
Infection Process and Genome Assembly Provide Insights into the Pathogenic Mechanism of Destructive Mycoparasite Calcarisporium cordycipiticola with Host Specificity



Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Culture Conditions
2.2. Agrobacterium tumefaciens-mediated Transformation (ATMT) of Calcarisporium cordycipiticola
2.3. Transformation of RFP-Tagged Calcarisporium cordycipiticola and Microscopy
2.4. Infection of Calcarisporium cordycipiticola to the Other Species of Cordycipitaceae
2.5. DNA/RNA Preparation
2.6. Genome Sequencing and Assembly
2.7. Assembly and Annotation of Mitogenomes
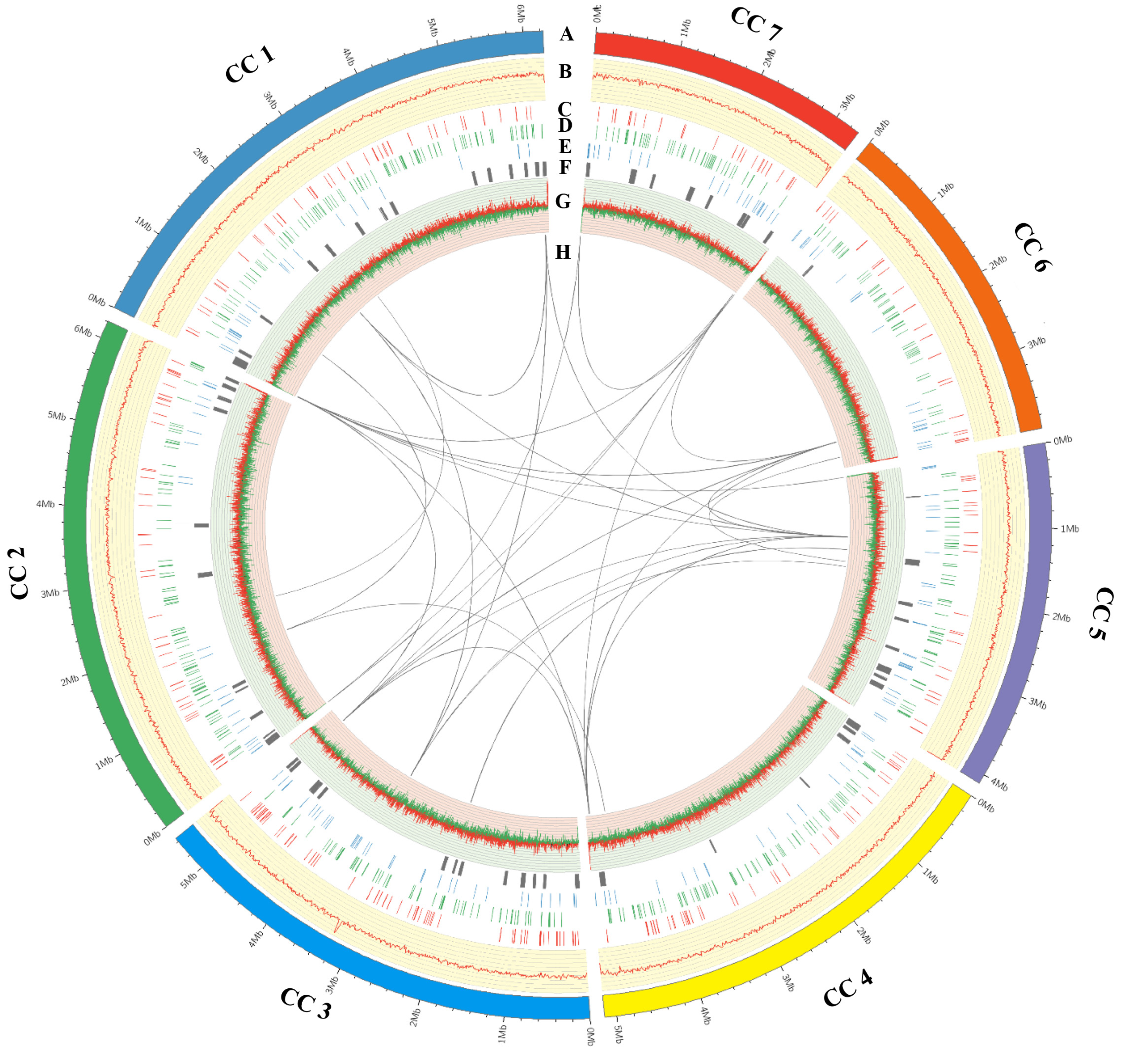
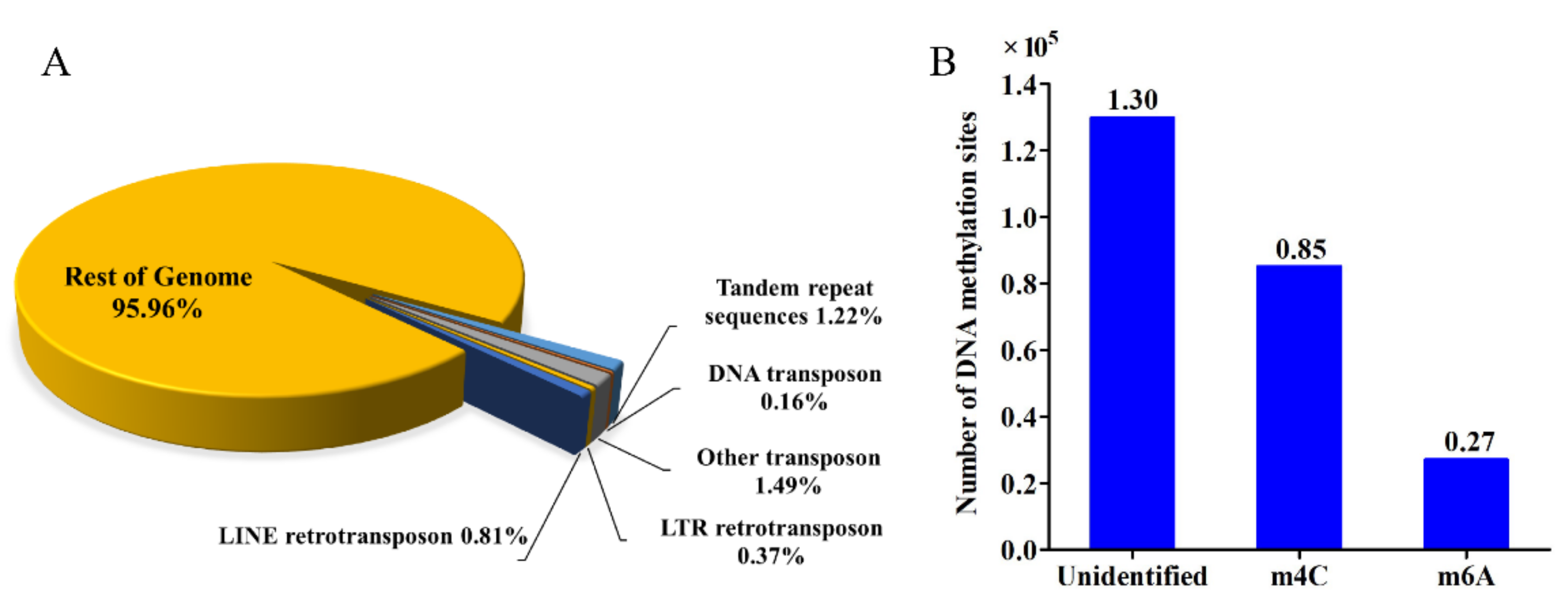
2.8. Repetitive Sequences, Transposases, Repeat-Induced Point Mutation and Whole Genome DNA Methylation Analysis
2.9. Genome Annotations
2.10. Functional Annotation of Pathogenicity-Related Genes
2.11. Phylogenomic Analysis
2.12. Comparison Analysis of Carbohydrate-Active Enzymes
2.13. Analysis of Genes Involved in Secondary Metabolism
2.14. Experimental Design and Statistical Analysis
3. Results
3.1. Development of ATMT for Efficient Transformation of Calcarisporium cordycipiticola and RFP-Labeling
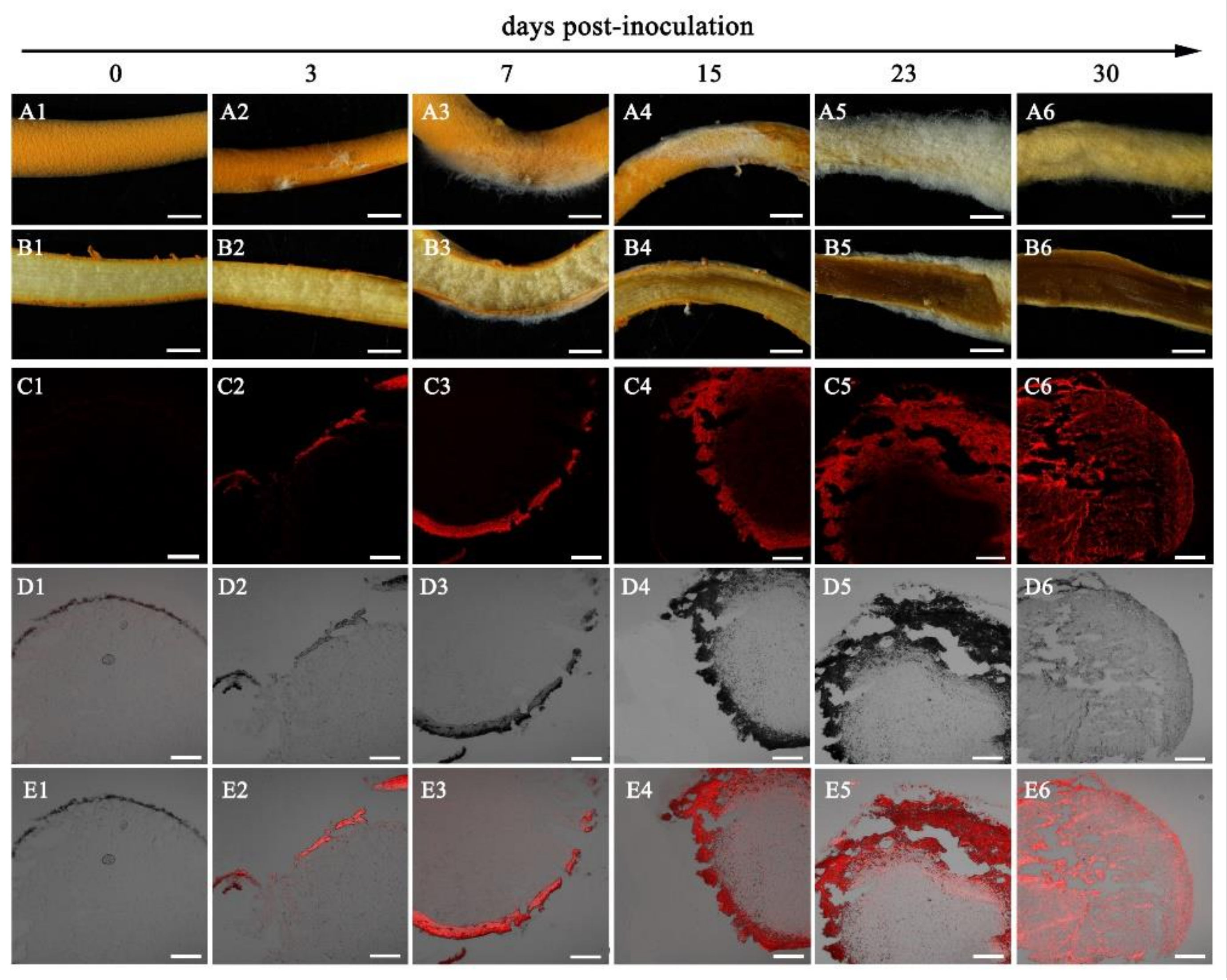
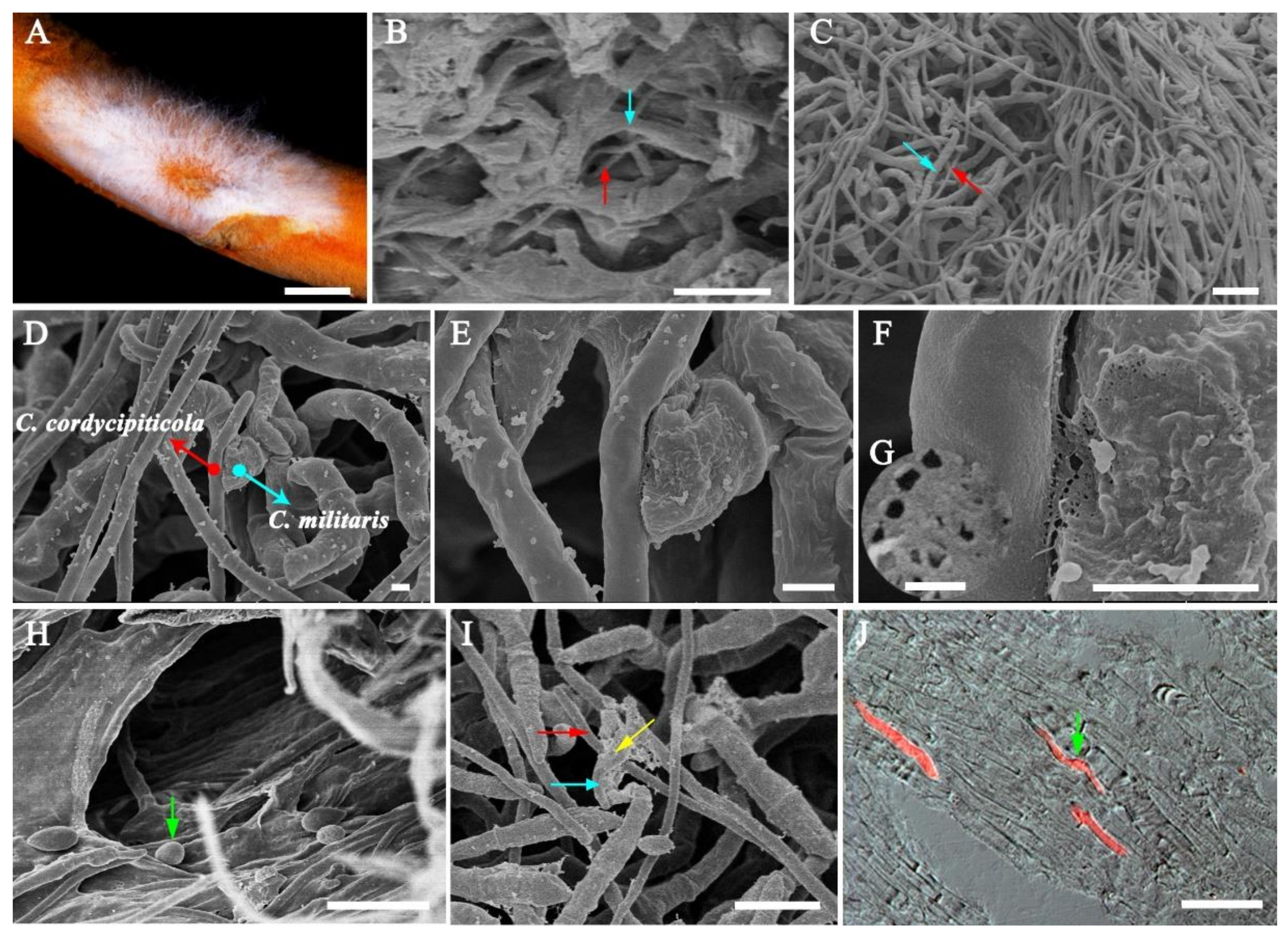
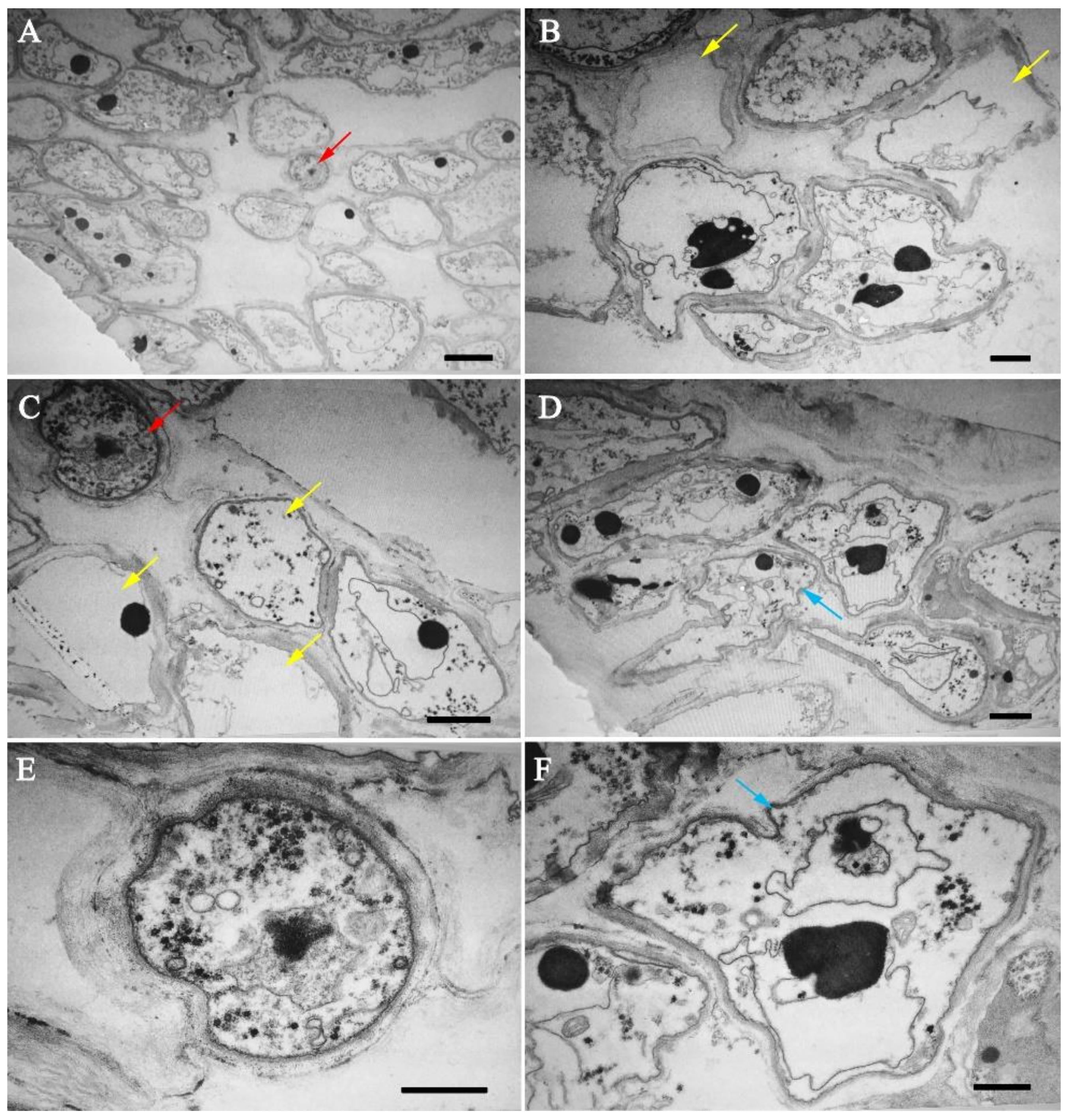
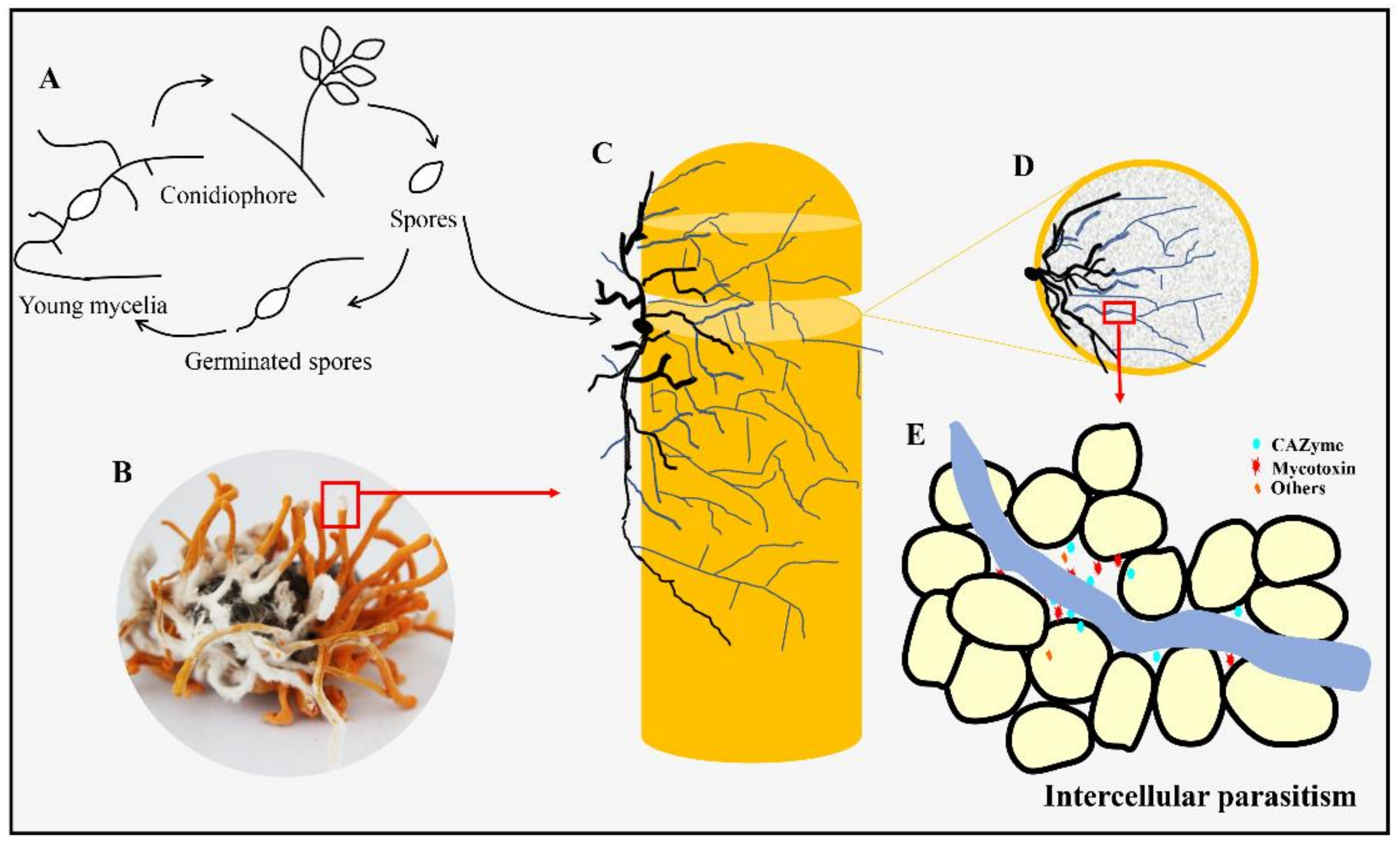
3.2. Infection Process of Calcarisporium cordycipiticola to the Fruiting Body of Cordyceps militaris
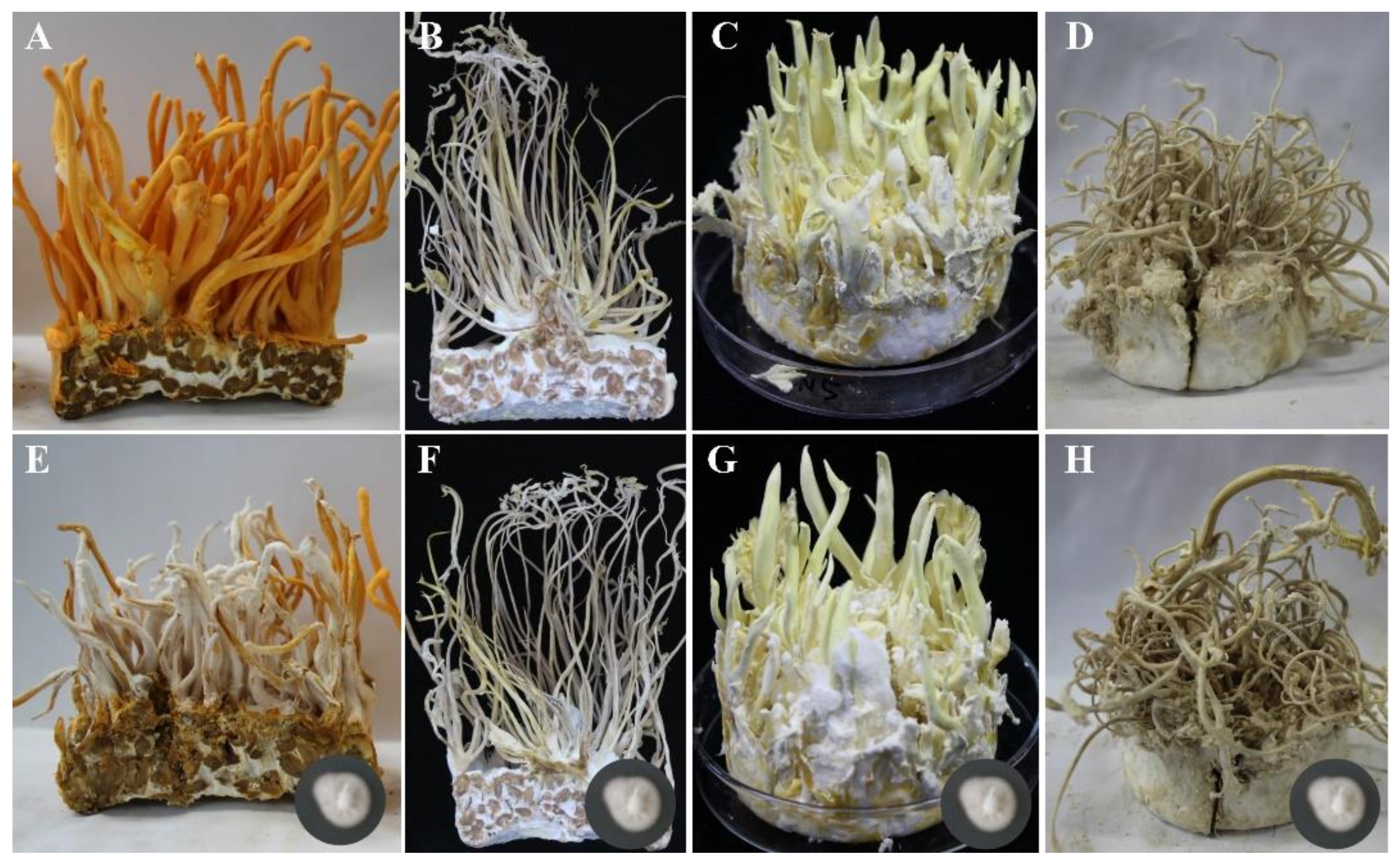
3.3. Host Specificity of Calcarisporium cordycipiticola
3.4. General Features of the Calcarisporium cordycipiticola Genome
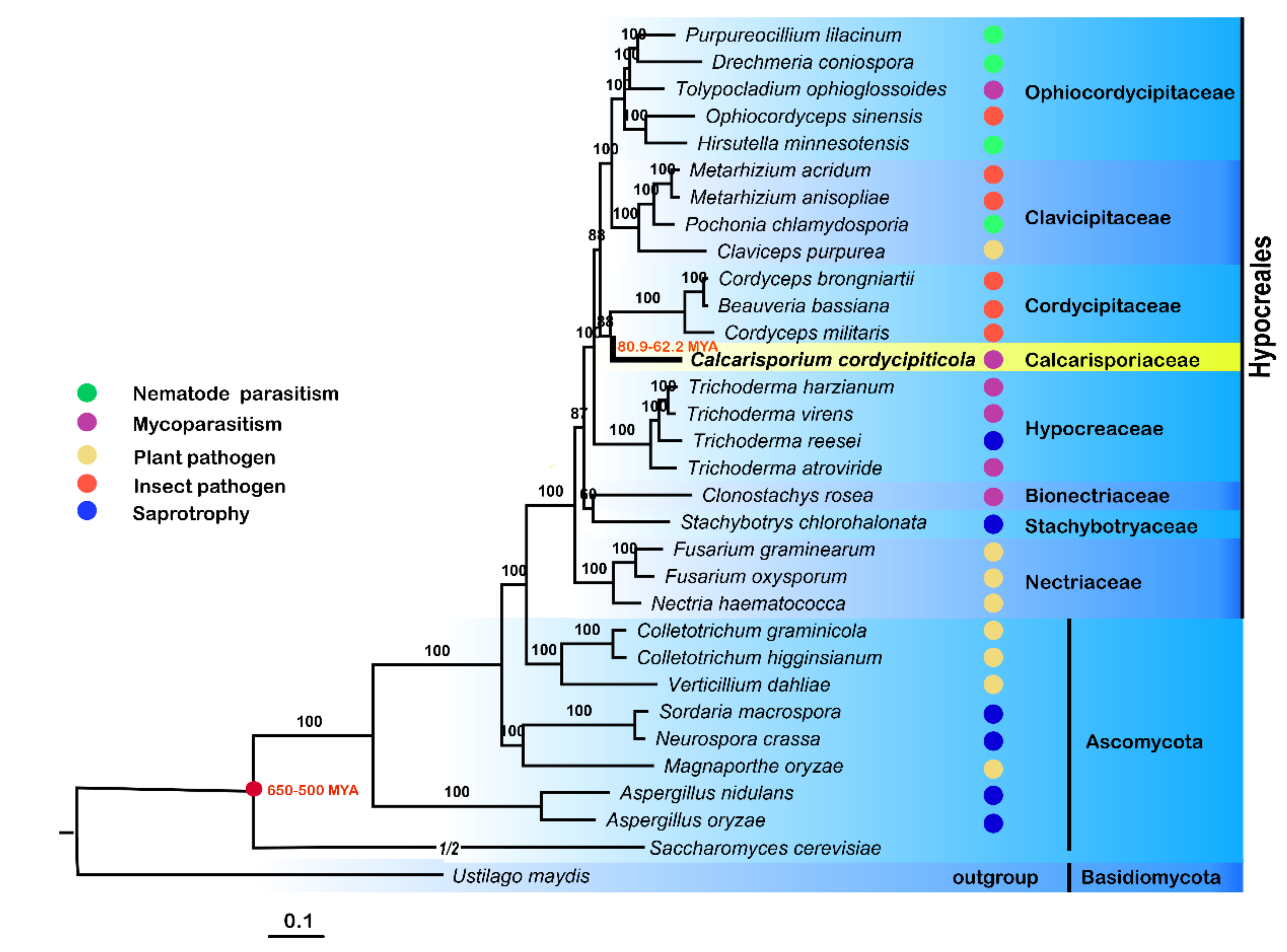
3.5. Phylogenomic and Evolution Analyses
3.6. The Secretome and Potential Eeffectors
3.7. Carbohydrate-active Enzymes (CAZymes)
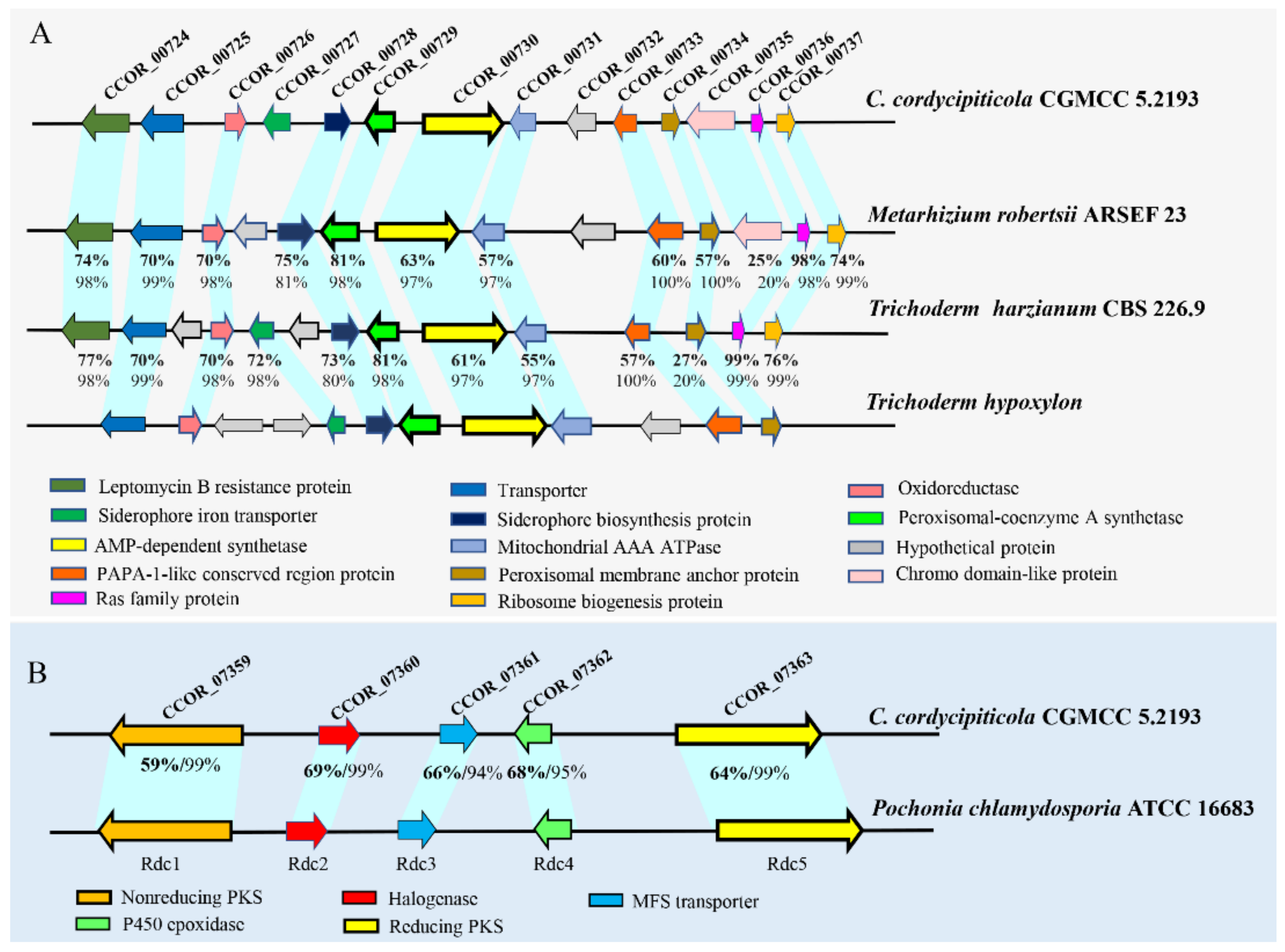
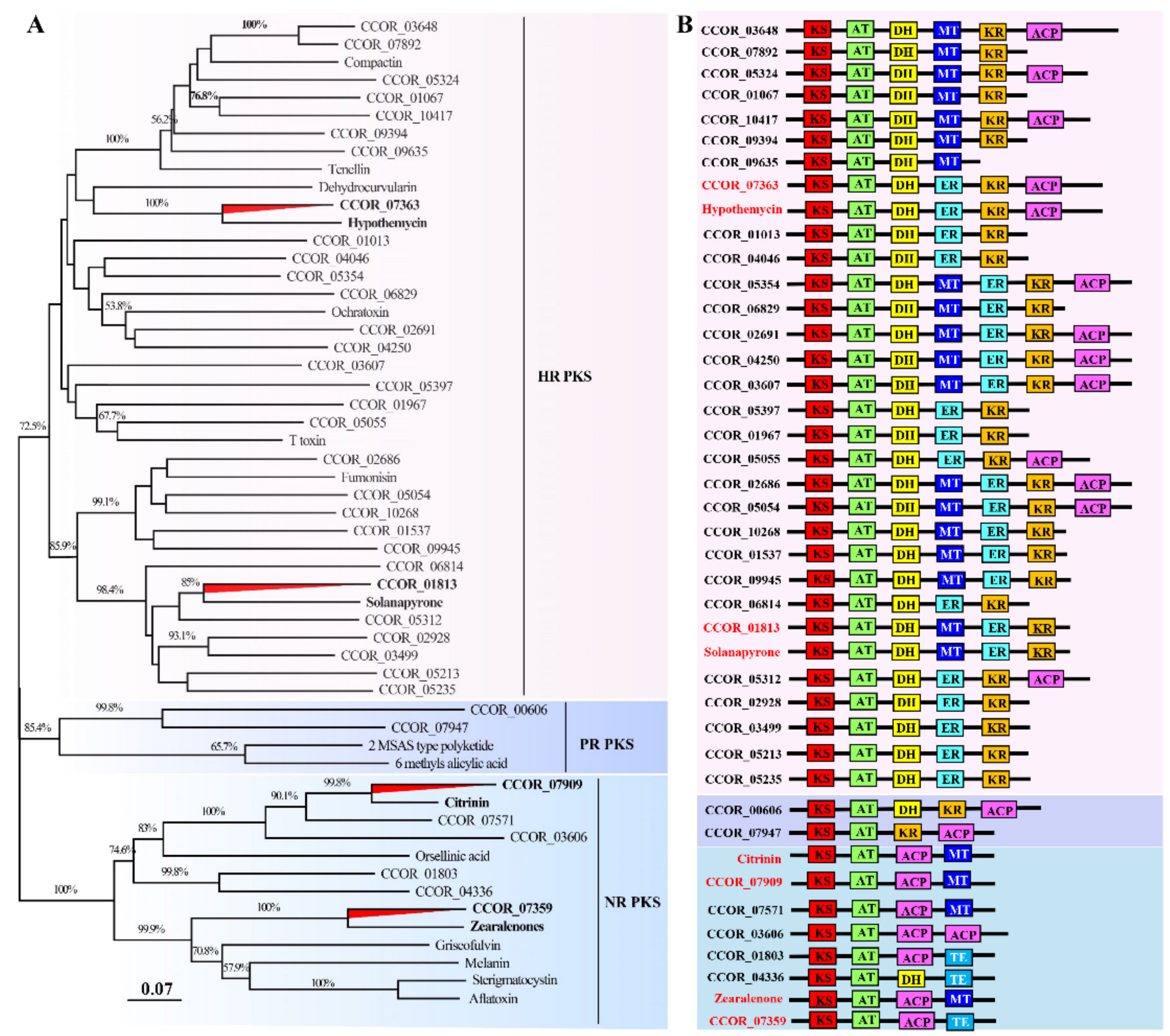
