
Triazole Derivatives Target 14α–Demethylase (LDM) Enzyme in Candida albicans Causing Ergosterol Biosynthesis Inhibition

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Candida Strains
2.2. Synthesis of Triazole Derivatives
2.3. In Silico Methodology
2.3.1. Molecular Docking
2.3.2. Molecular Dynamics (MD) Simulations
2.3.3. Post-Dynamics MD Analyses
2.3.4. Binding Free Energy Calculations
2.4. In Vitro Methodology
2.4.1. Antifungal Susceptibility
2.4.2. Ergosterol Biosynthesis Assay
2.4.3. Gene Expression
2.5. Effect on Biofilms
2.5.1. XTT Reduction Assay
2.5.2. Confocal Laser Scanning Microscopy
2.6. Statistics
3. Results and Discussion
3.1. In Silico Results
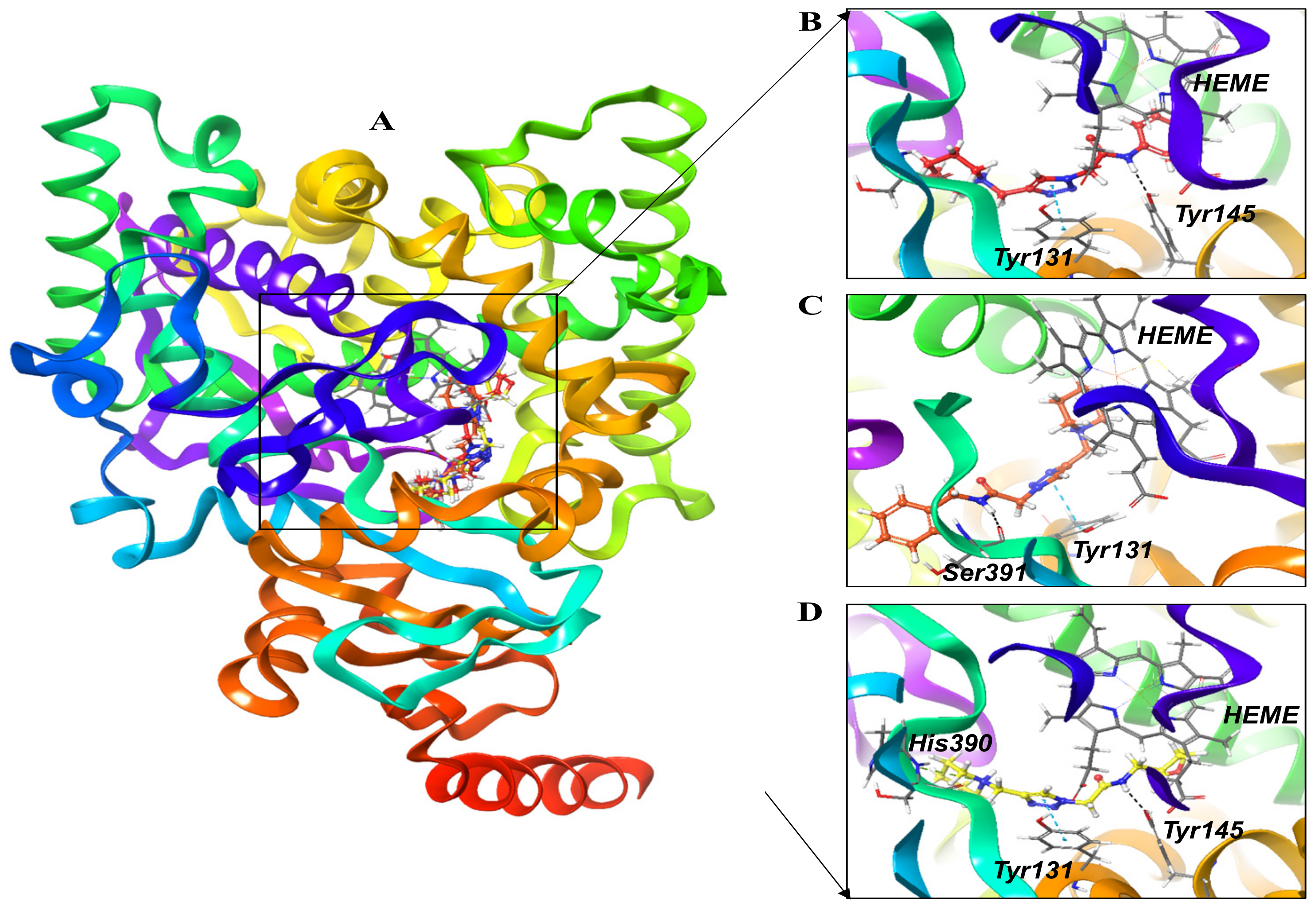
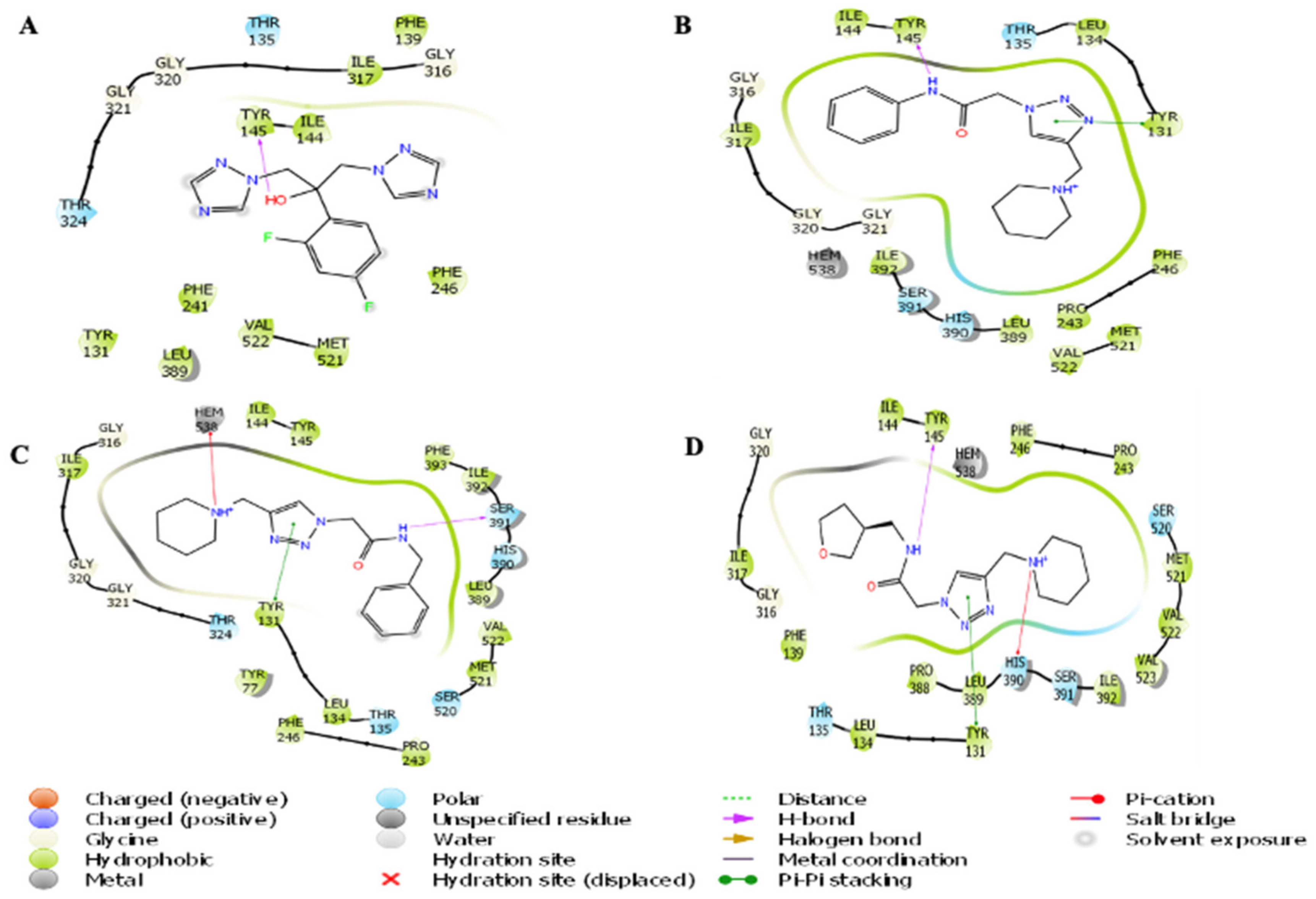
3.1.1. Molecular Docking Analysis
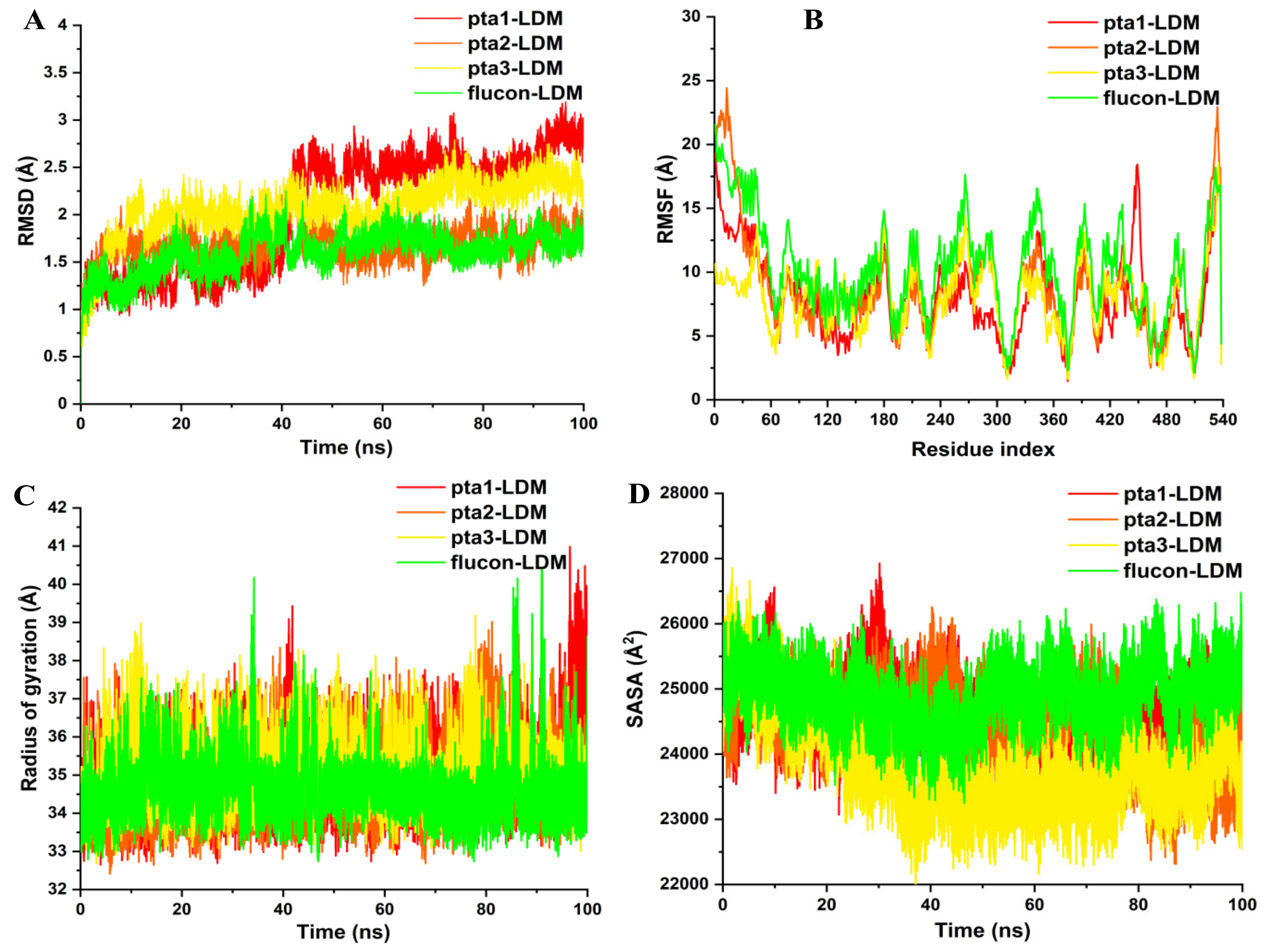
3.1.2. Post-Dynamics Trajectories Analysis
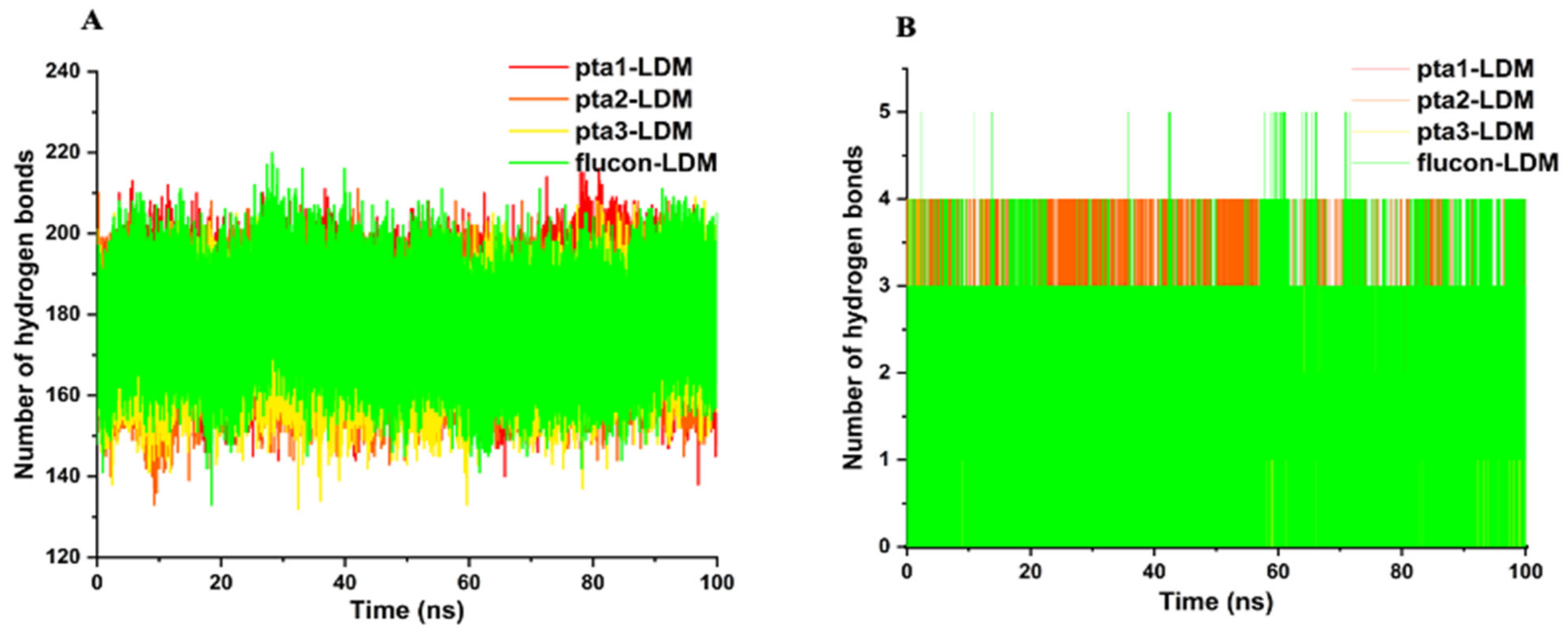
3.1.3. Hydrogen Bond Analysis
3.1.4. Mechanistic Insight into Free Binding Energy
3.2. In Vitro Results
3.2.1. Antifungal Susceptibility
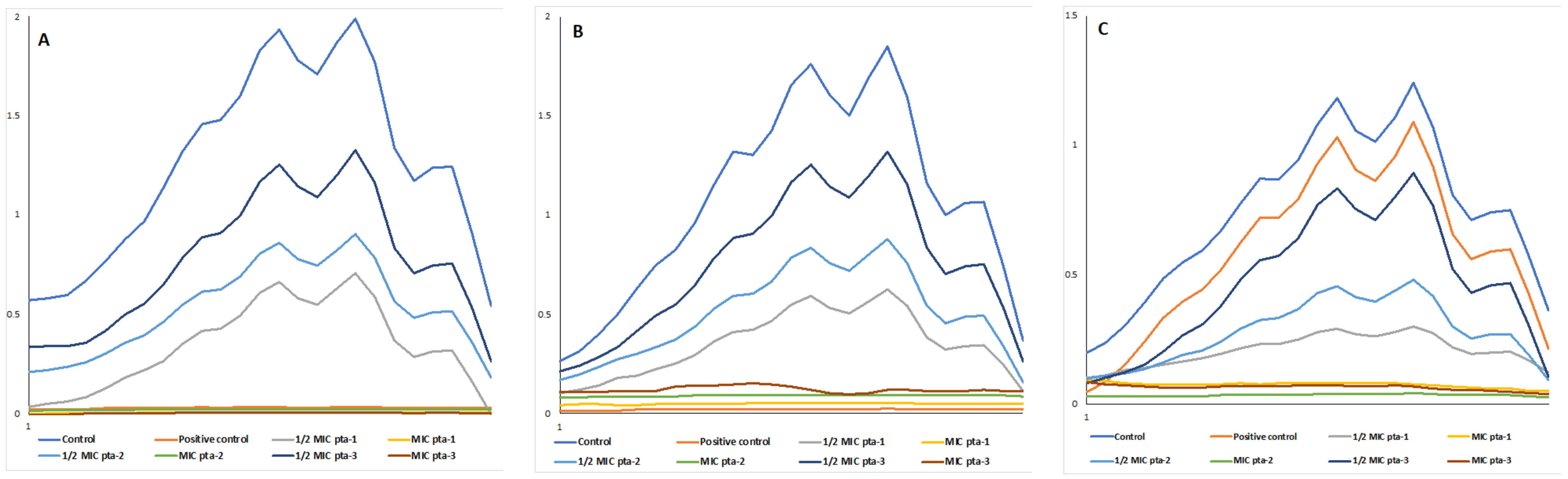
3.2.2. Ergosterol Biosynthesis
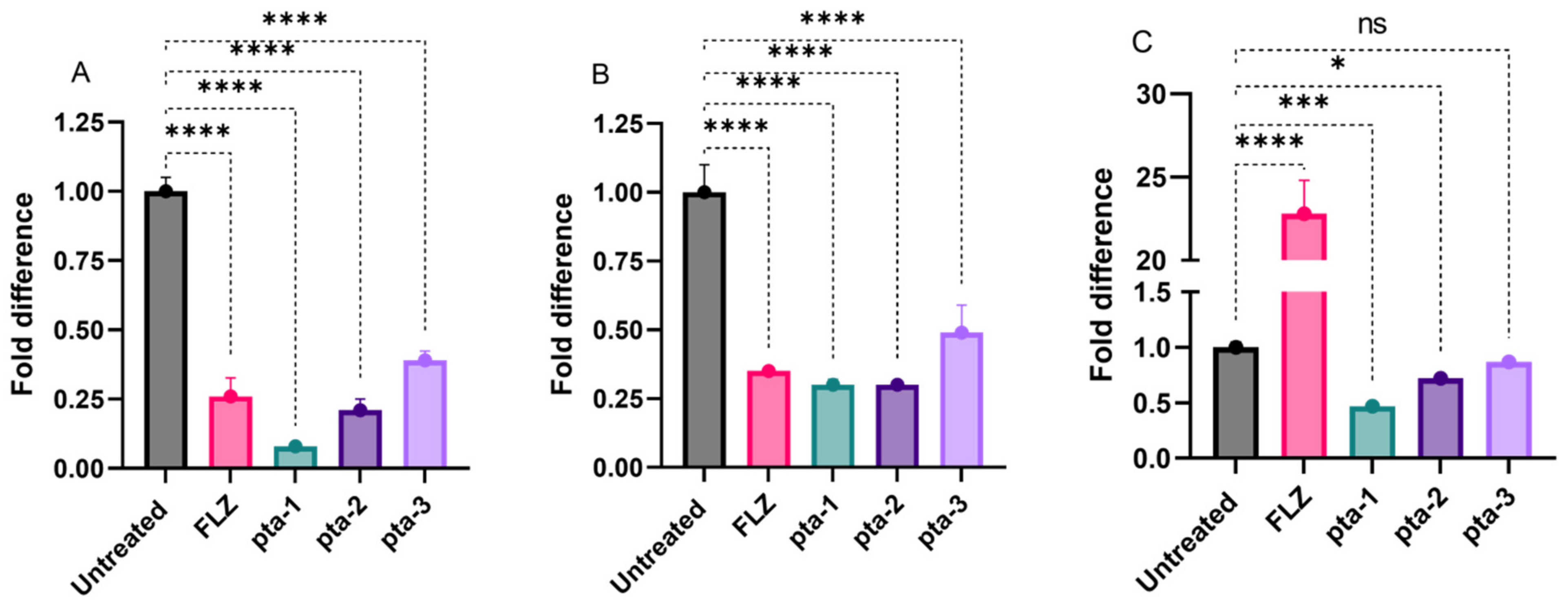
3.2.3. Gene Expression
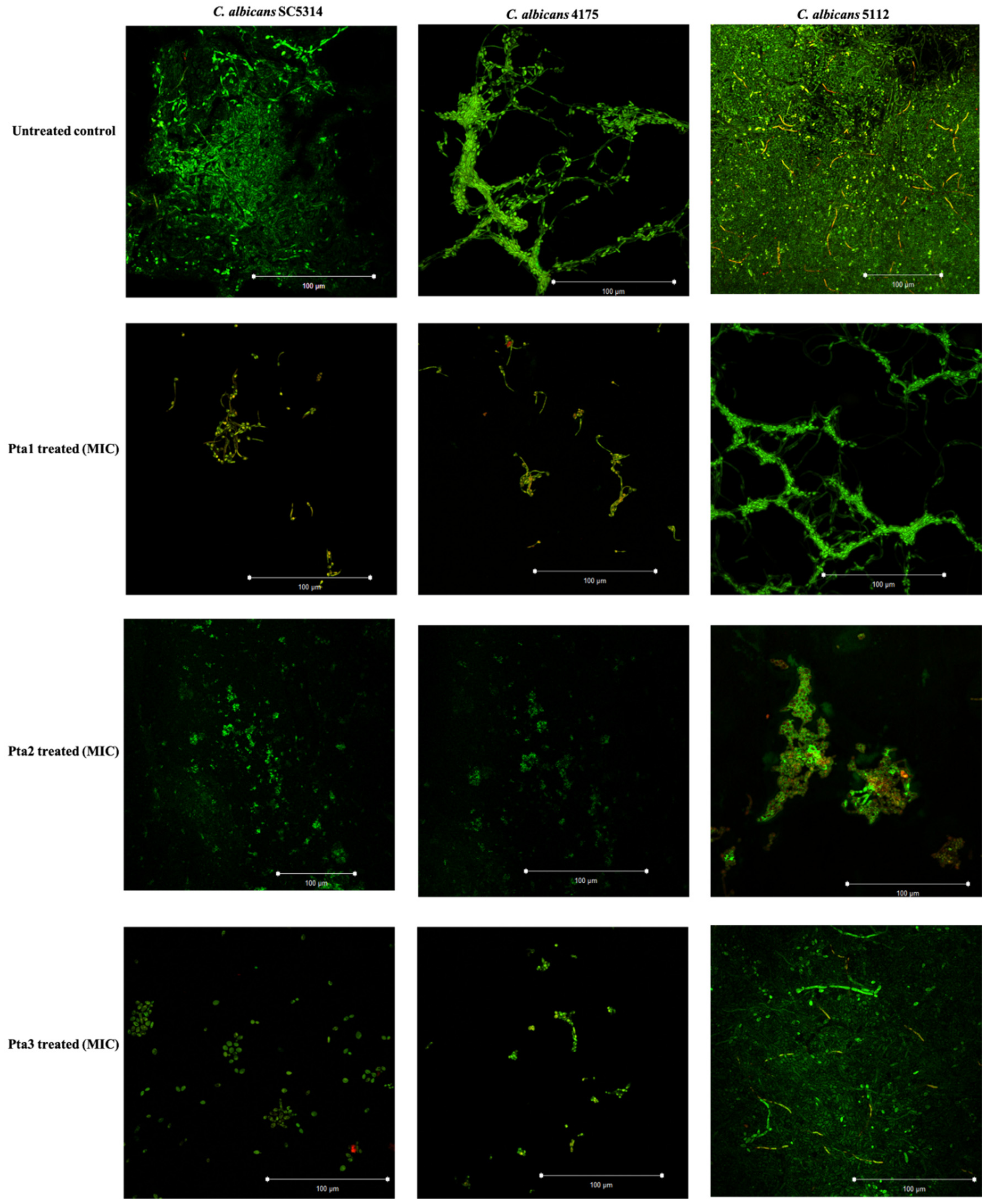
3.2.4. Biofilm Formation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Low, C.Y.; Rotstein, C. Emerging Fungal Infections in Immunocompromised Patients. F1000 Med. Rep. 2011, 3, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, G.D.; Denning, D.W.; Levitz, S.M. Tackling Human Fungal Infections. Science 2012, 336, 647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, G.D.; Denning, D.W.; Gow, N.A.R.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden Killers: Human Fungal Infections. Sci. Transl. Med. 2012, 4, 165rv13. [Google Scholar] [CrossRef] [Green Version]
