lipid metabolism, and mitochondrial biogenesis [78]. These disruptions potentially contribute to the development of pathological conditions. Moreover, advancements in sequencing technologies and genome-wide association studies (GWAS) have revealed that polymorphic variations in different autophagic genes, including AMPK subunits, are directly linked to a range of human diseases (Table 1) [79]. In this regard, the study and comprehension of AMPK modifications have identified it as a possible therapeutic target to address several human pathologies.
4.1. Type 2 Diabetes
Given the critical involvement of AMPK in the regulation of glucose metabolism, it is not surprising that alterations in AMPK have been found to be associated with the development of diabetes [91]. The prevalence of metabolic syndrome disorders, including diabetes, hypertension, and fatty liver, tends to increase with age on a global scale. In 2019, it was estimated that 463 million individuals were living with diabetes worldwide [92]. The relationship between diabetes development and AMPK dysregulation further highlights the significance of AMPK in maintaining proper glucose metabolism [93]. In this regard, type 2 diabetes, formerly known as noninsulin-dependent diabetes mellitus or adult-onset diabetes, is a metabolic disorder characterized by dysregulated glucose levels in the blood and abnormal metabolism of lipids. This condition occurs when the β cells in the pancreas, responsible for producing insulin, are unable to compensate for insulin resistance [94]. Insulin resistance is closely associated with elevated levels of triacylglycerols, particularly in the liver and skeletal muscle. It is widely recognized that individuals with a genetic predisposition are most susceptible to developing type 2 diabetes as a result of overnutrition, sedentary lifestyle, and consequent obesity. In fact, to effectively manage this disease, regular physical exercise and a suitable dietary regimen are considered fundamental measures [95].
Elevated blood glucose levels pose a significant challenge in the management of diabetes mellitus (DM). Thus, interventions that effectively lower blood glucose levels have the potential to improve DM outcomes. In this context, AMPK activation leads to an increase in GLUT4 expression, facilitating glucose uptake by muscle cells [96]. Furthermore, AMPK signaling also influences GLUT1, another key regulator of glucose uptake. GLUT1 modulation holds promise as a therapeutic target for DM, as downregulation of GLUT1 has been shown to alleviate diabetic retinopathy [97]. Consequently, targeting AMPK signaling and the glucose transporters situated on the cell membrane represents a viable target to enhance glucose uptake and ameliorate DM.
From a therapeutic perspective, given that skeletal muscle, liver, and adipose tissue are insulin-resistant peripheral tissues, chronic administration of metformin to activate AMPK would enhance insulin-mediated glucose uptake in muscle [98]. However, there is no evidence supporting this assertion in in vivo studies, as mice with AMPKβ1β2 knockout in muscle or mice overexpressing kinase-dead (KD) AMPKα2 in muscle exhibit normal insulin-stimulated glucose transport [99,100].
Furthermore, adipose tissue functions as the primary reservoir for plasma-free fatty acids (FFAs). The excessive accumulation of FFAs in skeletal muscle, liver, and adipocytes produces diacylglycerol (DAG) accumulation and the activation of protein kinase C (PKC) that can lead to insulin resistance [101]. Activation of AMPK in adipocytes inhibits lipogenesis by increasing phosphorylation of ACC and reducing the expression of lipogenic genes regulated by the transcription factor SREBP-1c, including stearoyl-CoA desaturase 1 (SCD1), fatty acid synthase (FAS), and ACC1 [102]. By stimulating fatty acid oxidation and suppressing fatty acid and triglyceride synthesis, interventions that activate AMPK are anticipated to reduce lipid accumulation in the liver and skeletal muscle, thereby improving insulin sensitivity. Additionally, these interventions are predicted to directly alleviate hyperglycemia associated with type 2 diabetes by promoting glucose uptake in skeletal muscle and inhibiting gluconeogenesis in the liver [103]. In this regard, the administration of AICAR, an adenosine analog that enters cells through adenosine transporters and is converted into the equivalent nucleotide called ZMP, which emulates all the activating effects of AMP on AMPK, has been shown to reverse various metabolic abnormalities in animal models of obesity and insulin resistance, including fa/fa rats, ob/ob mice, and rats fed a high-fat diet [104,105]. In addition, metformin indirectly activates AMPK both in cells and in vivo by inhibiting the mitochondrial respiratory chain. However, due to the indirect nature of AMPK activation, it was necessary to specifically analyze its effects on AMPK activity. To address this, studies using double knock-in (DKI) mice, in which the endogenous genes encoding acetyl-CoA carboxylase (ACC1 and ACC2) were replaced with genes carrying single amino acid substitutions that eliminate the AMPK sites, have provided valuable insights. In fact, these mice exhibited elevated ACC1/ACC2 activities and cellular malonyl-CoA levels in the liver, accompanied by reduced fatty acid oxidation [36]. Consequently, they displayed increased levels of hepatic di- and triacylglycerols and increased PKCε activity. Moreover, the mice showed elevated fasting glucose and insulin levels, glucose intolerance, insulin resistance, and reduced effectiveness of insulin in suppressing hepatic glucose production. Following long-term (6 weeks) treatment with metformin, the improvements in metabolic parameters such as fasting glucose, insulin suppression of hepatic glucose production, and expression of gluconeogenic enzymes observed in fat-fed wild-type animals were no longer evident in DKI mice. Therefore, these findings suggest that the insulin-sensitizing effects of metformin can primarily be attributed to its impact on fatty acid metabolism, mediated by AMPK.
Furthermore, type 2 diabetes is characterized by a reduction in the functional β cell population within the islets, which coincides with dysregulated glucose-induced insulin secretion. In this context, targeted inhibition of LKB1 expression in pancreatic β cells led to a significant enhancement in insulin secretion in response to glucose and improved glucose tolerance [106]. These effects were accompanied by notable alterations in β cell mass and polarity in vivo. Moreover, it has been found that LKB1 and AMPK may play distinct roles in the regulation of insulin secretion by islet β cells, with AMPK activation demonstrating beneficial effects on β cell function [107]. For example, the administration of AICAR has been shown to protect against the glucolipotoxicity-induced impairment of β-cell function [108].
4.2. Cardiovascular Disease
Cardiovascular disease (CVD) is a leading cause of death worldwide, especially among individuals over the age of 65, highlighting its association with aging [109]. During the normal aging process, the cardiac tissue undergoes remodeling, characterized by a gradual reduction in left ventricular mass and end-diastolic volume. However, in pathological conditions such as CVD, the left ventricle undergoes remodeling that leads to increased mass, often accompanied by cardiac hypertrophy [110].
The maintenance of cardiac health depends on various factors, including the activity of autophagy, which presents an important role in both the development of cardiac tissue and the preservation of cardiomyocyte homeostasis [111]. Autophagy, through the degradation of cellular components, contributes to the periodic renewal of cytosolic content, thereby ensuring cellular homeostasis. In CVD, dysregulation of cardiac homeostasis results in cardiomyocyte loss and an accumulation of extracellular matrix [112].
In the pathology context, a mouse model lacking AMPK subunits β1 and β2 (β1β2M-KO) exhibits dilated cardiomyopathy [113]. Cardiomyopathy can manifest as hypertrophy of the heart muscle, which, if untreated, can contribute to heart failure. In different animal models of myocardial hypertrophy, AMPK activation has shown beneficial effects. It increases the ejection fraction (EF), which is a measure of the heart’s pumping efficiency, and decreases the protein synthesis rate in myocardial cells. These effects help to counteract the detrimental effects of hypertrophy and improve cardiac function [114].
On the one hand, in the complex genetic pathway involved in the development of cardiomyopathies, it has been observed that the expression levels of SIRT2 are downregulated in hypertrophic hearts. In fact, knockout of SIRT2 exacerbates cardiac hypertrophy and fibrosis and impairs cardiac function in aged mice. On the other hand, overexpression of SIRT2, specifically in the heart, protects against cardiac hypertrophy and preserves cardiac function. From a genetic perspective, SIRT2 plays a crucial role in maintaining the activity of AMPK in the context of hypertrophic hearts [115]. This maintenance is accomplished through the involvement of LKB1. Additionally, AMPK inhibits myocardial hypertrophy by regulating myocardial energy metabolism through the SIRT1 signaling pathway.
On the other hand, an alteration in the production or availability of endothelium-derived nitric oxide (NO) leads to an impaired vasodilator response and the development of a prothrombotic and pro-inflammatory endothelium, ultimately resulting in endothelial dysfunction. AMPK plays a crucial role in maintaining vascular health by exerting beneficial effects on various aspects of vascular function. It promotes the production of NO and facilitates vascular smooth muscle (VSM) relaxation. Additionally, AMPK suppresses inflammation, reduces ROS production, and boosts antioxidant defenses within the vasculature. In this regard, high-fat diet-induced downregulation of the AMPK/PI3K/Akt/eNOS pathway and contributes to endothelial dysfunction in a rat model of diet-induced obesity [116]. Specifically, the loss of AMPKα1 in VSMCs promotes atherosclerotic calcification in vivo and endothelial cells from AMPKα2 knockout mice exhibit abnormal ER stress [117,118].
In the context of myocarditis, a cardiovascular disease that can progress to dilated cardiomyopathy (DCMI), using cardiac tissue samples from both younger and older patients with DCMI, it has been found that male DCMI patients present an increase in both the expression and phosphorylation of AMPK in the cardiac tissue [119]. However, there are no significant alterations in the expression of SIRT1 compared to control groups. According to the essential role of AMPK in regulating mitochondrial homeostasis, the upregulation of AMPK could have an impact on mitochondrial biology. In this sense, the reduction in mitochondrial mass observed in these patients could be attributed to an enhancement in mitochondrial clearance through autophagy, possibly driven by the heightened activity of AMPK [120].
As mentioned previously, AMPK plays an important role in maintaining normal structure and function of the atria due to the observed aberrant alterations in cardiomyocyte-specific AMPKβ1/β2 knockout mice, which exhibit significant remodeling of the left atria and spontaneous occurrence of atrial fibrillation (AF) [113]. In the specific context of heart failure with preserved ejection fraction (HFpEF), it has been found that the activity of AMPK is significantly reduced in the left atrial tissue of HFpEF mice, as indicated by decreased phosphorylation of AMPK. However, when HFpEF mice were treated with metformin, a significant increase in AMPK signaling was observed. This increase in AMPK signaling was associated with a reduction in the preponderance of AF, as evidenced by decreased inducibility and duration of AF episodes [121].
Furthermore, in cardiac tissue from HF patients and transverse aortic constriction (TAC)-induced mice, an isoform shift from AMPKα2 to AMPKα1 and a decrease in mitophagy and mitochondrial function have been described. However, overexpression of AMPKα2 in mouse hearts prevents the development of TAC-induced chronic HF, leading to increased mitophagy pathways and improved mitochondrial function. Conversely, AMPKα2−/− mutant mice showed aggravation of early progression of TAC-induced HF mediated by decreased cardiac mitophagy [122].
Therefore, induction of autophagy mediated by AMPK could serve as a potential therapeutic approach in different cardiac pathologies. By promoting autophagy, AMPK activation aims to preserve cellular homeostasis, ultimately contributing to the restoration and maintenance of proper cardiac function [123].
4.3. Atherosclerosis
Atherosclerosis is a genetically predisposed inflammatory disorder affecting elastic and musculoelastic arteries, characterized by the development of atheromatous plaques primarily composed of cholesterol [124]. These plaques can lead to arterial stenosis or thrombosis, particularly in cases where the plaque exhibits instability. This disease affects individuals as they age, and its associated complications, particularly coronary heart disease and stroke, are the leading causes of mortality in developed nations [125]. The main complications, coronary heart disease and stroke, are the predominant etiologies of mortality in developed countries. AMPK modulates atherosclerosis through its influence on macrophage cholesterol homeostasis, vascular dysfunction, and inflammation [126].