3.8. Great Biosynthetic Capabilities of Secondary Metabolites and Potential of Mycotoxin Biosynthesis in Calcarisporium cordycipiticola
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dong, C.H.; Li, W.J.; Li, Z.Z.; Yan, W.J.; Li, T.H.; Liu, X.Z.; Cai, L.; Ceng, W.P.; Chai, M.Q.; Chen, S.J.; et al. Cordyceps industry in China: Current status, challenges and perspectives-Jinhu declaration for Cordyceps industry development. Mycosystema 2016, 35, 1–15. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, F.; Xu, F.X.; Xu, Y.Y.; Dong, C.H. Study on fungal diseases of artificially cultivated Cordyceps militaris. Mycosystema 2021, 40, 1–19. [Google Scholar] [CrossRef]
- Liu, Q.; Wan, J.X.; Zhang, Y.C.; Dong, C.H. Biological characterization of the fungicolous fungus Calcarisporium cordycipiticola, a pathogen of Cordyceps militaris. Mycosystema 2018, 37, 1054–1062. [Google Scholar] [CrossRef]
- Sun, J.-Z.; Dong, C.-H.; Liu, X.-Z.; Hyde, K.D. Calcarisporium cordycipiticola sp. nov., an important fungal pathogen of Cordyceps militaris. Phytotaxa 2016, 268, 135. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.-Z.; Liu, X.-Z.; Hyde, K.D.; Zhao, Q.; Maharachchikumbura, S.; Camporesi, E.; Bhat, J.; Nilthong, S.; Lumyong, S. Calcarisporium xylariicola sp. nov. and introduction of Calcarisporiaceae fam. nov. in Hypocreales. Mycol. Prog. 2017, 16, 433–445. [Google Scholar] [CrossRef]
- Sun, J.-Z.; Liu, X.-Z.; McKenzie, E.H.C.; Jeewon, R.; Liu, J.-K.J.; Zhang, X.-L.; Zhao, Q.; Hyde, K.D. Correction to: Fungicolous fungi: Terminology, diversity, distribution, evolution, and species checklist. Fungal Divers. 2019, 95, 431–432. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, M.; Durling, M.B.; Choi, J.; Kosawang, C.; Lackner, G.; Tzelepis, G.D.; Nygren, K.; Dubey, M.K.; Kamou, N.; Levasseur, A.; et al. Insights on the Evolution of Mycoparasitism from the Genome of Clonostachys rosea. Genome Biol. Evol. 2015, 7, 465–480. [Google Scholar] [CrossRef]