- Sievert, D.M.; Ricks, P.; Edwards, J.R.; Schneider, A.; Patel, J.; Srinivasan, A.; Kallen, A.; Limbago, B.; Fridkin, S. Antimicrobial-Resistant Pathogens Associated with Healthcare-Associated Infections Summary of Data Reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009–2010. Infect. Control. Hosp. Epidemiol. 2013, 34, 1–14. [Google Scholar] [CrossRef]
- Rather, I.A.; Sabir, J.S.M.; Asseri, A.H.; Ali, S. Antifungal Activity of Human Cathelicidin LL-37, a Membrane Disrupting Peptide, by Triggering Oxidative Stress and Cell Cycle Arrest in Candida auris. J. Fungi 2022, 8, 204. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, E.; Hammarström, H.; Ataollahy, N.; Kondori, N. Species Distribution and Antifungal Drug Susceptibilities of Yeasts Isolated from the Blood Samples of Patients with Candidemia. Sci. Rep. 2019, 9, 3838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajput, S.B.; Mohan Karuppayil, S. Small Molecules Inhibit Growth, Viability and Ergosterol Biosynthesis in Candida albicans. SpringerPlus 2013, 2, 26. [Google Scholar] [CrossRef] [Green Version]
- Barrett-Bee, K.; Dixon, G. Ergosterol Biosynthesis Inhibition: A Target for Antifungal Agents. Acta Biochim. Pol. 1995, 42, 465–479. [Google Scholar] [CrossRef] [Green Version]
- Benkoő, I.; Hernádi, F.; Megyeri, A.; Kiss, A.; Somogyi, G.; Tegyey, Z.; Kraicsovits, F.; Kovács, P. Comparison of the toxicity of fluconazole and other azole antifungal drugs to murine and human granulocyte -macrophage progenitor cells in vitro. J. Antimicrob. Chemother. 1999, 43, 675–681. [Google Scholar] [CrossRef] [Green Version]