Vascular dysfunction, recognized as an early stage of atherosclerosis, arises primarily from immune cell infiltration into the vascular wall, inflammation, oxidative stress, impaired nitric oxide (NO) bioavailability, and endothelial cell apoptosis. These factors often coincide with a decrease in AMPK activity in the endothelium of the aorta [127]. Thereby, activating AMPK in vascular tissues may represent an effective approach to preserving cardiovascular health [8]. In fact, AMPK in cultured human aortic endothelial cells inhibits TNFα-stimulated leukocyte adhesion, accompanied by reduced secretion of monocyte chemotactic protein 1 (MCP-1) [128]. In this sense, activation of AMPK by adiponectin effectively counteracts palmitate-induced production of reactive oxygen species (ROS) and subsequent apoptosis mediated by p38 mitogen-activated protein kinase in endothelial cells.
Furthermore, the perturbation of macrophage cholesterol homeostasis plays a substantial role in the advancement of atherosclerosis through the process of monocyte infiltration during plaque formation [129]. This infiltration event induces the release of a variety of pro-inflammatory mediators and chemokines, ultimately leading to the generation of atherogenic foam cells due to the excessive uptake of modified low-density lipoprotein (LDL) particles, which further aggravates the progression of atherosclerosis. However, AMPK has demonstrated its ability to inhibit cholesterol buildup within macrophages by enhancing the efflux of cholesterol to high-density lipoprotein (HDL). This mechanism leads to a significant reduction in atherosclerotic plaque formation in ApoE-/- mice [130]. The favorable impact of AMPK is likely attributed to the upregulation of ATP-binding cassette sub-family G member 1 (ABCG1) and ATP-binding cassette transporter A1 (ABCA1) gene expression, accompanied by an increase in the expression of liver X receptor α (LXRα) among others [131].
On the other hand, AMPK could be involved in the release of inflammatory cytokines in macrophages, as reduced AMPK activity has been observed in lipopolysaccharide (LPS)-stimulated macrophages. In this context, activation of AMPK in macrophages through pharmacological agonists has been shown to attenuate LPS-induced pro-inflammatory factors while increasing the levels of anti-inflammatory cytokines [132]. From a molecular point of view, this effect may be mediated by the reduced acetylation and transcriptional activity of NF-κB, induced by SIRT1. In addition, AMPK has been shown to mediate the activation of the nucleotide-binding domain and leucine-rich repeat-containing protein 3 (NLRP3) inflammasome, which plays a significant role in both inflammation induced by saturated fatty acids in macrophages and the anti-inflammatory effects of monounsaturated fatty acids [133]. In the atherosclerosis context, NLRP3 inflammasome can be triggered by crystalline cholesterol, an endogenous factor and risk that contributes to the progression of the disease [134].
Moreover, metformin has been shown to alleviate oxidative stress and restore endothelial function through the activation of the AMPK/peroxisome proliferator-activated receptor δ (PPARδ) pathway. These findings highlight the potential of targeting AMPK signaling as a therapeutic strategy for mitigating vascular dysfunction and promoting cardiovascular health [135].
4.4. AMPK in Inflammatory Diseases
Inflammatory processes have been extensively linked to various diseases, such as diabetes, cancer, arthritis, and cardiovascular disorders. The initiation of inflammation occurs when the immune system, comprising bone marrow-derived cells like monocytes and macrophages, as well as non-bone marrow-derived cells, detects the presence of infection or tissue damage, serving as the body’s initial defensive response [136]. In this context, emerging evidence suggests AMPK possesses anti-inflammatory properties, and these effects may be mediated through its metabolic activities [137]. In resting immune cells, such as dendritic cells, neutrophils, and T cells (among others), oxidative metabolism is the primary means of ATP generation. However, upon activation, these cells undergo a metabolic shift towards aerobic glycolysis, similar to the metabolic alterations observed in tumor cells. In this regard, this metabolic switch in dendritic cells is associated with reduced AMPK activation, which can be inhibited by pharmacological activation of AMPK and promoted by downregulation of AMPK. Similar patterns are observed in macrophages, where classically activated (M1) macrophages involved in pro-inflammatory processes predominantly utilize aerobic glycolysis, while alternatively activated (M2) macrophages associated with inflammation resolution tend to rely on oxidative metabolism [138].
In fact, it has been shown that when using macrophages that present a downregulation in AMPK, AMPK normally attenuates the production of inflammatory cytokines. In this sense, AMPK-β1 knockout mice models present alterations in macrophages homeostasis due to a reduction in the phosphorylation of ACC, decreased mitochondrial content, and impaired fatty acid oxidation rates, leading to the accumulation of pro-inflammatory diacylglycerols [139].
From a therapeutic perspective, activation of AMPK using A-769662 exhibited an enhanced capacity for fatty acid oxidation in macrophages of wild-type phenotype, whereas macrophages lacking β1 subunit did not display a similar response [139]. Moreover, the suppressive effects of A-769662 on inflammation were compromised in the presence of blocked fatty acid oxidation. Thereby, AMPK’s anti-inflammatory mechanism could operate through its impact on fatty acid oxidation. In addition, direct activation of AMPK in the context of inflammation could be by salicylate, a natural compound that served as the precursor for the synthesis of aspirin (acetyl salicylate). Both salicylate and A-769662 exhibit binding affinity for the identical site on AMPK, acting as selective activators specifically targeting complexes that contain β1 receptors [6]. Moreover, both compounds demonstrated the ability to stimulate an increase in systemic fatty acid oxidation in mice with intact β1 receptors, whereas their impact on fat oxidation was absent in mice lacking β1 receptors [140].
4.5. AMPK in Neurodegenerative Disorders
Currently incurable diseases, such as Parkinson’s disease (PD), Huntington’s disease (HD), and Alzheimer’s disease (AD), exemplify well-known instances of neurodegenerative disorders. These pathologies are characterized by progressive degeneration of neuronal structure and function [141]. In fact, PD is predominantly recognized as a neurodegenerative condition primarily affecting motor function. The pathology of PD involves the depletion of dopaminergic neurons in the substantia nigra and the formation of aggregates of α-synuclein, known as Lewy bodies [142]. HD, on the other hand, is an age-related disorder that affects both movement and cognitive abilities. It arises from the expansion of CAG triplet repeats in exon 1 of the Htt gene [143]. The principal pathological change observed in HD is a pronounced reduction in the number of neurons involved in the synthesis of enkephalin and γ-aminobutyric acid (GABA). In addition, AD is the most prevalent form of dementia, typified by the presence of senile plaques and intracellular neurofibrillary tangles, primarily resulting from the accumulation of misfolded Aβ peptides and hyperphosphorylated tau in cortical and hippocampal regions of the brain [144]. It has been elucidated that the inability to effectively counteract the accumulation of misfolded proteins prone to aggregation impairs cellular viability and triggers progressive deterioration in central nervous system function. This phenomenon, observed across various neurodegenerative disorders, is closely associated with a dysfunction in the autophagic degradation of mitochondria, which is frequently compromised in these conditions. In the specific context of brain-specific AMPK activation, conflicting reports have emerged regarding the diverse roles of AMPK in either facilitating or mitigating the progression of neurodegeneration, depending on the specific circumstances [145,146].
In AD, the role of AMPK as a crucial regulator of Aβ generation has been well-established. In this regard, pharmacological activation of AMPK, leading to the induction of autophagy, has been shown to effectively prevent the accumulation of Aβ [147]. Moreover, the activation of AMPK has been associated with the impact of leptin on neuronal cells, including the reduction of tau phosphorylation, which is implicated in the formation of neurofibrillary tangles [148]. However, the effects of AMPK activation on neuronal cells may exhibit gender-dependent variations. In this context, chronic administration of metformin has been reported to exert beneficial effects, specifically in females, whereas in males, administration of metformin may lead to exacerbation of memory dysfunction [149].
On the other hand, in PD, AMPK activation has shown potential benefits in patients due to its interaction with parkin, a key regulator involved in maintaining mitochondrial homeostasis. This cooperative relationship between AMPK and parkin suggests a potential therapeutic role for AMPK activation in PD [150]. However, it has also been reported that poly(ADP-ribose) polymerase (PARP) activation can trigger the degeneration of dopaminergic neurons by activating AMPK. This additional role of AMPK in PD, mediated by PARP, adds complexity to the understanding of the several functions of AMPK in the context of this neurodegenerative disorder [151].
In addition, in a pathological context of HD, it has been demonstrated that the activation of AMPK by several pharmacological compounds, such as AICAR in striatal neurons, can promote neuronal loss and the formation of aggregates containing mutant huntingtin (Htt) protein [152]. However, the administration of metformin has shown promise as a potential protective intervention in HD [153]. The conflicting results observed in different studies could be elucidated by taking into account the timing of AMPK activation. It is postulated that AMPK activation may confer beneficial effects, particularly during the early stages of HD. This suggests that the therapeutic efficacy of AMPK activation may be contingent upon the stage of disease progression in HD.
4.6. Glaucoma
Glaucoma, an optic neuropathy disease characterized by the progressive apoptotic death of retinal ganglion cells (RGCs), poses a significant threat to vision as it damages the optic nerve irreversibly if left untreated [154]. This condition is marked by optic disc cupping, loss of axons, and degeneration of RGC bodies. In fact, several factors, such as elevated intraocular pressure, vascular abnormalities, oxidative damage, ischemia, excitotoxicity, and inflammation, have been found to contribute to the pathogenesis of glaucoma [155]. Phosphorylated AMPK levels have been found to increase in the optic nerves of glaucoma patients. This AMPK hyperactivation has been associated with synaptic elimination and dendrite retraction in RGCs [156]. Furthermore, increased deposition of extracellular matrix (ECM) components such as fibronectin, collagen, and matrix metalloproteinases have been observed in the trabecular meshwork cells of mammalian glaucoma models and the aqueous humor of patients with open-angle glaucoma [157,158]. In this regard, pharmacological activation of AMPK by AICAR treatment has been shown to downregulate cytoskeletal and ECM proteins in primary human trabecular meshwork cells by phosphorylating RhoA at Ser188 [159]. In addition, ocular hypertension, which is characterized by increased intraocular pressure, also triggers AMPK activation and contributes to RGC loss [160]. Furthermore, the spatiotemporal expression of MAPKs in the retina and optic nerve has also been implicated in the pathogenesis of glaucoma with increased intraocular pressure.
4.7. Hepatitis C Infection
The Hepatitis C virus (HCV) is responsible for causing acute and chronic hepatitis. The global burden of HCV infection is substantial, with over 170 million individuals diagnosed with HCV worldwide. Among them, approximately 71 million individuals experience the chronic form of the disease. In addition, the prevalence of HCV infection varies across different regions, with a rate of 1.5% in the European Union. In fact, the Eastern Mediterranean region exhibits the highest prevalence, with a rate of 2.3%. From a mechanistic point of view, it has been estimated that an infected hepatocyte can generate approximately 50 viral particles per day [161]. Considering that viral replication requires the synthesis of viral proteins, lipids, and RNA, it is plausible to assume that this process leads to heightened ATP turnover and subsequent activation of AMPK, which has the potential to hinder ongoing viral replication [162]. However, surprisingly, in a cell culture model from HCV infection, AMPK activation is downregulated. In this regard, this downregulation could be mediated by the viral protein NS5A, which interacts with and activates phosphoinositide 3-kinase (PI3K), leading to the activation of the PKB/Akt pathway [163]. The activation of this pathway promotes protein and lipid synthesis and cell survival but concurrently inhibits AMPK activity by phosphorylating AMPK-α1 at Ser487 within the ST loop.