- Li, D.; Sossah, F.L.; Sun, L.; Fu, Y.; Li, Y. Genome Analysis of Hypomyces perniciosus, the Causal Agent of Wet Bubble Disease of Button Mushroom (Agaricus bisporus). Genes 2019, 10, 417. [Google Scholar] [CrossRef] [Green Version]
- Banks, A.M.; Aminuddin, F.; Williams, K.; Batstone, T.; Barker, G.; Foster, G.D.; Bailey, A.M. Genome Sequence of Lecanicillium fungicola 150-1, the Causal Agent of Dry Bubble Disease. Microbiol. Resour. Announc. 2019, 8, e00340-19. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Liu, X.; Peng, B.; Liu, P.; Li, Z.; Dai, Y.; Xiao, S. Genomic Features of Cladobotryum dendroides, Which Causes Cobweb Disease in Edible Mushrooms, and Identification of Genes Related to Pathogenicity and Mycoparasitism. Pathogens 2020, 9, 232. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, J.T.; Gaze, R.H. Mushroom Pest and Disease Control: A Color Handbook; Manson Publishing Ltd. Academic Press: San Diego, CA, USA, 2008; p. 192. [Google Scholar]
- Zhou, C.Y. The Etiology and Pathogenic Mechanisms of Mycogone perniciosa Causing Wetbubble Disease on Agaricus bisporus. Ph.D. Thesis, Jilin Agricultural University, Jilin, China, 2014. [Google Scholar]
- Nunes, J.S.; de Brito, M.R.; Zied, D.C.; Leite, E.A.D.G.; Dias, E.S.; Alves, E. Evaluation of the infection process by Lecanicillium fungicola in Agaricus bisporus by scanning electron microscopy. Rev. Iberoam. Micol. 2017, 34, 36–42. [Google Scholar] [CrossRef] [Green Version]
- Kubicek, C.P.; Herrera-Estrella, A.; Seidl-Seiboth, V.A.; Martinez, D.; Druzhinina, I.S.; Thon, M.; Zeilinger, S.; Casas-Flores, S.A.; Horwitz, B.; Mukherjee, P.K.; et al. Comparative genome sequence analysis underscores mycoparasitism as the ancestral life style of Trichoderma. Genome Biol. 2011, 12, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Sossah, F.L.; Li, Z.; Hyde, K.D.; Li, D.; Xiao, S.; Fu, Y.; Yuan, X.; Li, Y. Genome-Wide Identification and Analysis of Chitinase GH18 Gene Family in Mycogone perniciosa. Front. Microbiol. 2021, 11, 596719. [Google Scholar] [CrossRef]