- Benitez, L.L.; Carver, P.L. Adverse Effects Associated with Long-Term Administration of Azole Antifungal Agents. Drugs 2019, 79, 833–853. [Google Scholar] [CrossRef]
- Peyton, L.R.; Gallagher, S.; Hashemzadeh, M. Triazole Antifungals: A Review. Drugs Today 2015, 51, 705–718. [Google Scholar]
- Berkow, E.L.; Lockhart, S. Fluconazole Resistance in Candida Species: A Current Perspective. Infect. Drug Resist. 2017, 10, 237–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wani, M.Y.; Ahmad, A.; Aqlan, F.M.; Al-Bogami, A.S. Azole Based Acetohydrazide Derivatives of Cinnamaldehyde Target and Kill Candida albicans by Causing Cellular Apoptosis. ACS Med. Chem. Lett. 2020, 11, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Gintjee, T.J.; Donnelley, M.A.; Thompson, G.R. Aspiring Antifungals: Review of Current Antifungal Pipeline Developments. J. Fungi 2020, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, V.; Wani, M.Y.; Al-Bogami, A.S.; Ahmad, A. Piperidine Based 1,2,3-Triazolylacetamide Derivatives Induce Cell Cycle Arrest and Apoptotic Cell Death in Candida auris. J. Adv. Res. 2021, 29, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput.—Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A Software Program for PKa Prediction and Protonation State Generation for Drug-like Molecules. J. Comput.—Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Chen, H.; Panagiotopoulos, A.Z. Molecular Modeling of Surfactant Micellization Using Solvent-Accessible Surface Area. Langmuir 2019, 35, 2443–2450. [Google Scholar] [CrossRef]