4.8. Cancer
Due to the role of AMPK in regulating cell proliferation by modulating cell energy, AMPK activation has emerged as a promising therapeutic target for various types of cancer [164]. The initial link between AMPK and cancer was established when it was discovered that LKB1 serves as the upstream kinase responsible for activating AMPK in response to energy stress and certain drugs like biguanides. LKB1 is described as a classical tumor suppressor, prompting investigations into whether its tumor-suppressive effects are mediated through AMPK [16,165]. LKB1 is also involved in phosphorylating and activating a family of twelve AMPK-related kinases that possess kinase domains closely related to AMPK. While one or more of these kinases may contribute to the tumor-suppressor properties of LKB1, AMPK remains the most plausible candidate due to its ability to inhibit mTORC1 and nearly all biosynthetic pathways required for cell growth, as well as its capacity to induce cell cycle arrest [50]. Phenotypically, AMPK can act as a tumor suppressor by impeding pro-tumorigenic metabolic processes and directly triggering cell cycle arrest in cancer cells. In this sense, AMPK activation initiates multiple mechanisms of cell death, influencing cell cycle checkpoints, autophagy, mitophagy, and apoptosis [166]. As mentioned earlier, AMPK promotes autophagy and mitophagy while initiating the apoptotic program through the activation of p53, p21, and p27. Furthermore, it induces cell cycle arrest by inhibiting HUR and concurrently activating Cyclins A, B1, and D1 [167].
In this regard, several studies have provided evidence for the anti-cancer effects of AMPK activation. For example, the treatment of hepatocellular carcinoma cells with AMPK activators, such as AICAR and metformin, has been shown to significantly inhibit cell proliferation and induce cell cycle arrest at the G1-S phase [168]. In fact, AMPK activation promotes autophagic and apoptotic cell death through the AMPK/JNK signaling pathway. In addition, the tumor-suppressor role of AMPK-α1 has been demonstrated by employing T-cell-specific knockouts of the PTEN and PRKAA1 genes, which encode AMPK-α1. In these models, the absence of PTEN and PRKAA1 genes led to early-onset lymphoma development, with significantly more aggressive tumors [169].
From a genetic perspective in the context of human cancers, biallelic loss-of-function mutations in LKB1 are frequently observed, occurring in up to 30% of non-small cell lung cancers, 20% of cervical cancers, and 10% of melanomas [170,171]. Loss of LKB1 function results in the failure of AMPK activation during energy stress. Furthermore, mutations in the subunits of AMPK itself appear to be less common in human cancer, possibly because each subunit, unlike LKB1, is encoded by multiple genes. However, downregulation of AMPK-α2 expression is relatively frequent in hepatocellular carcinoma and is associated with a poor prognosis [172].
Despite the evidence supporting the tumor-suppressing function of AMPK, there is ongoing debate regarding its potential pro-tumorigenic and pro-neoplastic properties. In this context, the tumor-suppressive effects of AMPK may be counteracted by the presence of intracellular stress or oncogenic signals within malignant cancer cells. These factors can disrupt the normal functioning of AMPK and impair its ability to inhibit tumor growth and progression [173]. However, the activation of AMPK in response to stress conditions such as glucose depletion or hypoxia can confer increased resistance to metabolic stress in tumor cells. For example, in the context of glioblastoma development, elevated levels of activated AMPK have been observed, and inhibiting AMPK has been shown to significantly decrease the growth rate of tumor cells. This suggests that AMPK plays a role not only in regulating ATP levels but also in modulating cell replication [174]. In this regard, AMPK is necessary for increased mitochondrial biogenesis in response to glucose limitation. However, studies propose that under conditions of glucose limitation, cancer cells achieve metabolic homeostasis and adapt to metabolic stress through the activation of the AMPK-p38-PGC-1α axis [175]. Moreover, under nutrient-starved conditions, AMPK may exhibit pro-tumorigenic effects and support tumor survival, whereas, in the presence of sufficient nutrients, AMPK displays tumor-suppressing effects [176]. Furthermore, AMPK-mediated autophagy provides a survival advantage to cancer cells by promoting cell growth and survival through the provision of metabolic substrates required for biosynthesis, meeting the metabolic demands of rapidly proliferating cancer cells. While mTORC1 inhibition prevents protein synthesis and cell proliferation, it has been suggested that mTORC2 may activate the PI3K-Akt signaling pathway, thereby promoting tumor cell survival [177].
The complex interplay between AMPK and cancer cells highlights the multifaceted nature of AMPK’s role in tumorigenesis. Its effects can vary depending on the specific metabolic and nutrient conditions within the tumor microenvironment. Moreover, AMPK-mediated autophagy and its interactions with other signaling pathways contribute to the intricate balance between tumor suppression and tumor promotion. Further research is needed to elucidate the precise mechanisms underlying these dual effects and their implications for cancer therapy [164].
5. Conclusions
AMPK acts as the main conserved regulator of cellular metabolism across several organisms, from yeast to mammals. Due to this reason, it is not surprising that disruptions in its function can contribute to the pathogenesis of several human diseases (Figure 3). In fact, AMPK has emerged as a promising therapeutic target for different human pathologies. In this context, with the advancements in NGS technology and the progress of personalized medicine, considerable efforts have been made to find new pharmacological agents capable of modulating AMPK (Table 2). Using computational approaches and high-throughput screening, it was possible to identify small molecules and compounds with the potential to activate AMPK directly or modulate its complex signaling network. In this sense, currently, the widely used AMPK-activating drug in human patients remains metformin. In fact, as previously mentioned, metformin has demonstrated promising outcomes in the treatment of diverse metabolic diseases, including type II diabetes, atherosclerosis, Alzheimer’s disease, Huntington’s disease, and various types of cancers.
Furthermore, due to the pivotal role of AMPK in autophagy regulation, which has recently been recognized as a hallmark of health, AMPK has the potential to act as a health-modifying agent. Thereby, the involvement of AMPK in the regulation of autophagy, coupled with the beneficial effects of autophagy in various physiological processes, suggests that the activation of autophagy could be a mechanistic link underlying some of the positive effects of AMPK activation during pathological situations.
Future studies focusing on unraveling the precise molecular mechanisms through which AMPK exerts its diverse beneficial effects on human health will provide further insights into these intriguing questions.
Author Contributions
I.T.-G. conceptualized the manuscript. V.R. and I.T.-G. contributed to researching the content for the review, discussing the content, and writing the review. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We are grateful to Juan Tornín and Aida Rodríguez for their helpful comments.
Conflicts of Interest
The authors declare no conflict of interest.
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic control of autophagy. Cell 2014, 159, 1263–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.C.; Hardie, D.G. AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab. 2018, 27, 299–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, M.; Osmond, B.C.; Botstein, D. Mutants of yeast defective in sucrose utilization. Genetics 1981, 98, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Grahame Hardie, D. AMP-activated protein kinase: A key regulator of energy balance with many roles in human disease. J. Intern. Med. 2014, 276, 543–559. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Yang, G.; Kim, Y.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhong, L.; Wang, F.; Zhu, H. Dissecting the role of AMP-activated protein kinase in human diseases. Acta Pharm. Sin. B 2017, 7, 249–259. [Google Scholar] [CrossRef]
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. AMPK: Positive and negative regulation, and its role in whole-body energy homeostasis. Curr. Opin. Cell Biol. 2015, 33, 1–7. [Google Scholar] [CrossRef]
- Willows, R.; Navaratnam, N.; Lima, A.; Read, J.; Carling, D. Effect of different γ-subunit isoforms on the regulation of AMPK. Biochem. J. 2017, 474, 1741–1754. [Google Scholar] [CrossRef] [Green Version]
- Ross, F.A.; Jensen, T.E.; Hardie, D.G. Differential regulation by AMP and ADP of AMPK complexes containing different γ subunit isoforms. Biochem. J. 2016, 473, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Stein, S.C.; Woods, A.; Jones, N.A.; Davison, M.D.; Carling, D. The regulation of AMP-activated protein kinase by phosphorylation. Biochem. J. 2000, 345 Pt 3, 437–443. [Google Scholar] [CrossRef]
- Xie, M.; Zhang, D.; Dyck, J.R.; Li, Y.; Zhang, H.; Morishima, M.; Mann, D.L.; Taffet, G.E.; Baldini, A.; Khoury, D.S.; et al. A pivotal role for endogenous TGF-beta-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 17378–17383. [Google Scholar] [CrossRef]
- Fogarty, S.; Hawley, S.A.; Green, K.A.; Saner, N.; Mustard, K.J.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta activates AMPK without forming a stable complex: Synergistic effects of Ca2+ and AMP. Biochem. J. 2010, 426, 109–118. [Google Scholar] [CrossRef]
