

Clinical Manifestations and Management of Fibrotic Pulmonary Sarcoidosis

1
Department of Thoracic Medicine and Surgery, Temple University Hospital, Philadelphia, PA 19140, USA
2
Department of Thoracic Medicine and Surgery, Lewis Katz School of Medicine, Temple University Hospital, Philadelphia, PA 19140, USA
*
Author to whom correspondence should be addressed.
J. Clin. Med. 2024, 13(1), 241; https://doi.org/10.3390/jcm13010241
Submission received: 13 November 2023
/
Revised: 22 December 2023
/
Accepted: 27 December 2023
/
Published: 31 December 2023
(This article belongs to the Special Issue Advances in the Diagnosis and Treatment of Pulmonary Sarcoidosis)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Fibrotic pulmonary sarcoidosis represents a distinct and relatively uncommon manifestation within the spectrum of sarcoidosis and has substantial morbidity and mortality. Due to the scarcity of research focused on this specific disease subtype, our current understanding of pathogenesis and optimal management remains constrained. This knowledge gap underscores the need for further investigation into areas such as targeted therapies, lung transplantation, and quality of life of patients with fibrotic pulmonary sarcoidosis. The primary aim of this review is to discuss recent developments within the realm of fibrotic pulmonary sarcoidosis to foster a more comprehensive understanding of the underlying mechanisms, prognosis, and potential treatment modalities.
1. Background
Sarcoidosis is a complex multisystem inflammatory disease characterized by the formation of noncaseating granulomas that predominantly affects the respiratory system [1,2,3]. While over 60% of sarcoidosis patients have resolution of disease in 2–5 years, the remaining experience chronic disease, including fibrotic change [4]. Sarcoidosis generally exhibits notable demographic disparities, with the highest incidence and prevalence seen in Black patients, particularly among females [5]. A population-based study in the United States found that African Americans with sarcoidosis had a 20% higher rate of pulmonary fibrosis, and African-American women with sarcoidosis had a higher mortality rate at a younger age when contrasted with their Caucasian counterparts [6].
The Scadding staging system is used to assess radiographic stages of pulmonary sarcoidosis, with stage 4 denoting advanced fibrotic changes [7]. Approximately 5.4–19.9% of patients may present with fibrotic disease initially [7,8]. Patients with chronic disease experience increased breathlessness and decreased quality of life as radiographic disease worsens [9]. Advanced pulmonary sarcoidosis (APS) is used to denote the forms of sarcoidosis that cause significant risk of loss of lung function, respiratory failure, or death, and include advanced fibrosis and associated complications as well as pulmonary hypertension [10,11]. Although mortality in sarcoidosis is reported to be less than 5%, mortality in APS ranges from 11–21% [4,11,12,13,14,15]. Most of the poor outcomes attributed to APS are due to fibrotic pulmonary sarcoidosis, an entity that needs to be understood better. In this article, we aim to review recent advances in pathogenesis, clinical presentation, evaluation, and management of fibrotic pulmonary sarcoidosis.
2. Pathobiology
2.1. Basic Pathophysiology: An Interplay between Genetic and Environmental Factors
The pathobiology underlying sarcoidosis and its development into fibrotic disease remains a subject of ongoing research. Sarcoidosis is largely believed to result from a culmination of abnormal immunologic responses following antigen exposure (Figure 1) in a genetically predisposed host. Air pollutants, infectious agents such as mycobacteria and Cutibacterium acnes, and exposure to inorganic dust such as silica have been implicated in pulmonary sarcoidosis [16,17,18,19]. Genetic factors have been implicated in the susceptibility and manifestation of the disease, such as HLA-DRB1 on chromosome 6, 5q11.2, 1p22, 3p21-14, 11p15, and 17q21 [5,20,21,22]. Specific alleles of HLA-DRB1 on chromosome 6 may have race-specific associations with varying phenotypes and confer protective effects against disease, while others may be associated with increased disease severity [23,24].
2.2. Evolving Knowledge of Pathophysiology in Fibrotic Pulmonary Sarcoidosis
Several genes have been linked to the pathogenesis of fibrotic disease. GREM1 on chromosome 15q13-q15 encodes a glycoprotein, gremlin, that inhibits bone morphogenic proteins (BMPs) from the TGF-B family [25]. TGF-B, a cytokine secreted by macrophages, T-lymphocytes, and bronchial epithelial cells, promotes extracellular matrix accumulation and inhibits matrix degradation [26]. A study examining GREM1 variations among sarcoidosis patients with and without fibrosis on chest radiography revealed that carriers of the GREM1 CC genotype exhibited elevated gremlin levels and were at a 6.4-fold higher risk of developing fibrosis [27]. Genetic variations of TGF-B3 are notably greater in fibrotic patients, and may be associated with the development of pulmonary fibrosis in sarcoidosis [28].