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpí, J.L. Molecular Dynamics Simulations: Advances and Applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37. [Google Scholar]
- Lee, T.S.; Cerutti, D.S.; Mermelstein, D.; Lin, C.; Legrand, S.; Giese, T.J.; Roitberg, A.; Case, D.A.; Walker, R.C.; York, D.M. GPU-Accelerated Molecular Dynamics and Free Energy Methods in Amber18: Performance Enhancements and New Features. J. Chem. Inf. Modeling 2018, 58, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Perez, A.; MacCallum, J.L.; Brini, E.; Simmerling, C.; Dill, K.A. Grid-Based Backbone Correction to the Ff12SB Protein Force Field for Implicit-Solvent Simulations. J. Chem. Theory Comput. 2015, 11, 4770–4779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrach, M.F.; Drossel, B. Structure and Dynamics of TIP3P, TIP4P, and TIP5P Water near Smooth and Atomistic Walls of Different Hydroaffinity. J. Chem. Phys. 2014, 140, 174501. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Pan, D.; Li, J.; Zhang, L.; Shao, X. Application of Berendsen Barostat in Dissipative Particle Dynamics for Nonequilibrium Dynamic Simulation. J. Chem. Phys. 2017, 146, 124108. [Google Scholar] [CrossRef]
- Gonnet, P. P-SHAKE: A Quadratically Convergent SHAKE in O (N2). J. Comput. Phys. 2007, 220, 740–750. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Seifert, E. OriginPro 9.1: Scientific Data Analysis and Graphing Software—Software Review. J. Chem. Inf. Modeling 2014, 54, 1552. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Modification of the Generalized Born Model Suitable for Macromolecules. J. Phys. Chem. B 2000, 104, 3712–3720. [Google Scholar] [CrossRef] [Green Version]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate Atomic Surfaces from Linear Combinations of Pairwise Overlaps (LCPO). J. Comput. Chem. 1999, 20, 217–230. [Google Scholar] [CrossRef]