- Neumann, D. Is TAK1 a Direct Upstream Kinase of AMPK? Int. J. Mol. Sci. 2018, 19, 2412. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Boudeau, J.; Reid, J.L.; Mustard, K.J.; Udd, L.; Mäkelä, T.P.; Alessi, D.R.; Hardie, D.G. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2003, 2, 28. [Google Scholar] [CrossRef] [Green Version]
- Oakhill, J.S.; Steel, R.; Chen, Z.P.; Scott, J.W.; Ling, N.; Tam, S.; Kemp, B.E. AMPK is a direct adenylate charge-regulated protein kinase. Science 2011, 332, 1433–1435. [Google Scholar] [CrossRef]
- Gowans, G.J.; Hawley, S.A.; Ross, F.A.; Hardie, D.G. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 2013, 18, 556–566. [Google Scholar] [CrossRef] [Green Version]
- Soltys, C.L.; Kovacic, S.; Dyck, J.R. Activation of cardiac AMP-activated protein kinase by LKB1 expression or chemical hypoxia is blunted by increased Akt activity. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2472–H2479. [Google Scholar] [CrossRef] [Green Version]
- Ning, J.; Xi, G.; Clemmons, D.R. Suppression of AMPK activation via S485 phosphorylation by IGF-I during hyperglycemia is mediated by AKT activation in vascular smooth muscle cells. Endocrinology 2011, 152, 3143–3154. [Google Scholar] [CrossRef]
- Hawley, S.A.; Ross, F.A.; Gowans, G.J.; Tibarewal, P.; Leslie, N.R.; Hardie, D.G. Phosphorylation by Akt within the ST loop of AMPK-α1 down-regulates its activation in tumour cells. Biochem. J. 2014, 459, 275–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horman, S.; Vertommen, D.; Heath, R.; Neumann, D.; Mouton, V.; Woods, A.; Schlattner, U.; Wallimann, T.; Carling, D.; Hue, L.; et al. Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase alpha-subunits in heart via hierarchical phosphorylation of Ser485/491. J. Biol. Chem. 2006, 281, 5335–5340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heathcote, H.R.; Mancini, S.J.; Strembitska, A.; Jamal, K.; Reihill, J.A.; Palmer, T.M.; Gould, G.W.; Salt, I.P. Protein kinase C phosphorylates AMP-activated protein kinase α1 Ser487. Biochem. J. 2016, 473, 4681–4697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagon, Y.; Hur, E.; Zheng, B.; Wellenstein, K.; Cantley, L.C.; Kahn, B.B. p70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin's effect on food intake. Cell Metab. 2012, 16, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Anand, S.K.; Singh, N.; Dwivedi, U.N.; Kakkar, P. AMP-activated protein kinase: An energy sensor and survival mechanism in the reinstatement of metabolic homeostasis. Exp. Cell Res. 2023, 428, 113614. [Google Scholar] [CrossRef]
- Gowans, G.J.; Hardie, D.G. AMPK: A cellular energy sensor primarily regulated by AMP. Biochem. Soc. Trans. 2014, 42, 71–75. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [Green Version]
- Tamargo-Gómez, I.; Mariño, G. AMPK: Regulation of Metabolic Dynamics in the Context of Autophagy. Int. J. Mol. Sci. 2018, 19, 3812. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; Zheng, B.; Shaywitz, A.; Dagon, Y.; Tower, C.; Bellinger, G.; Shen, C.H.; Wen, J.; Asara, J.; McGraw, T.E.; et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell 2013, 49, 1167–1175. [Google Scholar] [CrossRef] [Green Version]
- Chavez, J.A.; Roach, W.G.; Keller, S.R.; Lane, W.S.; Lienhard, G.E. Inhibition of GLUT4 translocation by Tbc1d1, a Rab GTPase-activating protein abundant in skeletal muscle, is partially relieved by AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 9187–9195. [Google Scholar] [CrossRef] [Green Version]
- Doménech, E.; Maestre, C.; Esteban-Martínez, L.; Partida, D.; Pascual, R.; Fernández-Miranda, G.; Seco, E.; Campos-Olivas, R.; Pérez, M.; Megias, D.; et al. AMPK and PFKFB3 mediate glycolysis and survival in response to mitophagy during mitotic arrest. Nat. Cell Biol. 2015, 17, 1304–1316. [Google Scholar] [CrossRef]
- Bultot, L.; Guigas, B.; Von Wilamowitz-Moellendorff, A.; Maisin, L.; Vertommen, D.; Hussain, N.; Beullens, M.; Guinovart, J.J.; Foretz, M.; Viollet, B.; et al. AMP-activated protein kinase phosphorylates and inactivates liver glycogen synthase. Biochem. J. 2012, 443, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Janzen, N.R.; Whitfield, J.; Hoffman, N.J. Interactive Roles for AMPK and Glycogen from Cellular Energy Sensing to Exercise Metabolism. Int. J. Mol. Sci. 2018, 19, 3344. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Seo, W.Y.; Song, K.H.; Chanda, D.; Kim, Y.D.; Kim, D.K.; Lee, M.W.; Ryu, D.; Kim, Y.H.; Noh, J.R.; et al. AMPK-dependent repression of hepatic gluconeogenesis via disruption of CREB.CRTC2 complex by orphan nuclear receptor small heterodimer partner. J. Biol. Chem. 2010, 285, 32182–32191. [Google Scholar] [CrossRef] [Green Version]
- Guttzeit, S.; Backs, J. Post-translational modifications talk and crosstalk to class IIa histone deacetylases. J. Mol. Cell. Cardiol. 2022, 162, 53–61. [Google Scholar] [CrossRef]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.P.; O'Neill, H.M.; Ford, R.J.; Palanivel, R.; O'Brien, M.; et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef] [Green Version]
- Ahmadian, M.; Abbott, M.J.; Tang, T.; Hudak, C.S.; Kim, Y.; Bruss, M.; Hellerstein, M.K.; Lee, H.Y.; Samuel, V.T.; Shulman, G.I.; et al. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab. 2011, 13, 739–748. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.; Pan, J.; Qu, N.; Lei, Y.; Han, J.; Zhang, J.; Han, D. The AMPK pathway in fatty liver disease. Front. Physiol. 2022, 13, 970292. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, Q.; Fu, J.; Ren, R. Polysaccharides derived from natural sources regulate triglyceride and cholesterol metabolism: A review of the mechanisms. Food Funct. 2019, 10, 2330–2339. [Google Scholar] [CrossRef]
- Sato, S.; Jung, H.; Nakagawa, T.; Pawlosky, R.; Takeshima, T.; Lee, W.R.; Sakiyama, H.; Laxman, S.; Wynn, R.M.; Tu, B.P.; et al. Metabolite Regulation of Nuclear Localization of Carbohydrate-response Element-binding Protein (ChREBP): ROLE OF AMP AS AN ALLOSTERIC INHIBITOR. J. Biol. Chem. 2016, 291, 10515–10527. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Tsuyama, T.; Sato, C.; Karim, M.F.; Yoshizawa, T.; Inoue, M.; Yamagata, K. Hypoxia reduces HNF4α/MODY1 protein expression in pancreatic β-cells by activating AMP-activated protein kinase. J. Biol. Chem. 2017, 292, 8716–8728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balamurugan, K.; Chandra, K.; Sai Latha, S.; Swathi, M.; Joshi, M.B.; Misra, P.; Parsa, K.V.L. PHLPPs: Emerging players in metabolic disorders. Drug Discov. Today 2022, 27, 103317. [Google Scholar] [CrossRef] [PubMed]
- Dudley, L.J.; Makar, A.N.; Gammoh, N. Membrane targeting of core autophagy players during autophagosome biogenesis. FEBS J. 2020, 287, 4806–4821. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.A.; Ktistakis, N.T. Autophagosome Biogenesis Machinery. J. Mol. Biol. 2020, 432, 2449–2461. [Google Scholar] [CrossRef]
- Deleyto-Seldas, N.; Efeyan, A. The mTOR-Autophagy Axis and the Control of Metabolism. Front. Cell Dev. Biol. 2021, 9, 655731. [Google Scholar] [CrossRef]
- Inoki, K.; Kim, J.; Guan, K.L. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 381–400. [Google Scholar] [CrossRef]
- Russell, R.C.; Yuan, H.X.; Guan, K.L. Autophagy regulation by nutrient signaling. Cell Res. 2014, 24, 42–57. [Google Scholar] [CrossRef] [Green Version]
- Wirth, M.; Joachim, J.; Tooze, S.A. Autophagosome formation--the role of ULK1 and Beclin1-PI3KC3 complexes in setting the stage. Semin. Cancer Biol. 2013, 23, 301–309. [Google Scholar] [CrossRef]
- Mizushima, N. The ATG conjugation systems in autophagy. Curr. Opin. Cell Biol. 2020, 63, 1–10. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell. 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Weerasekara, V.K.; Panek, D.J.; Broadbent, D.G.; Mortenson, J.B.; Mathis, A.D.; Logan, G.N.; Prince, J.T.; Thomson, D.M.; Thompson, J.W.; Andersen, J.L. Metabolic-stress-induced rearrangement of the 14-3-3ζ interactome promotes autophagy via a ULK1- and AMPK-regulated 14-3-3ζ interaction with phosphorylated Atg9. Mol. Cell. Biol. 2014, 34, 4379–4388. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.; Su, H.; Zhang, D.; Wang, Y.; Shen, Q.; Liu, B.; Huang, R.; Zhou, T.; Peng, C.; Wong, C.C.; et al. AMPK-Dependent Phosphorylation of GAPDH Triggers Sirt1 Activation and Is Necessary for Autophagy upon Glucose Starvation. Mol. Cell 2015, 60, 930–940. [Google Scholar] [CrossRef] [Green Version]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef] [Green Version]
- Fulco, M.; Cen, Y.; Zhao, P.; Hoffman, E.P.; McBurney, M.W.; Sauve, A.A.; Sartorelli, V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell 2008, 14, 661–673. [Google Scholar] [CrossRef] [Green Version]
- Costford, S.R.; Bajpeyi, S.; Pasarica, M.; Albarado, D.C.; Thomas, S.C.; Xie, H.; Church, T.S.; Jubrias, S.A.; Conley, K.E.; Smith, S.R. Skeletal muscle NAMPT is induced by exercise in humans. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E117–E126. [Google Scholar] [CrossRef] [Green Version]
- Sacitharan, P.K.; Bou-Gharios, G.; Edwards, J.R. SIRT1 directly activates autophagy in human chondrocytes. Cell Death Discov. 2020, 6, 41. [Google Scholar] [CrossRef]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; Depinho, R.A.; Sadoshima, J. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ. Res. 2010, 107, 1470–1482. [Google Scholar] [CrossRef]
- Young, N.P.; Kamireddy, A.; Van Nostrand, J.L.; Eichner, L.J.; Shokhirev, M.N.; Dayn, Y.; Shaw, R.J. AMPK governs lineage specification through Tfeb-dependent regulation of lysosomes. Genes Dev. 2016, 30, 535–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H.; Kim, G.; Han, D.H.; Lee, M.; Kim, I.; Kim, B.; Kim, K.H.; Song, Y.M.; Yoo, J.E.; Wang, H.J.; et al. Ezetimibe ameliorates steatohepatitis via AMP activated protein kinase-TFEB-mediated activation of autophagy and NLRP3 inflammasome inhibition. Autophagy 2017, 13, 1767–1781. [Google Scholar] [CrossRef] [PubMed]