Fibrotic pulmonary sarcoidosis has also been linked to specific variants, such as caspase recruitment domain 15 (CARD15) 2104T (702W), CARD15 1761G (587R), and C-C chemokine receptor 5 (CCR-5) [29]. Additionally, a promoter variation in prostaglandin-endoperoxide synthase 2 (PTGS2), −765G>C, has been identified as another potential risk factor for fibrotic disease in sarcoidosis. PTGS2 serves as a regulatory enzyme responsible for synthesizing prostaglandin E2, which is known for its antifibrotic properties. Carriers of the −765C allele were found to exhibit increased susceptibility to sarcoidosis, poorer prognosis, and an increased predisposition to fibrotic disease [30].
The pathogenesis of fibrotic pulmonary sarcoidosis remains unclear, but current investigations have identified several potential mechanisms that could elucidate both the fibrotic and inflammatory reactions observed. Pulmonary sarcoidosis is a granulomatous disease characterized by accumulation of lymphocytes and macrophages, inducing granuloma formation. An unknown antigen is first presented to CD4+ T-lymphocytes that trigger T-helper 17 (Th17)-related cytokines, interleukin-17A (IL-17A), regulatory T-cells, and tumor necrosis factor (TNF), a proinflammatory cytokine, to produce granulomas [31]. Granulomas may spontaneously resolve or persist, and may progress to fibrosis via high levels of TNF and mononuclear phagocytes (MNPs) and activation of fibroblasts, myofibroblasts, and collagen formation [32].
Chronic fibrosis is thought to be the culmination of increased Th17 cells and primed monocyte-derived macrophages (toll-like receptor-3 (TLR3) polymorphism, type 1 interferon signaling) responding disproportionately to an insult [33]. In particular, monomorphisms in TLR3 have been implicated in fibrotic pulmonary sarcoidosis, resulting in reduced TLR3 function in innate immune responses and reduced apoptosis of fibroblasts [33,34]. This response drives production of chemokine ligand 18 (CCL-18), which induces fibrogenesis [33]. CCL-18 is associated with fibrotic pathogenesis in IPF, with increased mortality and fibrotic burden on imaging [35,36]. Another protein, annexin A11 (ANXA11), is a calcium-dependent protein involved in innate immunity and cell apoptosis. A small study found a correlation between a minor allele in the ANXA11 gene and African Americans with fibrotic pulmonary sarcoidosis, and suggested ANXA11 polymorphism may lead to persistence of Th1 and Th17 cells, resistance to apoptosis, and persistence of granuloma [24,33].
Increased production of CCL-18 from macrophages attracts activated CD4 T-cells and increases transforming growth factor-beta (TGF-β) secretion, enhancing Th17-mediated inflammation. Th17 expresses IL-17A, a proinflammatory cytokine that drives fibrosis and causes corticosteroid resistance [37]. One study found higher bronchoalveolar lavage (BAL) IL-17 levels in patients with pulmonary sarcoidosis without disease resolution, but this was not studied in patients with or without fibrotic disease [38]. Another acute phase reactant is serum amyloid antigen (SAA), which has been shown to induce Th17 response, chronic inflammation, and fibrosis [39]. It can stimulate the production of Th1-mediated granulomatous inflammation via TNG, IL-10, and IL-18, and has been shown to correlate positively with fibrotic disease in chronic fibrotic sarcoidosis [40,41].
Regulatory T-cells (Tregs) are a specialized subset of CD4+ T-cells involved in immunosuppression via production of inhibitory cytokines such as interleukin-10 (IL-10), inhibitory receptors such as cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), and deplete interleukin-2 (IL-2) [42]. Prior studies have found lower numbers of Tregs in BAL and Treg dysfunction in patients with active sarcoidosis [43,44,45]. In both active and fibrotic sarcoidosis patients compared with IPF patients, a recent study found an imbalance of Tregs and Th17.1 cells in peripheral blood and BAL fluid, with lower frequency of Tregs but high Th17.1 in BAL and higher frequency of Tregs but low Th17.1 in peripheral blood [46]. The authors suggest that an increased proportion of circulating Tregs was associated with fibrotic disease on radiography, and the lung microenvironment may affect immunological pathogenesis of sarcoidosis [46].