- Alexander, B.D.; Procop, G.W.; Dufresne, P.; Fuller, J.; Ghannoum, M.A.; Hanson, K.E.; Holliday, D.; Holliday, N.M.; Kovanda, L.; Lockhart, S.R.; et al. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts, 4th Informational Supplement; M27–S4; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2017; Volume 28. [Google Scholar]
- Ahmad, A.; Khan, A.; Akhtar, F.; Yousuf, S.; Xess, I.; Khan, L.A.; Manzoor, N. Fungicidal Activity of Thymol and Carvacrol by Disrupting Ergosterol Biosynthesis and Membrane Integrity against Candida. Eur. J. Clin. Microbiol. Infect. Dis. 2011, 30, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Wani, M.Y.; Khan, A.; Manzoor, N.; Molepo, J. Synergistic Interactions of Eugenol-Tosylate and Its Congeners with Fluconazole against Candida albicans. PLoS ONE 2015, 10, e0145053. [Google Scholar] [CrossRef]
- Srivastava, V.; Ahmad, A. Abrogation of Pathogenic Attributes in Drug Resistant Candida auris Strains by Farnesol. PLoS ONE 2020, 15, e0233102. [Google Scholar] [CrossRef]
- Khan, S.; Bjij, I.; Soliman, M.E.S. Selective Covalent Inhibition of “Allosteric Cys121” Distort the Binding of PTP1B Enzyme: A Novel Therapeutic Approach for Cancer Treatment. Cell Biochem. Biophys. 2019, 77, 203–211. [Google Scholar] [CrossRef]
- Khan, S.; Bjij, I.; Betz, R.M.; Soliman, M.E.S. Reversible versus Irreversible Inhibition Modes of ERK2: A Comparative Analysis for ERK2 Protein Kinase in Cancer Therapy. Future Med. Chem. 2018, 10, 1003–1015. [Google Scholar] [CrossRef]