- Bailey, A.M.; Collopy, P.D.; Thomas, D.J.; Sergeant, M.; Costa, A.; Barker, G.; Mills, P.R.; Challen, M.P.; Foster, G. Transcriptomic analysis of the interactions between Agaricus bisporus and Lecanicillium fungicola. Fungal Genet. Biol. 2013, 55, 67–76. [Google Scholar] [CrossRef]
- Gallo, A.; Ferrara, M.; Perrone, G. Phylogenetic Study of Polyketide Synthases and Nonribosomal Peptide Synthetases Involved in the Biosynthesis of Mycotoxins. Toxins 2013, 5, 717–742. [Google Scholar] [CrossRef] [Green Version]
- Zheng, P.; Xia, Y.; Xiao, G.; Xiong, C.; Hu, X.; Zhang, S.; Zheng, H.; Huang, Y.; Zhou, Y.; Wang, S.; et al. Genome sequence of the insect pathogenic fungus Cordyceps militaris, a valued traditional chinese medicine. Genome Biol. 2011, 12, R116. [Google Scholar] [CrossRef] [Green Version]
- Cao, F.; Cheng, J.-T.; Chen, X.-A.; Li, Y.-Q.; Mao, X.-M. Development of an efficient genetic system in a gene cluster-rich endophytic fungus Calcarisporium arbuscula NRRL 3705. J. Microbiol. Methods 2018, 151, 1–6. [Google Scholar] [CrossRef]
- Zhan, Y.; Dong, C.-H.; Yao, Y.-J. Antioxidant Activities of Aqueous Extract from Cultivated Fruit-bodies of Cordyceps militaris (L.) Link In Vitro. J. Integr. Plant Biol. 2006, 48, 1365–1370. [Google Scholar] [CrossRef]
- Ayache, J.; Beaunier, L.; Boumendil, J.; Ehret, G.; Laub, D. Web sample preparation guide for transmission electron microscopy (TEM). In Proceedings of the 14th European Microscopy Congress, Aachen, Germany, 1–5 September 2008. [Google Scholar]