- Villena, J.A. New insights into PGC-1 coactivators: Redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015, 282, 647–672. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Pan, R.; Li, R.; Niemann, B.; Aurich, A.C.; Chen, Y.; Rohrbach, S. Mitochondrial biogenesis and peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) deacetylation by physical activity: Intact adipocytokine signaling is required. Diabetes 2011, 60, 157–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanna, A.; Henson, S.M.; Escors, D.; Akbar, A.N. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat. Immunol. 2014, 15, 965–972. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.Y.; Liang, J.; Yang, B.C.; Leung, P.S. SIRT1 Activation Promotes β-Cell Regeneration by Activating Endocrine Progenitor Cells via AMPK Signaling-Mediated Fatty Acid Oxidation. Stem Cells 2019, 37, 1416–1428. [Google Scholar] [CrossRef]
- Vancura, A.; Nagar, S.; Kaur, P.; Bu, P.; Bhagwat, M.; Vancurova, I. Reciprocal Regulation of AMPK/SNF1 and Protein Acetylation. Int. J. Mol. Sci. 2018, 19, 3314. [Google Scholar] [CrossRef] [Green Version]
- Abu Shelbayeh, O.; Arroum, T.; Morris, S.; Busch, K.B. PGC-1α Is a Master Regulator of Mitochondrial Lifecycle and ROS Stress Response. Antioxidants 2023, 12, 1075. [Google Scholar] [CrossRef]
- Mansueto, G.; Armani, A.; Viscomi, C.; D'Orsi, L.; De Cegli, R.; Polishchuk, E.V.; Lamperti, C.; Di Meo, I.; Romanello, V.; Marchet, S.; et al. Transcription Factor EB Controls Metabolic Flexibility during Exercise. Cell Metab. 2017, 25, 182–196. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Green, A.; Hossain, T.; Eckmann, D.M. Mitochondrial dynamics involves molecular and mechanical events in motility, fusion and fission. Front. Cell Dev. Biol. 2022, 10, 1010232. [Google Scholar] [CrossRef]
- Seabright, A.P.; Fine, N.H.F.; Barlow, J.P.; Lord, S.O.; Musa, I.; Gray, A.; Bryant, J.A.; Banzhaf, M.; Lavery, G.G.; Hardie, D.G.; et al. AMPK activation induces mitophagy and promotes mitochondrial fission while activating TBK1 in a PINK1-Parkin independent manner. FASEB J. 2020, 34, 6284–6301. [Google Scholar] [CrossRef] [Green Version]
- Zerihun, M.; Sukumaran, S.; Qvit, N. The Drp1-Mediated Mitochondrial Fission Protein Interactome as an Emerging Core Player in Mitochondrial Dynamics and Cardiovascular Disease Therapy. Int. J. Mol. Sci. 2023, 24, 5785. [Google Scholar] [CrossRef]
- Qiao, H.; Ren, H.; Du, H.; Zhang, M.; Xiong, X.; Lv, R. Liraglutide repairs the infarcted heart: The role of the SIRT1/Parkin/mitophagy pathway. Mol. Med. Rep. 2018, 17, 3722–3734. [Google Scholar] [CrossRef] [Green Version]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [Green Version]
- Laker, R.C.; Drake, J.C.; Wilson, R.J.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J.; et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017, 8, 548. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, G.R.; Hardie, D.G. New insights into activation and function of the AMPK. Nat. Rev. Mol. Cell Biol. 2023, 24, 255–272. [Google Scholar] [CrossRef]
- Tamargo-Gómez, I.; Fernández, Á.; Mariño, G. Pathogenic Single Nucleotide Polymorphisms on Autophagy-Related Genes. Int. J. Mol. Sci. 2020, 21, 8196. [Google Scholar] [CrossRef]
- Qiu, L.; Qu, X.; He, J.; Cheng, L.; Zhang, R.; Sun, M.; Yang, Y.; Wang, J.; Wang, M.; Zhu, X.; et al. Predictive model for risk of gastric cancer using genetic variants from genome-wide association studies and high-evidence meta-analysis. Cancer Med. 2020, 9, 7310–7316. [Google Scholar] [CrossRef]
- Tokunaga, R.; Cao, S.; Naseem, M.; Battaglin, F.; Lo, J.H.; Arai, H.; Loupakis, F.; Stintzing, S.; Puccini, A.; Berger, M.D.; et al. AMPK variant, a candidate of novel predictor for chemotherapy in metastatic colorectal cancer: A meta-analysis using TRIBE, MAVERICC and FIRE3. Int. J. Cancer 2019, 145, 2082–2090. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Zhang, Y.; Yan, F.T.; Zheng, X. Association of PRKAA1 gene polymorphisms with chronic hepatitis B virus infection in Chinese Han population. Braz. J. Infect. Dis. 2016, 20, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Jiang, B.; He, B.; Tang, M.; Wang, P.; Chen, L.; Lu, J.; Lu, P. Genetic variations in PRKAA1 predict the risk and progression of gastric Cancer. BMC Cancer 2018, 18, 923. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Mu, Y.; Wang, W.; Tan, B.T.; Chen, Y.H.; Li, Y.P.; Zhu, D.; Li, W.; Cui, J.; Yu, L.H. Effect of AMPK Subunit Alpha 2 Polymorphisms on Postherpetic Pain Susceptibility in Southwestern Han Chinese. J. Pain Res. 2022, 15, 3319–3326. [Google Scholar] [CrossRef]
- Li, Q.; Li, C.; Li, H.; Zeng, L.; Kang, Z.; Mao, Y.; Tang, X.; Zheng, P.; He, L.; Luo, F.; et al. Effect of AMP-activated protein kinase subunit alpha 2 (PRKAA2) genetic polymorphisms on susceptibility to type 2 diabetes mellitus and diabetic nephropathy in a Chinese population. J. Diabetes 2018, 10, 43–49. [Google Scholar] [CrossRef]
- Shen, J.Z.; Ge, W.H.; Fang, Y.; Liu, H. A novel polymorphism in protein kinase AMP-activated catalytic subunit alpha 2 (PRKAA2) is associated with type 2 diabetes in the Han Chinese population. J. Diabetes 2017, 9, 606–612. [Google Scholar] [CrossRef]
- Mo, X.; Zhang, H.; Zhou, Z.; Zhu, Z.; HuangFu, X.; Xu, T.; Wang, A.; Guo, Z.; Zhang, Y. SNPs rs10224002 in PRKAG2 may disturb gene expression and consequently affect hypertension. Mol. Biol. Rep. 2019, 46, 1617–1624. [Google Scholar] [CrossRef]
- Thevenon, J.; Laurent, G.; Ader, F.; Laforêt, P.; Klug, D.; Duva Pentiah, A.; Gouya, L.; Maurage, C.A.; Kacet, S.; Eicher, J.C.; et al. High prevalence of arrhythmic and myocardial complications in patients with cardiac glycogenosis due to PRKAG2 mutations. Europace 2017, 19, 651–659. [Google Scholar] [CrossRef]
- Slattery, M.L.; Herrick, J.S.; Lundgreen, A.; Fitzpatrick, F.A.; Curtin, K.; Wolff, R.K. Genetic variation in a metabolic signaling pathway and colon and rectal cancer risk: mTOR, PTEN, STK11, RPKAA1, PRKAG2, TSC1, TSC2, PI3K and Akt1. Carcinogenesis 2010, 31, 1604–1611. [Google Scholar] [CrossRef] [Green Version]
- Jablonski, K.A.; McAteer, J.B.; de Bakker, P.I.; Franks, P.W.; Pollin, T.I.; Hanson, R.L.; Saxena, R.; Fowler, S.; Shuldiner, A.R.; Knowler, W.C.; et al. Common variants in 40 genes assessed for diabetes incidence and response to metformin and lifestyle intervention in the diabetes prevention program. Diabetes 2010, 59, 2672–2681. [Google Scholar] [CrossRef] [Green Version]
- Misra, P.; Chakrabarti, R. The role of AMP kinase in diabetes. Indian J. Med. Res. 2007, 125, 389–398. [Google Scholar]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [Green Version]
- Mohseni, R.; Teimouri, M.; Safaei, M.; Arab Sadeghabadi, Z. AMP-activated protein kinase is a key regulator of obesity-associated factors. Cell Biochem. Funct. 2023, 41, 20–32. [Google Scholar] [CrossRef]
- Prasad, M.K.; Mohandas, S.; Ramkumar, K.M. Dysfunctions, molecular mechanisms, and therapeutic strategies of pancreatic β-cells in diabetes. Apoptosis 2023, 28, 958–976. [Google Scholar] [CrossRef]
- Cao, R.; Tian, H.; Zhang, Y.; Liu, G.; Xu, H.; Rao, G.; Tian, Y.; Fu, X. Signaling pathways and intervention for therapy of type 2 diabetes mellitus. MedComm 2023, 4, e283. [Google Scholar] [CrossRef]
- McGee, S.L.; van Denderen, B.J.; Howlett, K.F.; Mollica, J.; Schertzer, J.D.; Kemp, B.E.; Hargreaves, M. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes 2008, 57, 860–867. [Google Scholar] [CrossRef]
- Holoman, N.C.; Aiello, J.J.; Trobenter, T.D.; Tarchick, M.J.; Kozlowski, M.R.; Makowski, E.R.; De Vivo, D.C.; Singh, C.; Sears, J.E.; Samuels, I.S. Reduction of Glut1 in the Neural Retina But Not the RPE Alleviates Polyol Accumulation and Normalizes Early Characteristics of Diabetic Retinopathy. J. Neurosci. 2021, 41, 3275–3299. [Google Scholar] [CrossRef]
- Coughlan, K.A.; Valentine, R.J.; Ruderman, N.B.; Saha, A.K. AMPK activation: A therapeutic target for type 2 diabetes? Diabetes Metab. Syndr. Obes. 2014, 7, 241–253. [Google Scholar] [CrossRef] [Green Version]
- Choi, R.H.; McConahay, A.; Johnson, M.B.; Jeong, H.W.; Koh, H.J. Adipose tissue-specific knockout of AMPKα1/α2 results in normal AICAR tolerance and glucose metabolism. Biochem. Biophys. Res. Commun. 2019, 519, 633–638. [Google Scholar] [CrossRef]
- Hingst, J.R.; Kjøbsted, R.; Birk, J.B.; Jørgensen, N.O.; Larsen, M.R.; Kido, K.; Larsen, J.K.; Kjeldsen, S.A.S.; Fentz, J.; Frøsig, C.; et al. Inducible deletion of skeletal muscle AMPKα reveals that AMPK is required for nucleotide balance but dispensable for muscle glucose uptake and fat oxidation during exercise. Mol. Metab. 2020, 40, 101028. [Google Scholar] [CrossRef]
- Li, M.; Chi, X.; Wang, Y.; Setrerrahmane, S.; Xie, W.; Xu, H. Trends in insulin resistance: Insights into mechanisms and therapeutic strategy. Signal Transduct. Target. Ther. 2022, 7, 216. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Sun, J.; Liu, M.; Zhou, Y.; Zhang, L.; Li, Y. The New Role of AMP-Activated Protein Kinase in Regulating Fat Metabolism and Energy Expenditure in Adipose Tissue. Biomolecules 2021, 11, 1757. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Young, J.L.; Wang, K.; Qian, Y.; Cai, L. Diabetic-induced alterations in hepatic glucose and lipid metabolism: The role of type 1 and type 2 diabetes mellitus (Review). Mol. Med. Rep. 2020, 22, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Višnjić, D.; Lalić, H.; Dembitz, V.; Tomić, B.; Smoljo, T. AICAr, a Widely Used AMPK Activator with Important AMPK-Independent Effects: A Systematic Review. Cells 2021, 10, 1095. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Lantier, L.; Devin-Leclerc, J.; Hebrard, S.; Amouyal, C.; Mounier, R.; Foretz, M.; Andreelli, F. Targeting the AMPK pathway for the treatment of Type 2 diabetes. Front. Biosci. 2009, 14, 3380–3400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swisa, A.; Granot, Z.; Tamarina, N.; Sayers, S.; Bardeesy, N.; Philipson, L.; Hodson, D.J.; Wikstrom, J.D.; Rutter, G.A.; Leibowitz, G.; et al. Loss of Liver Kinase B1 (LKB1) in Beta Cells Enhances Glucose-stimulated Insulin Secretion Despite Profound Mitochondrial Defects. J. Biol. Chem. 2015, 290, 20934–20946. [Google Scholar] [CrossRef] [Green Version]