- Martínez, L. Automatic Identification of Mobile and Rigid Substructures in Molecular Dynamics Simulations and Fractional Structural Fluctuation Analysis. PLoS ONE 2015, 10, e0119264. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Khan, S.; Fakhar, Z.; Hussain, A.; Rehman, M.T.; Alajmi, M.F.; Islam, A.; Ahmad, F.; Hassan, M.I. Identification of Potential Inhibitors of Calcium/Calmodulin-Dependent Protein Kinase IV from Bioactive Phytoconstituents. Oxidative Med. Cell. Longev. 2020, 2020, 2094635. [Google Scholar] [CrossRef]
- Hata, M.; Ishii, Y.; Watanabe, E.; Uoto, K.; Kobayashi, S.; Yoshida, K.I.; Otani, T.; Ando, A. Inhibition of Ergosterol Synthesis by Novel Antifungal Compounds Targeting C-14 Reductase. Med. Mycol. 2010, 48, 613–621. [Google Scholar] [CrossRef] [Green Version]
- Suchodolski, J.; Muraszko, J.; Bernat, P.; Krasowska, A. Lactate like Fluconazole Reduces Ergosterol Content in the Plasma Membrane and Synergistically Kills Candida albicans. Int. J. Mol. Sci. 2021, 22, 5219. [Google Scholar] [CrossRef] [PubMed]
- Abe, F.; Hiraki, T. Mechanistic Role of Ergosterol in Membrane Rigidity and Cycloheximide Resistance in Saccharomyces Cerevisiae. Biochim. Biophys. Acta—Biomembr. 2009, 1788, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Sheng, F.; Zhao, J.; Chen, L.; Li, C. ERG11 Mutations and Expression of Resistance Genes in Fluconazole-Resistant Candida albicans Isolates. Arch. Microbiol. 2015, 197, 1087–1093. [Google Scholar] [CrossRef]