- Liu, K.; Wang, F.; Liu, G.; Dong, C. Effect of Environmental Conditions on Synnema Formation and Nucleoside Production in Cicada Flower, Isaria cicadae (Ascomycetes). Int. J. Med. Mushrooms 2019, 21, 59–69. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Chin, C.-S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptivek-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Gardner, P.P.; Daub, J.; Tate, J.G.; Nawrocki, E.P.; Kolbe, D.L.; Lindgreen, S.; Wilkinson, A.C.; Finn, R.D.; Griffiths-Jones, S.; Eddy, S.R.; et al. Rfam: Updates to the RNA families database. Nucleic Acids Res. 2009, 37, D136–D140. [Google Scholar] [CrossRef] [Green Version]
- Lassmann, T.; Sonnhammer, E. Kalign, Kalignvu and Mumsa: Web servers for multiple sequence alignment. Nucleic Acids Res. 2006, 34, W596–W599. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N. Using Repeat Masker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinform. 2004, 5, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Hane, J.K.; Oliver, R.P. RIPCAL: A tool for alignment-based analysis of repeat-induced point mutations in fungal genomic sequences. BMC Bioinform. 2008, 9, 478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blow, M.J.; Clark, T.; Daum, C.G.; Deutschbauer, A.M.; Fomenkov, A.; Fries, R.; Froula, J.; Kang, D.D.; Malmstrom, R.; Morgan, R.D.; et al. The Epigenomic Landscape of Prokaryotes. PLoS Genet. 2016, 12, e1005854. [Google Scholar] [CrossRef] [Green Version]
- Ter-Hovhannisyan, V. Unsupervised and Semi-Supervised Training Methods for Eukaryotic Gene Prediction. Ph.D. Thesis, Georgia Institute of Technology, Atlanta, GA, USA, 2008. [Google Scholar]
- Salamov, A.A.; Solovyev, V.V. Ab initio Gene Finding in Drosophila Genomic DNA. Genome Res. 2000, 10, 516–522. [Google Scholar] [CrossRef] [Green Version]