- Kone, M.; Pullen, T.J.; Sun, G.; Ibberson, M.; Martinez-Sanchez, A.; Sayers, S.; Nguyen-Tu, M.S.; Kantor, C.; Swisa, A.; Dor, Y.; et al. LKB1 and AMPK differentially regulate pancreatic β-cell identity. FASEB J. 2014, 28, 4972–4985. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; You, Y.H.; Ham, D.S.; Yang, H.K.; Yoon, K.H. The Paradoxical Effects of AMPK on Insulin Gene Expression and Glucose-Induced Insulin Secretion. J. Cell. Biochem. 2016, 117, 239–246. [Google Scholar] [CrossRef]
- GBD 2017 Causes of Death Collaborators. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef] [Green Version]
- Peverill, R.E. Changes in left ventricular size, geometry, pump function and left heart pressures during healthy aging. Rev. Cardiovasc. Med. 2021, 22, 717–729. [Google Scholar] [CrossRef]
- Abdellatif, M.; Ljubojevic-Holzer, S.; Madeo, F.; Sedej, S. Autophagy in cardiovascular health and disease. Prog. Mol. Biol. Transl. Sci. 2020, 172, 87–106. [Google Scholar] [CrossRef]
- Gatica, D.; Chiong, M.; Lavandero, S.; Klionsky, D.J. Molecular mechanisms of autophagy in the cardiovascular system. Circ. Res. 2015, 116, 456–467. [Google Scholar] [CrossRef]
- Sung, M.M.; Zordoky, B.N.; Bujak, A.L.; Lally, J.S.; Fung, D.; Young, M.E.; Horman, S.; Miller, E.J.; Light, P.E.; Kemp, B.E.; et al. AMPK deficiency in cardiac muscle results in dilated cardiomyopathy in the absence of changes in energy metabolism. Cardiovasc. Res. 2015, 107, 235–245. [Google Scholar] [CrossRef] [Green Version]
- Gundewar, S.; Calvert, J.W.; Jha, S.; Toedt-Pingel, I.; Ji, S.Y.; Nunez, D.; Ramachandran, A.; Anaya-Cisneros, M.; Tian, R.; Lefer, D.J. Activation of AMP-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ. Res. 2009, 104, 403–411. [Google Scholar] [CrossRef]
- Tang, X.; Chen, X.F.; Wang, N.Y.; Wang, X.M.; Liang, S.T.; Zheng, W.; Lu, Y.B.; Zhao, X.; Hao, D.L.; Zhang, Z.Q.; et al. SIRT2 Acts as a Cardioprotective Deacetylase in Pathological Cardiac Hypertrophy. Circulation 2017, 136, 2051–2067. [Google Scholar] [CrossRef]
- García-Prieto, C.F.; Hernández-Nuño, F.; Rio, D.D.; Ruiz-Hurtado, G.; Aránguez, I.; Ruiz-Gayo, M.; Somoza, B.; Fernández-Alfonso, M.S. High-fat diet induces endothelial dysfunction through a down-regulation of the endothelial AMPK-PI3K-Akt-eNOS pathway. Mol. Nutr. Food Res. 2015, 59, 520–532. [Google Scholar] [CrossRef]
- Cai, Z.; Ding, Y.; Zhang, M.; Lu, Q.; Wu, S.; Zhu, H.; Song, P.; Zou, M.H. Ablation of Adenosine Monophosphate-Activated Protein Kinase α1 in Vascular Smooth Muscle Cells Promotes Diet-Induced Atherosclerotic Calcification In Vivo. Circ. Res. 2016, 119, 422–433. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, M.; Liang, B.; Xie, Z.; Zhao, Z.; Asfa, S.; Choi, H.C.; Zou, M.H. Reduction of AMP-activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation 2010, 121, 792–803. [Google Scholar] [CrossRef] [Green Version]
- Barcena, M.L.; Tonini, G.; Haritonow, N.; Breiter, P.; Milting, H.; Baczko, I.; Müller-Werdan, U.; Ladilov, Y.; Regitz-Zagrosek, V. Sex and age differences in AMPK phosphorylation, mitochondrial homeostasis, and inflammation in hearts from inflammatory cardiomyopathy patients. Aging Cell 2023, e13894. [Google Scholar] [CrossRef]
- Wu, S.; Zou, M.H. AMPK, Mitochondrial Function, and Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 4987. [Google Scholar] [CrossRef]
- Tong, D.; Schiattarella, G.G.; Jiang, N.; Daou, D.; Luo, Y.; Link, M.S.; Lavandero, S.; Gillette, T.G.; Hill, J.A. Impaired AMP-Activated Protein Kinase Signaling in Heart Failure With Preserved Ejection Fraction-Associated Atrial Fibrillation. Circulation 2022, 146, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Nie, J.; Wu, L.; Hu, Y.; Wen, Z.; Dong, L.; Zou, M.H.; Chen, C.; Wang, D.W. AMPKα2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation. Circ. Res. 2018, 122, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Bairwa, S.C.; Parajuli, N.; Dyck, J.R. The role of AMPK in cardiomyocyte health and survival. Biochim. Biophys. Acta 2016, 1862, 2199–2210. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef]
- Gusev, E.; Sarapultsev, A. Atherosclerosis and Inflammation: Insights from the Theory of General Pathological Processes. Int. J. Mol. Sci. 2023, 24, 7910. [Google Scholar] [CrossRef]
- Ou, H.; Liu, C.; Feng, W.; Xiao, X.; Tang, S.; Mo, Z. Role of AMPK in atherosclerosis via autophagy regulation. Sci. China Life Sci. 2018, 61, 1212–1221. [Google Scholar] [CrossRef]
- Weikel, K.A.; Cacicedo, J.M.; Ruderman, N.B.; Ido, Y. Glucose and palmitate uncouple AMPK from autophagy in human aortic endothelial cells. Am. J. Physiol. Cell Physiol. 2015, 308, C249–C263. [Google Scholar] [CrossRef] [Green Version]
- Ewart, M.A.; Kennedy, S. AMPK and vasculoprotection. Pharmacol. Ther. 2011, 131, 242–253. [Google Scholar] [CrossRef]
- Remmerie, A.; Scott, C.L. Macrophages and lipid metabolism. Cell. Immunol. 2018, 330, 27–42. [Google Scholar] [CrossRef]
- Ma, A.; Wang, J.; Yang, L.; An, Y.; Zhu, H. AMPK activation enhances the anti-atherogenic effects of high density lipoproteins in apoE. J. Lipid Res. 2017, 58, 1536–1547. [Google Scholar] [CrossRef] [Green Version]
- Kemmerer, M.; Wittig, I.; Richter, F.; Brüne, B.; Namgaladze, D. AMPK activates LXRα and ABCA1 expression in human macrophages. Int. J. Biochem. Cell Biol. 2016, 78, 1–9. [Google Scholar] [CrossRef]
- Sag, D.; Carling, D.; Stout, R.D.; Suttles, J. Adenosine 5'-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J. Immunol. 2008, 181, 8633–8641. [Google Scholar] [CrossRef] [Green Version]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Jin, Y.; Fu, J. Novel Insights Into the NLRP 3 Inflammasome in Atherosclerosis. J. Am. Heart Assoc. 2019, 8, e012219. [Google Scholar] [CrossRef] [Green Version]
- Cheang, W.S.; Tian, X.Y.; Wong, W.T.; Lau, C.W.; Lee, S.S.; Chen, Z.Y.; Yao, X.; Wang, N.; Huang, Y. Metformin protects endothelial function in diet-induced obese mice by inhibition of endoplasmic reticulum stress through 5' adenosine monophosphate-activated protein kinase-peroxisome proliferator-activated receptor δ pathway. Arter. Thromb. Vasc. Biol. 2014, 34, 830–836. [Google Scholar] [CrossRef] [Green Version]
- Park, M.D.; Silvin, A.; Ginhoux, F.; Merad, M. Macrophages in health and disease. Cell 2022, 185, 4259–4279. [Google Scholar] [CrossRef]
- Noor, H.B.; Mou, N.A.; Salem, L.; Shimul, M.F.A.; Biswas, S.; Akther, R.; Khan, S.; Raihan, S.; Mohib, M.M.; Sagor, M.A.T. Anti-inflammatory Property of AMP-activated Protein Kinase. Antiinflamm. Antiallergy Agents Med. Chem. 2020, 19, 2–41. [Google Scholar] [CrossRef]
- Kelly, B.; O'Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [Green Version]
- Galic, S.; Fullerton, M.D.; Schertzer, J.D.; Sikkema, S.; Marcinko, K.; Walkley, C.R.; Izon, D.; Honeyman, J.; Chen, Z.P.; van Denderen, B.J.; et al. Hematopoietic AMPK β1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J. Clin. Investig. 2011, 121, 4903–4915. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Fullerton, M.D.; Ross, F.A.; Schertzer, J.D.; Chevtzoff, C.; Walker, K.J.; Peggie, M.W.; Zibrova, D.; Green, K.A.; Mustard, K.J.; et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 2012, 336, 918–922. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Kang, J.H.; Lee, S. Autophagy in Neurodegenerative Diseases: A Hunter for Aggregates. Int. J. Mol. Sci. 2020, 21, 3369. [Google Scholar] [CrossRef] [PubMed]
- Mahul-Mellier, A.L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iennaco, R.; Formenti, G.; Trovesi, C.; Rossi, R.L.; Zuccato, C.; Lischetti, T.; Bocchi, V.D.; Scolz, A.; Martínez-Labarga, C.; Rickards, O.; et al. The evolutionary history of the polyQ tract in huntingtin sheds light on its functional pro-neural activities. Cell Death Differ. 2022, 29, 293–305. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer's disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Vicente, M. Autophagy in neurodegenerative diseases: From pathogenic dysfunction to therapeutic modulation. Semin. Cell Dev. Biol. 2015, 40, 115–126. [Google Scholar] [CrossRef]
- Martinez-Vicente, M. Neuronal Mitophagy in Neurodegenerative Diseases. Front. Mol. Neurosci. 2017, 10, 64. [Google Scholar] [CrossRef] [Green Version]
- Assefa, B.T.; Tafere, G.G.; Wondafrash, D.Z.; Gidey, M.T. The Bewildering Effect of AMPK Activators in Alzheimer's Disease: Review of the Current Evidence. Biomed Res. Int. 2020, 2020, 9895121. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Haapasalo, A.; Soininen, H.; Hiltunen, M. AMP-activated protein kinase: A potential player in Alzheimer's disease. J. Neurochem. 2011, 118, 460–474. [Google Scholar] [CrossRef]
- DiTacchio, K.A.; Heinemann, S.F.; Dziewczapolski, G. Metformin treatment alters memory function in a mouse model of Alzheimer's disease. J. Alzheimers Dis. 2015, 44, 43–48. [Google Scholar] [CrossRef]
- Hang, L.; Thundyil, J.; Lim, K.L. Mitochondrial dysfunction and Parkinson disease: A Parkin-AMPK alliance in neuroprotection. Ann. N. Y. Acad. Sci. 2015, 1350, 37–47. [Google Scholar] [CrossRef]
- Curry, D.W.; Stutz, B.; Andrews, Z.B.; Elsworth, J.D. Targeting AMPK Signaling as a Neuroprotective Strategy in Parkinson's Disease. J. Park. Dis. 2018, 8, 161–181. [Google Scholar] [CrossRef]
- Ju, T.C.; Chen, H.M.; Lin, J.T.; Chang, C.P.; Chang, W.C.; Kang, J.J.; Sun, C.P.; Tao, M.H.; Tu, P.H.; Chang, C.; et al. Nuclear translocation of AMPK-alpha1 potentiates striatal neurodegeneration in Huntington's disease. J. Cell Biol. 2011, 194, 209–227. [Google Scholar] [CrossRef]
- Hervás, D.; Fornés-Ferrer, V.; Gómez-Escribano, A.P.; Sequedo, M.D.; Peiró, C.; Millán, J.M.; Vázquez-Manrique, R.P. Metformin intake associates with better cognitive function in patients with Huntington's disease. PLoS ONE 2017, 12, e0179283. [Google Scholar] [CrossRef] [Green Version]
- Hsu, E.; Desai, M. Glaucoma and Systemic Disease. Life 2023, 13, 1018. [Google Scholar] [CrossRef]