- Alvarez-Rueda, N.; Fleury, A.; Logé, C.; Pagniez, F.; Robert, E.; Morio, F.; Le Pape, P. The Amino Acid Substitution N136Y in Candida albicans Sterol 14alpha-Demethylase Is Involved in Fluconazole Resistance. Med. Mycol. 2016, 54, 764–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddouzi, J.; Parker, J.E.; Vale-Silva, L.A.; Coste, A.; Ischer, F.; Kelly, S.; Manai, M.; Sanglard, D. Molecular Mechanisms of Drug Resistance in Clinical Candida Species Isolated from Tunisian Hospitals. Antimicrob. Agents Chemother. 2013, 57, 3182–3193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.H.; Wei, X.; Ma, M.; Chen, X.J.; Xu, S.B. Possible Inhibitory Molecular Mechanism of Farnesol on the Development of Fluconazole Resistance in Candida albicans Biofilm. Antimicrob. Agents Chemother. 2012, 56, 770–775. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | ΔEvdW | ΔEele | ΔGgas | ΔGpolar | ΔGnonpolar | ΔGsol | ΔGbind |
|---|---|---|---|---|---|---|---|
| pta1-LDM | −41.52 | −19.39 | −67.92 | 41.03 | −4.58 | 36.45 | −43.66 |
| pta2-LDM | −41.98 | −18.17 | −61.67 | 39.07 | −6.31 | −32.76 | −40.10 |
| pta3-LDM | −39.80 | −21.64 | −61.45 | 30.43 | −4.26 | 26.18 | −42.66 |
| FLZ-LDM | −43.98 | −14.32 | −58.37 | 32.21 | −5.16 | 27.04 | −31.27 |
| Isolates | Fluconazole Susceptibility | pta1 (µg/mL) | pta2 (µg/mL) | pta3 (µg/mL) | FLZ | |||
|---|---|---|---|---|---|---|---|---|
| MIC | MFC | MIC | MFC | MIC | MFC | |||
| C. albicans SC5314 | Susceptible | 0.024 | 0.097 | 0.097 | 0.390 | 0.781 | 1.560 | 0.250 |
| C. albicans 4175 | Susceptible | 0.048 | 0.097 | 0.097 | 0.390 | 1.560 | 3.125 | 0.125 |
| C. albicans 5112 | Resistant | 0.390 | 0.781 | 1.560 | 1.560 | 3.125 | 6.250 | 64.00 |
| Candida Strains | Test Compounds | Mean Ergosterol Content * | |
|---|---|---|---|
| C. albicans SC5314 | Negative control | 0.0231 | |