- Allen, J.; Majoros, W.H.; Pertea, M.; Salzberg, S.L. JIGSAW, GeneZilla, and GlimmerHMM: Puzzling out the features of human genes in the ENCODE regions. Genome Biol. 2006, 7, S9. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Zeng, Q.; Pearson, M.D.; Cuomo, C.A.; Wortman, J.R. Approaches to Fungal Genome Annotation. Mycology 2011, 2, 118–141. [Google Scholar]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [Green Version]
- Prakash, A.; Jeffryes, M.; Bateman, A.; Finn, R.D. The HMMER Web Server for Protein Sequence Similarity Search. Curr. Protoc. Bioinform. 2017, 60, 3–15. [Google Scholar] [CrossRef]
- Armenteros, J.J.A.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Emanuelsson, O.; Brunak, S.; Von Heijne, G.; Nielsen, H.A. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2007, 2, 953–971. [Google Scholar] [CrossRef]
- Sonnhammer, E.L.; von Heijne, G.; Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. In Proceedings of the 16th International Conference on Intelligent System for Molecular Biology, Menlo Park, CA, USA, 1–28 July 1998. [Google Scholar]
- Horton, P.; Park, K.-J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef] [Green Version]
- Sperschneider, J.; Dodds, P.N.; Gardiner, D.M.; Singh, K.B.; Taylor, J.M. Improved prediction of fungal effector proteins from secretomes with EffectorP 2.0. Mol. Plant Pathol. 2018, 19, 2094–2110. [Google Scholar] [CrossRef] [Green Version]
- Urban, M.; Cuzick, A.; Rutherford, K.; Irvine, A.; Pedro, H.; Pant, R.; Sadanadan, V.; Khamari, L.; Billal, S.; Mohanty, S.; et al. PHI-base: A new interface and further additions for the multi-species pathogen–host interactions database. Nucleic Acids Res. 2017, 45, D604–D610. [Google Scholar] [CrossRef]
- Vaidya, G.; Lohman, D.J.; Meier, R. SequenceMatrix: Concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 2011, 27, 171–180. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree v. 1.4.3. 2016. Available online: http://tree.bio.ed.ac.uk/software/figtree (accessed on 14 May 2020).