- Dada, T.; Verma, S.; Gagrani, M.; Bhartiya, S.; Chauhan, N.; Satpute, K.; Sharma, N. Ocular and Systemic Factors Associated with Glaucoma. J. Curr. Glaucoma Pract. 2022, 16, 179–191. [Google Scholar] [CrossRef]
- Belforte, N.; Agostinone, J.; Alarcon-Martinez, L.; Villafranca-Baughman, D.; Dotigny, F.; Cueva Vargas, J.L.; Di Polo, A. AMPK hyperactivation promotes dendrite retraction, synaptic loss, and neuronal dysfunction in glaucoma. Mol. Neurodegener. 2021, 16, 43. [Google Scholar] [CrossRef]
- Kasetti, R.B.; Maddineni, P.; Millar, J.C.; Clark, A.F.; Zode, G.S. Increased synthesis and deposition of extracellular matrix proteins leads to endoplasmic reticulum stress in the trabecular meshwork. Sci. Rep. 2017, 7, 14951. [Google Scholar] [CrossRef] [Green Version]
- Kasetti, R.B.; Patel, P.D.; Maddineni, P.; Zode, G.S. Ex-vivo cultured human corneoscleral segment model to study the effects of glaucoma factors on trabecular meshwork. PLoS ONE 2020, 15, e0232111. [Google Scholar] [CrossRef]
- Chatterjee, A.; Villarreal, G.; Oh, D.J.; Kang, M.H.; Rhee, D.J. AMP-activated protein kinase regulates intraocular pressure, extracellular matrix, and cytoskeleton in trabecular meshwork. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3127–3139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukal, D.K.; Malaviya, P.B.; Sharma, T. Role of the AMPK signalling pathway in the aetiopathogenesis of ocular diseases. Hum. Exp. Toxicol. 2022, 41, 9603271211063165. [Google Scholar] [CrossRef] [PubMed]
- Stasi, C.; Silvestri, C.; Voller, F. Update on Hepatitis C Epidemiology: Unaware and Untreated Infected Population Could Be the Key to Elimination. SN Compr. Clin. Med. 2020, 2, 2808–2815. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Altamirano, M.M.B.; Kolstoe, S.E.; Sánchez-García, F.J. Virus Control of Cell Metabolism for Replication and Evasion of Host Immune Responses. Front. Cell. Infect. Microbiol. 2019, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Street, A.; Macdonald, A.; Crowder, K.; Harris, M. The Hepatitis C virus NS5A protein activates a phosphoinositide 3-kinase-dependent survival signaling cascade. J. Biol. Chem. 2004, 279, 12232–12241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keerthana, C.K.; Rayginia, T.P.; Shifana, S.C.; Anto, N.P.; Kalimuthu, K.; Isakov, N.; Anto, R.J. The role of AMPK in cancer metabolism and its impact on the immunomodulation of the tumor microenvironment. Front. Immunol. 2023, 14, 1114582. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef]
- Luo, Z.; Zang, M.; Guo, W. AMPK as a metabolic tumor suppressor: Control of metabolism and cell growth. Future Oncol. 2010, 6, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Saikia, R.; Joseph, J. AMPK: A key regulator of energy stress and calcium-induced autophagy. J. Mol. Med. 2021, 99, 1539–1551. [Google Scholar] [CrossRef]
- Jiang, X.; Tan, H.Y.; Teng, S.; Chan, Y.T.; Wang, D.; Wang, N. The Role of AMP-Activated Protein Kinase as a Potential Target of Treatment of Hepatocellular Carcinoma. Cancers 2019, 11, 647. [Google Scholar] [CrossRef] [Green Version]
- Vara-Ciruelos, D.; Dandapani, M.; Russell, F.M.; Grzes, K.M.; Atrih, A.; Foretz, M.; Viollet, B.; Lamont, D.J.; Cantrell, D.A.; Hardie, D.G. Phenformin, But Not Metformin, Delays Development of T Cell Acute Lymphoblastic Leukemia/Lymphoma via Cell-Autonomous AMPK Activation. Cell Rep. 2019, 27, 690–698.e694. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Cespedes, M.; Parrella, P.; Esteller, M.; Nomoto, S.; Trink, B.; Engles, J.M.; Westra, W.H.; Herman, J.G.; Sidransky, D. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002, 62, 3659–3662. [Google Scholar]
- Wingo, S.N.; Gallardo, T.D.; Akbay, E.A.; Liang, M.C.; Contreras, C.M.; Boren, T.; Shimamura, T.; Miller, D.S.; Sharpless, N.E.; Bardeesy, N.; et al. Somatic LKB1 mutations promote cervical cancer progression. PLoS ONE 2009, 4, e5137. [Google Scholar] [CrossRef]
- Hardie, D.G. Molecular Pathways: Is AMPK a Friend or a Foe in Cancer? Clin. Cancer Res. 2015, 21, 3836–3840. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Mills, G.B. AMPK: A contextual oncogene or tumor suppressor? Cancer Res. 2013, 73, 2929–2935. [Google Scholar] [CrossRef] [Green Version]
- Chhipa, R.R.; Fan, Q.; Anderson, J.; Muraleedharan, R.; Huang, Y.; Ciraolo, G.; Chen, X.; Waclaw, R.; Chow, L.M.; Khuchua, Z.; et al. AMP kinase promotes glioblastoma bioenergetics and tumour growth. Nat. Cell Biol. 2018, 20, 823–835. [Google Scholar] [CrossRef]
- Chaube, B.; Malvi, P.; Singh, S.V.; Mohammad, N.; Viollet, B.; Bhat, M.K. AMPK maintains energy homeostasis and survival in cancer cells via regulating p38/PGC-1α-mediated mitochondrial biogenesis. Cell Death Discov. 2015, 1, 15063. [Google Scholar] [CrossRef] [Green Version]
- Sadria, M.; Seo, D.; Layton, A.T. The mixed blessing of AMPK signaling in Cancer treatments. BMC Cancer 2022, 22, 105. [Google Scholar] [CrossRef]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef] [Green Version]
- Allen, K.M.; Coughlan, K.A.; Mahmood, F.N.; Valentine, R.J.; Ruderman, N.B.; Saha, A.K. The effects of troglitazone on AMPK in HepG2 cells. Arch. Biochem. Biophys. 2017, 623–624, 49–57. [Google Scholar] [CrossRef]
- Karunakaran, U.; Elumalai, S.; Moon, J.S.; Won, K.C. Pioglitazone-induced AMPK-Glutaminase-1 prevents high glucose-induced pancreatic β-cell dysfunction by glutathione antioxidant system. Redox Biol. 2021, 45, 102029. [Google Scholar] [CrossRef]
- Li, J.; Feng, Z.; Lu, B.; Fang, X.; Huang, D.; Wang, B. Resveratrol alleviates high glucose-induced oxidative stress and apoptosis in rat cardiac microvascular endothelial cell through AMPK/Sirt1 activation. Biochem. Biophys. Rep. 2023, 34, 101444. [Google Scholar] [CrossRef]
- Jin, T.; Zhang, Y.; Botchway, B.O.A.; Huang, M.; Lu, Q.; Liu, X. Quercetin activates the Sestrin2/AMPK/SIRT1 axis to improve amyotrophic lateral sclerosis. Biomed. Pharmacother. 2023, 161, 114515. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Wang, H.; Yang, Y.; Yao, Y.; Ma, H. Genistein activated SIRT1-AMPK signaling pathway mediated by ERβ-FOXO1-Nampt to reduce fat accumulation in chicken hepatocytes. Life Sci. 2023, 312, 121259. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Ding, Y.W.; Wang, L.L.; Jiang, J.D. Berberine stimulates lysosomal AMPK independent of PEN2 and maintains cellular AMPK activity through inhibiting the dephosphorylation regulator UHRF1. Front. Pharmacol. 2023, 14, 1148611. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Joshi, S.; Semwal, D.; Bisht, A.; Paliwal, S.; Dwivedi, J.; Sharma, S. Curcumin: An Insight into Molecular Pathways Involved in Anticancer Activity. Mini Rev. Med. Chem. 2021, 21, 2420–2457. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Pan, Y.; Wang, S.; Liu, Y.; Chen, G.; Zhou, L.; Ni, W.; Wang, A.; Lu, Y. Cryptotanshinone activates AMPK-TSC2 axis leading to inhibition of mTORC1 signaling in cancer cells. BMC Cancer 2017, 17, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Göransson, O.; McBride, A.; Hawley, S.A.; Ross, F.A.; Shpiro, N.; Foretz, M.; Viollet, B.; Hardie, D.G.; Sakamoto, K. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J. Biol. Chem. 2007, 282, 32549–32560. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.Y.; Chen, F.L.; Ji, F.; Fei, H.D.; Xie, Y.; Wang, S.G. Activation of AMP-activated protein kinase by compound 991 protects osteoblasts from dexamethasone. Biochem. Biophys. Res. Commun. 2018, 495, 1014–1021. [Google Scholar] [CrossRef]
- Takaya, K.; Okabe, K.; Sakai, S.; Aramaki-Hattori, N.; Asou, T.; Kishi, K. Compound 13 Promotes Epidermal Healing in Mouse Fetuses via Activation of AMPK. Biomedicines 2023, 11, 1013. [Google Scholar] [CrossRef]
- Jensen, T.E.; Ross, F.A.; Kleinert, M.; Sylow, L.; Knudsen, J.R.; Gowans, G.J.; Hardie, D.G.; Richter, E.A. PT-1 selectively activates AMPK-γ1 complexes in mouse skeletal muscle, but activates all three γ subunit complexes in cultured human cells by inhibiting the respiratory chain. Biochem. J. 2015, 467, 461–472. [Google Scholar] [CrossRef]
- Zadra, G.; Photopoulos, C.; Tyekucheva, S.; Heidari, P.; Weng, Q.P.; Fedele, G.; Liu, H.; Scaglia, N.; Priolo, C.; Sicinska, E.; et al. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol. Med. 2014, 6, 519–538. [Google Scholar] [CrossRef]
Figure 1.
The domain structure of AMPK heterotrimer. Functional AMPK complexes consist of one catalytic and two regulatory subunits. Upon activation, AMPK exerts its effects by decreasing energy-consuming anabolic processes, including lipid synthesis, glycogen storage, gluconeogenesis, and protein synthesis. Simultaneously, it enhances energy-providing catabolic processes that generate ATP, such as glucose metabolism, lipid oxidation, mitochondrial biogenesis, and autophagy. Arrows represent the increase (green) or decrease (red) in AMPK activity.
Figure 1.
The domain structure of AMPK heterotrimer. Functional AMPK complexes consist of one catalytic and two regulatory subunits. Upon activation, AMPK exerts its effects by decreasing energy-consuming anabolic processes, including lipid synthesis, glycogen storage, gluconeogenesis, and protein synthesis. Simultaneously, it enhances energy-providing catabolic processes that generate ATP, such as glucose metabolism, lipid oxidation, mitochondrial biogenesis, and autophagy. Arrows represent the increase (green) or decrease (red) in AMPK activity.

Figure 2.
AMPK plays a regulatory role in several metabolic targets. These metabolic pathways governed by AMPK can be classified into four main categories: Glucose metabolism, lipid metabolism, autophagy, and mitochondrial metabolism.
Figure 2.
AMPK plays a regulatory role in several metabolic targets. These metabolic pathways governed by AMPK can be classified into four main categories: Glucose metabolism, lipid metabolism, autophagy, and mitochondrial metabolism.

Figure 3.
Representative links between AMPK and human pathologies.