| Positive control | 0.00027 (99) ** | ||
| pta1 | ½ MIC | 0.00834 (64) ** | |
| MIC | 0.00085 (96) ** | ||
| pta2 | ½ MIC | 0.01035 (55) ** | |
| MIC | 0.00131(94) ** | ||
| pta3 | ½ MIC | 0.01543 (33) ** | |
| MIC | 0.00208 (91) ** | ||
| C. albicans 4175 | Negative control | 0.01911 | |
| Positive control | 0.00037 (98) ** | ||
| pta1 | ½ MIC | 0.00647 (66) ** | |
| MIC | 0.00055 (97) ** | ||
| pta2 | ½ MIC | 0.00915 (52) ** | |
| MIC | 0.00093 (95) ** | ||
| pta3 | ½ MIC | 0.0137 (28) ** | |
| MIC | 0.0013 (93) ** | ||
| C. albicans 5112 | Negative control | 0.01311 | |
| Positive control | 0.01141 (13) ** | ||
| pta1 | ½ MIC | 0.00341 (74) ** | |
| MIC | 0.00054 (96) ** | ||
| pta2 | ½ MIC | 0.0055 (58) ** | |
| MIC | 0.00089 (93) ** | ||
| pta3 | ½ MIC | 0.01007 (23) ** | |
| MIC | 0.0012 (91) ** | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rather, I.A.; Sabir, J.S.M.; Asseri, A.H.; Wani, M.Y.; Ahmad, A. Triazole Derivatives Target 14α–Demethylase (LDM) Enzyme in Candida albicans Causing Ergosterol Biosynthesis Inhibition. J. Fungi 2022, 8, 688. https://doi.org/10.3390/jof8070688
Rather IA, Sabir JSM, Asseri AH, Wani MY, Ahmad A. Triazole Derivatives Target 14α–Demethylase (LDM) Enzyme in Candida albicans Causing Ergosterol Biosynthesis Inhibition. Journal of Fungi. 2022; 8(7):688. https://doi.org/10.3390/jof8070688
Chicago/Turabian StyleRather, Irfan A., Jamal S. M. Sabir, Amer H. Asseri, Mohmmad Younus Wani, and Aijaz Ahmad. 2022. "Triazole Derivatives Target 14α–Demethylase (LDM) Enzyme in Candida albicans Causing Ergosterol Biosynthesis Inhibition" Journal of Fungi 8, no. 7: 688. https://doi.org/10.3390/jof8070688