- Sanderson, M.J. r8s: Inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 2003, 19, 301–302. [Google Scholar] [CrossRef] [Green Version]
- Lücking, R.; Huhndorf, S.; Pfister, D.H.; Plata, E.R.; Lumbsch, H.T. Fungi evolved right on track. Mycology 2009, 101, 810–822. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Zhang, H.; Wu, P.; Entwistle, S.; Li, X.; Yohe, T.; Yi, H.; Yang, Z.; Yin, Y. dbCAN-seq: A database of carbohydrate-active enzyme (CAZyme) sequence and annotation. Nucleic Acids Res. 2018, 46, D516–D521. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Kornerup, A.; Wanscher, J.H. Methuen Handbook of Colour, 3rd ed.; Eyre Methuen: London, UK, 1978; p. 225. [Google Scholar]
- Zhang, Y.B.; Zhuang, W.Y. First step evaluation of Trichoderma antagonism against plant pathogenic fungi in dual culture. Mycosystema 2017, 36, 1251–1259. [Google Scholar] [CrossRef]
- Feng, H.L.; Zhu, H.T. Histological studies on Cordyceps militaris. Acta Mycol. Sin. 1990, 9, 1–5. [Google Scholar] [CrossRef]
- Kumar, A.; Sørensen, J.L.; Hansen, F.T.; Arvas, M.; Syed, M.F.; Hassan, L.; Benz, J.P.; Record, E.; Henrissat, B.; Pöggeler, S.; et al. Genome Sequencing and analyses of Two Marine Fungi from the North Sea Unraveled a Plethora of Novel Biosynthetic Gene Clusters. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.T.; Cao, F.; Chen, X.N.; Li, Y.Q.; Mao, X.M. Genomic and transcriptomic survey of an endophytic fungus Calcarisporium arbuscula NRRL 3705 and potential overview of its secondary metabolites. BMC Genom. 2020, 21, 424. [Google Scholar] [CrossRef]
- Kramer, G.J.; Nodwell, J.R. Chromosome level assembly and secondary metabolite potential of the parasitic fungus Cordyceps militaris. BMC Genom. 2017, 18, 912. [Google Scholar] [CrossRef] [Green Version]
- Galagan, J.E.; Selker, E.U. RIP: The evolutionary cost of genome defense. Trends Genet. 2004, 20, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Chater-Diehl, E.; Sarah, J.G.; Cytrynbaum, C.; Turinsky, A.L.; Choufani, S.; Weksberg, R. Anatomy of DNA methylation signatures: Emerging insights and applications. Am. J. Hum. Genet. 2021, 108, 1359–1366. [Google Scholar] [CrossRef]
- Xiao, G.; Ying, S.-H.; Zheng, P.; Wang, Z.-L.; Zhang, S.; Xie, X.-Q.; Shang, Y.; Leger, R.J.S.; Zhao, G.-P.; Wang, C.; et al. Genomic perspectives on the evolution of fungal entomopathogenicity in Beauveria bassiana. Sci. Rep. 2012, 2, 483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McElwain, J.C.; Punyasena, S. Mass extinction events and the plant fossil record. Trends Ecol. Evol. 2007, 22, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Quandt, C.A.; Kepler, R.M.; Gams, W.; Araújo, J.P.M.; Ban, S.; Evans, H.C.; Hughes, D.; Humber, R.; Hywel-Jones, N.; Li, Z.; et al. Phylogenetic-based nomenclatural proposals for Ophiocordycipitaceae (Hypocreales) with new combinations in Tolypocladium. IMA Fungus 2014, 5, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Cuomo, C.A.; Güldener, U.; Xu, J.-R.; Trail, F.; Turgeon, B.G.; Di Pietro, A.; Walton, J.D.; Ma, L.-J.; Baker, S.; Rep, M.; et al. The Fusarium graminearum Genome Reveals a Link Between Localized Polymorphism and Pathogen Specialization. Science 2007, 317, 1400–1402. [Google Scholar] [CrossRef] [Green Version]
- Asai, S.; Shirasu, K. Plant cells under siege: Plant immune system versus pathogen effectors. Curr. Opin. Plant Biol. 2015, 28, 1–8. [Google Scholar] [CrossRef]
- Marton, K.; Flajšman, M.; Radišek, S.; Košmelj, K.; Jakše, J.; Javornik, B.; Berne, S. Comprehensive analysis of Verticillium nonalfalfae in silico secretome uncovers putative effector proteins expressed during hop invasion. PLoS ONE 2018, 13, e0198971. [Google Scholar] [CrossRef] [Green Version]
- Druzhinina, I.S.; Seidl-Seiboth, V.; Herrera-Estrella, A.; Horwitz, B.A.; Kenerley, C.M.; Monte, E.; Mukherjee, P.K.; Zeilinger, S.; Grigoriev, I.; Kubicek, C.P. Trichoderma: The genomics of opportunistic success. Nat. Rev. Genet. 2011, 9, 749–759. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, X.; Liu, L.; Xiang, M.; Wang, W.; Sun, X.; Che, Y.; Guo, L.; Liu, G.; Guo, L.; et al. Genomic and transcriptomic analysis of the endophytic fungus Pestalotiopsis fici reveals its lifestyle and high potential for synthesis of natural products. BMC Genom. 2015, 16, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Qi, L.; Chen, G.; Yin, W.-B. Discovery and genetic identification of amphiphilic coprogen siderophores from Trichoderm hypoxylon. Appl. Microbiol. Biotechnol. 2021, 105, 2831–2839. [Google Scholar] [CrossRef]
- Qin, F.; Li, Y.; Lin, R.; Zhang, X.; Mao, Z.; Ling, J.; Yang, Y.; Zhuang, X.; Du, S.; Cheng, X.; et al. Antibacterial Radicicol Analogues from Pochonia chlamydosporia and Their Biosynthetic Gene Cluster. J. Agric. Food Chem. 2019, 67, 7266–7273. [Google Scholar] [CrossRef]
- Nam, S.; Ga, Y.J.; Lee, J.-Y.; Hwang, W.-Y.; Jung, E.; Shin, J.S.; Chen, W.; Choi, G.; Zhou, B.; Yeh, J.-Y.; et al. Radicicol Inhibits Chikungunya Virus Replication by Targeting Nonstructural Protein 2. Antimicrob. Agents Chemother. 2021, 65. [Google Scholar] [CrossRef]
- Turbyville, T.J.; Wijeratne, E.M.K.; Liu, M.X.; Burns, A.M.; Seliga, C.J.; Luevano, L.A.; David, C.L.; Faeth, S.H.; Whitesell, L.; Gunatilaka, A.A.L. Search for Hsp90 Inhibitors with Potential Anticancer Activity: Isolation and SAR Studies of Radicicol and Monocillin I from Two Plant-Associated Fungi of the Sonoran Desert. J. Nat. Prod. 2006, 69, 178–184. [Google Scholar] [CrossRef] [Green Version]
- Awakawa, T.; Yang, X.-L.; Wakimoto, T.; Abe, I. Pyranonigrin E: A PKS-NRPS Hybrid Metabolite from Aspergillus niger Identified by Genome Mining. ChemBioChem 2013, 14, 2095–2099. [Google Scholar] [CrossRef]
- Kishimoto, S.; Tsunematsu, Y.; Sato, M.; Watanabe, K. Elucidation of Biosynthetic Pathways of Natural Products. Chem. Rec. 2017, 17, 1095–1108. [Google Scholar] [CrossRef]
- Wight, W.D.; Kim, K.-H.; Lawrence, C.B.; Walton, J.D. Biosynthesis and Role in Virulence of the Histone Deacetylase Inhibitor Depudecin from Alternaria brassicicola. Mol. Plant-Microbe Interactions 2009, 22, 1258–1267. [Google Scholar] [CrossRef] [Green Version]
- Kasahara, K.; Miyamoto, T.; Fujimoto, T.; Oguri, H.; Tokiwano, T.; Oikawa, H.; Ebizuka, Y.; Fujii, I. Solanapyrone Synthase, a Possible Diels-Alderase and Iterative Type I Polyketide Synthase Encoded in a Biosynthetic Gene Cluster from Alternaria solani. ChemBioChem 2010, 11, 1245–1252. [Google Scholar] [CrossRef]
- Reeves, C.D.; Hu, Z.; Reid, R.; Kealey, J.T. Genes for the Biosynthesis of the Fungal Polyketides Hypothemycin from Hypomyces subiculosus and Radicicol from Pochonia chlamydosporia. Appl. Environ. Microbiol. 2008, 74, 5121–5129. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Cox, R.J. The molecular steps of citrinin biosynthesis in fungi. Chem. Sci. 2016, 7, 2119–2127. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, M.; Atanasova, L.; Jensen, D.F.; Zeilinger, S. Necrotrophic Mycoparasites and Their Genomes. Microbiology 2017, 5, 1–21. [Google Scholar] [CrossRef]