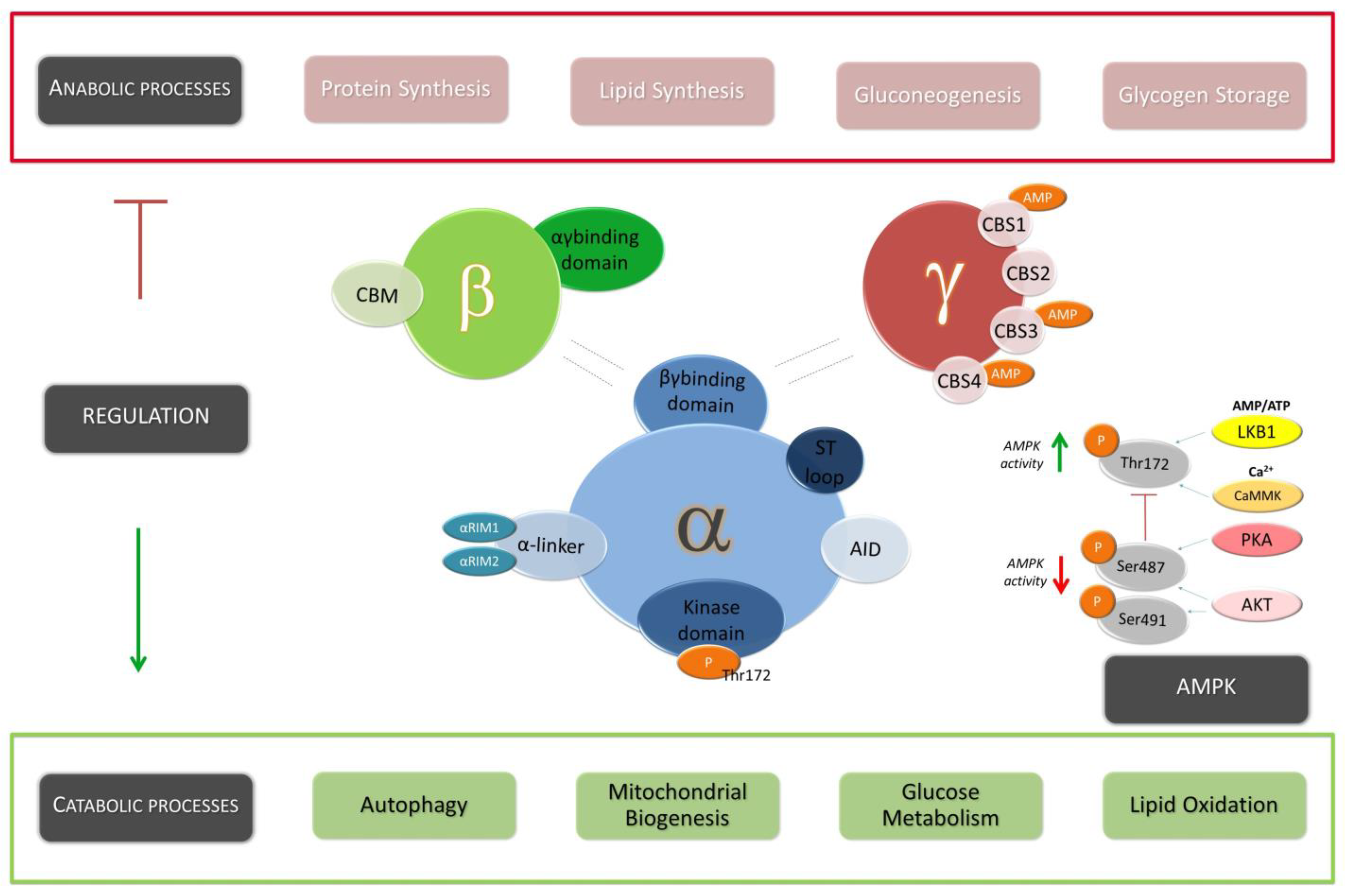{kind=link}
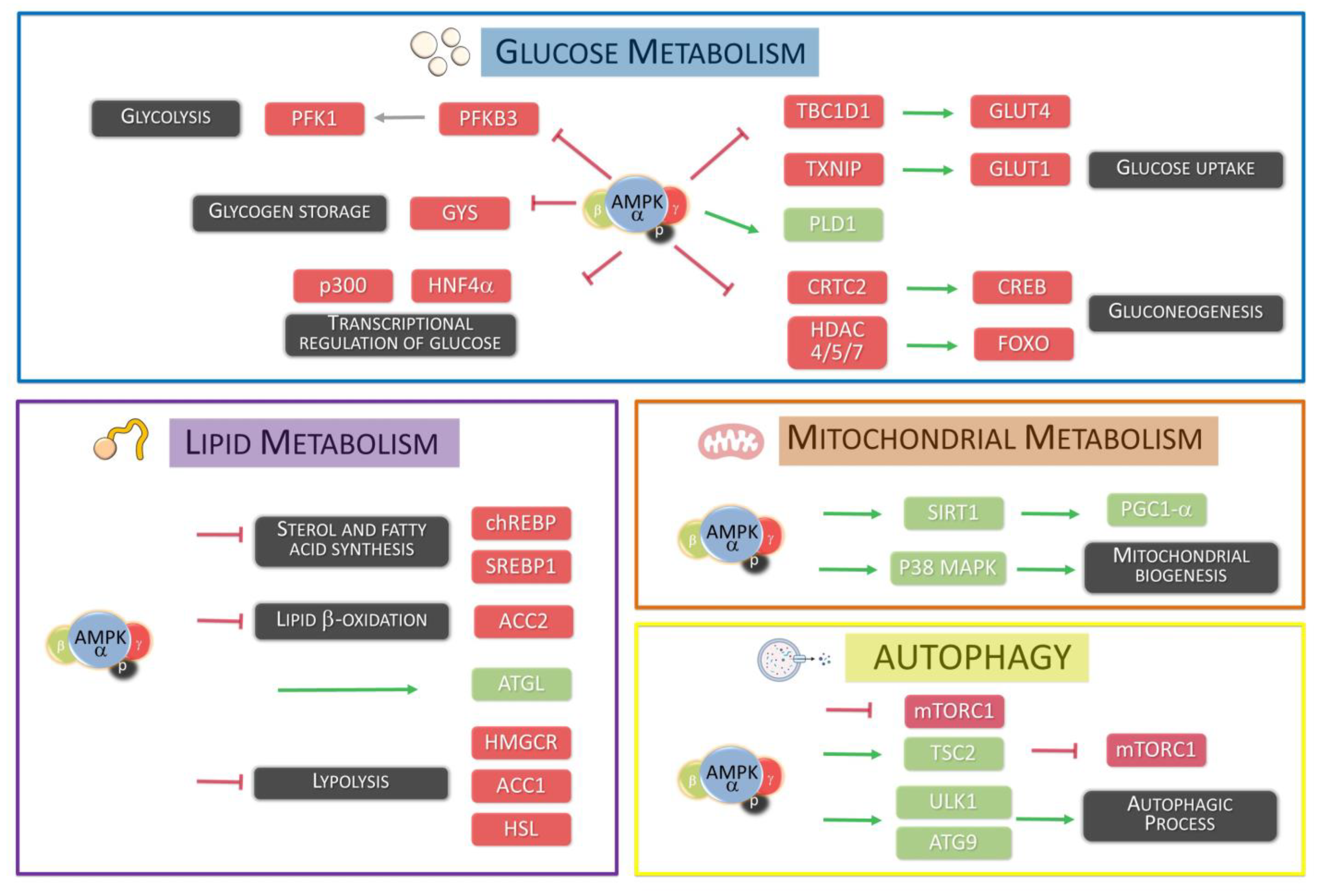{kind=link}
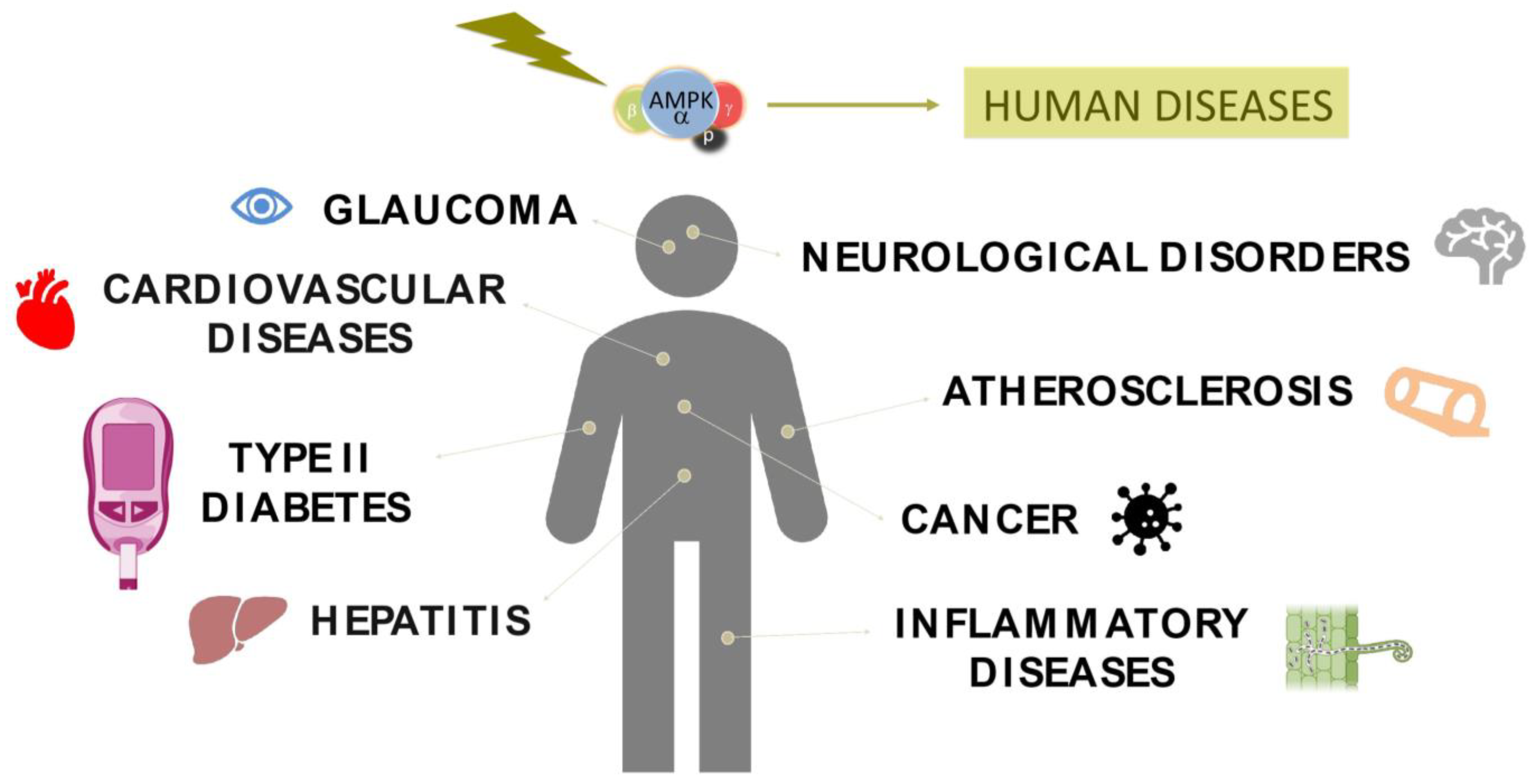{kind=link}
Table 1.
Clinically relevant SNPs in AMPK.
| Gen | dbSNP rsID | Disease | Ref. |
|---|---|---|---|
| PRKAA1 | rs13361707 | Gastric cancer | [80] |
| rs13361707 | Colorectal cancer | [81] | |
| rs10074991 | Colorectal cancer | [81] | |
| rs3792822 | Hepatitis B | [82] | |
| rs10074991 | Gastric cancer | [83] | |
| rs461404 | Gastric cancer | [83] | |
| rs154268 | Gastric cancer | [83] | |
| PRKAA2 | rs10789038 | Neuropathic pain | [84] |
| rs10789038 | Type 2 diabetes | [85] | |
| rs2796498 | Type 2 diabetes | [85] | |
| rs2746342 | Type 2 diabetes | [86] | |
| rs10224002 | Hypertension | [87] | |
| rs121908987 | Cardiac glycogenosis | [88] | |
| PRKAG1 | rs1138908 | Colorectal cancer | [81] |
| PRKAG2 | rs1029947 | Colorectal cancer | [89] |
| rs1104897 | Colorectal cancer | [89] | |
| rs5017427 | Type 2 diabetes | [90] | |
| rs954482 | Type 2 diabetes | [90] | |
| rs2727537 | Type 2 diabetes | [90] |
Table 2.
Potential AMPK activators.
| Compound | Activation Mechanism | Target | Ref. |
|---|---|---|---|
| Metformin | Increase of AMP-ATP ratio | Complex I (mitochondrial respiratory chain) | [80,135] |
| Troglitazone | Increase of AMP-ATP ratio | Complex I (mitochondrial respiratory chain) | [178] |
| Pioglitazone | Increase of AMP-ATP ratio | Complex I (mitochondrial respiratory chain) | [179] |
| Resveratrol | Increase of AMP-ATP ratio | F1F0-ATPase-ATP synthase | [180] |
| Quercetin | Increase of AMP-ATP ratio | F1F0-ATPase-ATP synthase | [181] |
| Genistein | Increase of AMP-ATP ratio | F1F0-ATPase-ATP synthase | [182] |
| Berberine | Increase of AMP-ATP ratio | Complex I (mitochondrial respiratory chain) | [183] |
| Curcumin | Increase of AMP-ATP ratio | F1F0-ATPase-ATP synthase | [184] |
| Cryptotanshinone | Increase of ROS | unknown | [185] |
| AICAR | Direct AMPK activation | No isoform specificity | [104] |
| A-769662 | Direct AMPK activation | β1 subunit specificity | [186] |
| Benzimidazole (Compound 911) | Direct AMPK activation | β1 subunit specificity | [187] |
| Compound 13 | Direct AMPK activation | α1 subunit specificity | [188] |
| PT-1 | Direct AMPK activation | y1 subunit specificity | [189] |
| MT 63–78 | Direct AMPK activation | β1 subunit specificity | [190] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rey, V.; Tamargo-Gómez, I. From Kinases to Diseases: Investigating the Role of AMPK in Human Pathologies. Kinases Phosphatases 2023, 1, 181-205. https://doi.org/10.3390/kinasesphosphatases1030012
AMA Style
Rey V, Tamargo-Gómez I. From Kinases to Diseases: Investigating the Role of AMPK in Human Pathologies. Kinases and Phosphatases. 2023; 1(3):181-205. https://doi.org/10.3390/kinasesphosphatases1030012
Chicago/Turabian StyleRey, Verónica, and Isaac Tamargo-Gómez. 2023. "From Kinases to Diseases: Investigating the Role of AMPK in Human Pathologies" Kinases and Phosphatases 1, no. 3: 181-205. https://doi.org/10.3390/kinasesphosphatases1030012