- Barnett, H.L. A new Calcarisporium parasitic on other fungi. Mycologia 1958, 50, 497–500. [Google Scholar] [CrossRef]
- Lv, Z.; He, Z.; Hao, L.; Kang, X.; Ma, B.; Li, H.; Luo, Y.; Yuan, J.; He, N. Genome Sequencing Analysis of Scleromitrula shiraiana, a Causal Agent of Mulberry Sclerotial Disease With Narrow Host Range. Front. Microbiol. 2021, 11, 603927. [Google Scholar] [CrossRef]
- Alexopoulos, C.J.; Blackwell, M.; Mims, C.W. Introductory Mycology, 4th ed.; John Wiley and Sons Inc.: Manhattan, NY, USA, 1996; p. 164. [Google Scholar]
- Zeng, W.; Melotto, M.; He, S.Y. Plant stomata: A checkpoint of host immunity and pathogen virulence. Curr. Opin. Biotechnol. 2010, 21, 599–603. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Vujanovic, V. Relationship between mycoparasites lifestyles and biocontrol behaviors against Fusarium spp. and mycotoxins production. Appl. Microbiol. Biotechnol. 2016, 100, 5257–5272. [Google Scholar] [CrossRef]
- Lu, Z.; Tombolini, R.; Woo, S.; Zeilinger, S.; Lorito, M.; Jansson, J.K. In Vivo Study of Trichoderma -Pathogen-Plant Interactions, Using Constitutive and Inducible Green Fluorescent Protein Reporter Systems. Appl. Environ. Microbiol. 2004, 70, 3073–3081. [Google Scholar] [CrossRef] [Green Version]
- Hoch, H.C. Mycoparasitic relationships. III. Parasitism of Physalospora obtuse by Calcarisporium parasiticum. Can. J. Bot. 1977, 55, 198–207. [Google Scholar] [CrossRef]
- Boddy, L.; Hiscox, J. Fungal Ecology: Principles and Mechanisms of Colonization and Competition by Saprotrophic Fungi. Microbiol. Spectr. 2016, 4, 293–308. [Google Scholar] [CrossRef] [Green Version]
- Grooters, A.M. Deep Fungal Infections. Saunders Manual of Small Animal Practice, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2006; pp. 435–444. [Google Scholar]
- Dos Santos, T.L.; Belan, L.L.; Zied, D.C.; Dias, E.S.; Alves, E. Essential oils in the control of dry bubble disease in white button mushroom. Ciência Rural 2017, 47. [Google Scholar] [CrossRef]
- Huang, Q.G.; Wang, S.; Zhang, Y. The interactions between Mycogone perniciosa and Agaricus bisporus. Mycosystema 2014, 33, 440–448. [Google Scholar] [CrossRef]
- Junker, K.; Chailyan, A.; Hesselbart, A.; Forster, J.; Wendland, J. Multi-omics characterization of the necrotrophic mycoparasite Saccharomycopsis schoenii. PLOS Pathog. 2019, 15, e1007692. [Google Scholar] [CrossRef] [Green Version]
- Quandt, C.A.; Bushley, K.E.; Spatafora, J.W. The genome of the truffle-parasite Tolypocladium ophioglossoides and the evolution of antifungal peptaibiotics. BMC Genom. 2015, 16, 1–14. [Google Scholar] [CrossRef] [Green Version]
- de Man, T.J.B.; Stajich, J.E.; Kubicek, C.P.; Teiling, C.; Chenthamara, K.; Atanasova, L.; Druzhinina, I.S.; Levenkova, N.; Birnbaum, S.S.L.; Barribeau, S.; et al. Small genome of the fungus Escovopsis weberi, a specialized disease agent of ant agriculture. Proc. Natl. Acad. Sci. USA 2016, 113, 3567–3572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Dong, Y.; Liao, W.; Zhang, X.; Wang, Q.; Li, G.; Xu, J.-R.; Liu, H. High-Quality Genome Resource of Clonostachys rosea strain CanS41 by Oxford Nanopore Long-Read Sequencing. Plant Dis. 2021, PDIS12202615A. [Google Scholar] [CrossRef]
- Aguileta, G.; Hood, M.E.; Refrégier, G.; Giraud, T. Chapter 3 Genome Evolution in Plant Pathogenic and Symbiotic Fungi. Adv. Bot. Res. 2009, 49, 151–193. [Google Scholar] [CrossRef]
- Keller, N.P. Translating biosynthetic gene clusters into fungal armor and weaponry. Nat. Chem. Biol. 2015, 11, 671–677. [Google Scholar] [CrossRef] [Green Version]
- Sung, G.-H.; Hywel-Jones, N.L.; Sung, J.-M.; Luangsa-Ard, J.J.; Shrestha, B.; Spatafora, J.W. Phylogenetic classification of Cordyceps and the clavicipitaceous fungi. Stud. Mycol. 2007, 57, 5–59. [Google Scholar] [CrossRef] [Green Version]
- Spatafora, J.W.; Sung, G.-H.; Sung, J.-M.; Hywel-Jones, N.L.; White, J. Phylogenetic evidence for an animal pathogen origin of ergot and the grass endophytes. Mol. Ecol. 2007, 16, 1701–1711. [Google Scholar] [CrossRef]
- Harman, G.E.; Kubicek, C.P. Trichoderma and Gliocladium; Taylor and Francis: London, UK, 1998; Volume 2, pp. 243–270. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | C. cordycipiticola CGMCC 5.2193 | C. militaris Cm01 |
|---|---|---|
| Size (Mbp) * | 34.51 | 32.2 |
| Coverage (fold) | 170× | 147× |
| (G + C) percentage (%) | 51.7 | 51.4 |
| N50 (Mbp) | 5.45 | 4.6 |
| Percentage repeat rate | 4.04 | 3.04 |
| Protein-coding genes | 10,443 | 9684 |
| Average gene length (bp) | 1692 | 1742 |
| Percentage of secreted proteins (%) | 9.02 | 8.00 |
| Gene density (no. gene per Mbp) | 302 | 257 |
| tRNA genes | 155 | 136 |
| Pseudogenes | 343 | 102 |
| NCBI accession | PRJNA766243 | AEVU00000000 |
| Life-Style | Species | AA | CBM | CBM1 | CE | GH | GT | PL | All |
|---|---|---|---|---|---|---|---|---|---|
| mycoparasitic fungi | C. cordycipiticola | 67 | 26 | 4 | 56 | 211 | 71 | 6 | 441 |
| C. rosea | 103 | 29 | 13 | 97 | 310 | 73 | 31 | 656 | |
| T. harzianum | 61 | 49 | 21 | 58 | 245 | 76 | 6 | 516 | |
| T. atroviride | 51 | 41 | 22 | 43 | 234 | 74 | 8 | 473 | |
| nematode parasitic fungi | P. chlamydosporia | 63 | 33 | 9 | 43 | 278 | 86 | 7 | 519 |
| H. minnesotensis | 54 | 28 | 0 | 32 | 137 | 80 | 2 | 333 | |
| entomopathogenic fungi | B. bassiana | 51 | 30 | 3 | 33 | 160 | 79 | 2 | 358 |
| C. militaris | 38 | 30 | 1 | 30 | 148 | 69 | 3 | 319 | |
| O. sinensis | 35 | 24 | 0 | 23 | 95 | 64 | 2 | 243 | |
| plant pathogenic fungus | F. graminearum | 82 | 42 | 12 | 74 | 237 | 78 | 22 | 547 |
| M. oryzae | 100 | 51 | 22 | 72 | 244 | 82 | 6 | 577 | |
| V. dahliae | 85 | 31 | 28 | 65 | 241 | 69 | 34 | 553 | |
| B. cinerea | 82 | 38 | 23 | 71 | 260 | 84 | 10 | 568 |
| Feature | Calcarisporium cordycipiticola CGMCC 5.2193 | Calcarisporium arbuscula NRRL 3705 | Calcarisporium sp. 525 | Cordyceps militaris Cm01 |
|---|---|---|---|---|
| NRPS | 14 | 12 | 17 | 8 |
| T1PKS | 18 | 23 | 24 | 6 |
| Terpene | 4 | 11 | 7 | 4 |
| NRPS-like | 12 | 0 | 0 | 6 |
| PKS-NRPS-Hybrid (NRPS-PKS-Hybrid) | 9 | 7 | 3 | 5 |
| other | 9 | 12 | 9 | 1 |
| Total | 66 | 65 | 60 | 30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Xu, Y.; Zhang, X.; Li, K.; Li, X.; Wang, F.; Xu, F.; Dong, C. Infection Process and Genome Assembly Provide Insights into the Pathogenic Mechanism of Destructive Mycoparasite Calcarisporium cordycipiticola with Host Specificity. J. Fungi 2021, 7, 918. https://doi.org/10.3390/jof7110918
Liu Q, Xu Y, Zhang X, Li K, Li X, Wang F, Xu F, Dong C. Infection Process and Genome Assembly Provide Insights into the Pathogenic Mechanism of Destructive Mycoparasite Calcarisporium cordycipiticola with Host Specificity. Journal of Fungi. 2021; 7(11):918. https://doi.org/10.3390/jof7110918
Chicago/Turabian StyleLiu, Qing, Yanyan Xu, Xiaoling Zhang, Kuan Li, Xiao Li, Fen Wang, Fangxu Xu, and Caihong Dong. 2021. "Infection Process and Genome Assembly Provide Insights into the Pathogenic Mechanism of Destructive Mycoparasite Calcarisporium cordycipiticola with Host Specificity" Journal of Fungi 7, no. 11: 918. https://doi.org/10.3390/jof7110918