Another pathway that may contribute to granuloma formation and Th17 differentiation is a dysregulation of the mammalian target of rapamycin (mTOR) pathway. mTOR regulates autophagy and growth in response to stressors [47]. Defects in mTOR-related pathways may inactivate autophagy, decrease pathogen clearance, and cause granulomatous formation and persistence [48]. In fibrotic pulmonary sarcoidosis, mTOR complex 1 (mTORC1) remains upregulated, impairing antigen clearance and promoting excess granulomatous formation [33,49,50].
Recently, the hypoxia-induced factor 1-alpha (HIF1α) pathway has garnered attention. A recent investigation found that when exposed to hypoxic conditions, monocyte-derived macrophages increase their proinflammatory response and reduce antigen presentation, leading to a reduction in T-cell response [51]. Through the secretion of profibrotic factor plasminogen activator inhibitor-1 (PAI-1), this process may promote development and persistence of granulomas in active sarcoidosis, reduce fibrinolytic activity, and ultimately contribute to the development of fibrotic disease [51].
3. Clinical Manifestations
Prior studies have found that the average age of presentation with fibrotic pulmonary sarcoidosis is in the fourth decade of life [7,12]. Up to 20% of patients can present with fibrotic disease at initial presentation, but chronic disease may develop in 20–25% of patients with a prior diagnosis of sarcoidosis [7,52,53]. Clinical symptoms are nonspecific and include dyspnea (80%), cough (51.4%), hemoptysis (2.8%), sputum production (18.3%), crackles (28.2%), digital clubbing (6.3%), and wheezing (5.6%) [12].
3.1. Imaging
On chest radiography (CXR), Scadding stage 4 is defined by the presence of pulmonary fibrosis, as mentioned above (Figure 2) [7]. Patients may have upper-lobe-predominant linear opacities projecting from the hilum with dilated airways [54]. High-resolution computed tomography (HRCT) gives a more comprehensive understanding of anatomic changes. Three major patterns of fibrotic sarcoidosis can be identified: central bronchial distortion, peripheral upper zone honeycombing, and diffuse hilar linear opacities (Figure 2) [11,55,56]. Fibrocystic opacities may track along the airways from the hilum to peribronchovascular and fissural regions [11]. HRCT may show subpleural honeycombing, fibrocystic lesions larger than traditional honeycombing, paracicatricial emphysema, and development of mycetomas [11,57]. Granulomatous infiltration of the airways will cause airway distortion, airway angulation, and diffuse wall thickening [7,56]. HRCT can also help screen for sarcoidosis-associated pulmonary hypertension by using a ratio of main pulmonary artery diameter/ascending aorta diameter (MPAD/AAD) greater than 1, evaluating for a dilated pulmonary artery greater than 30 mm (Figure 2), and using a ratio of the diameter of the main pulmonary artery/body surface area (MPA/BSA) greater than 16 [3,11,58].
Fluorodeoxyglucose positron emission tomography integrated with computed tomography (FDG-PET/CT) in combination with cardiac MRI is predominantly used for the diagnosis and management of cardiac sarcoidosis [3,59]. In pulmonary sarcoidosis, FDG-PET/CT has exhibited a high sensitivity rate ranging between 94% and 100% in identification of ongoing inflammatory processes (Figure 3) [60,61]. Few studies have investigated its diagnostic use in pulmonary sarcoidosis [62,63,64]. One retrospective study involving 95 patients, with 85% demonstrating signs of fibrotic disease, found that the severity of pulmonary involvement as assessed by HRCT and lung function parameters was associated with increased FDG uptake at a threshold standardized uptake value (SUVmax) of greater than or equal to 2.5 [63]. Another study assessed the role of FDG-PET/CT in comparison with HRCT to identify sarcoidosis activity, and found a discordance rate of greater than 50% between FDG uptake and pathologic changes on HRCT. The presence of active nodal disease, active parenchymal changes, and disease recurrence in extrapulmonary regions were additional findings noted on FDG-PET/CT not discernible on HRCT [64].
In the context of disease activity monitoring, a limited number of studies found that patients who exhibited reductions in SUVmax values following glucocorticoid therapy experienced lower rates of relapse, in contrast to individuals without reduction in SUVmax, who notably had higher relapse rates (Figure 3) [65,66]. To date, there is no SUVmax threshold that is validated to denote disease activity or recommendations for use of FDG-PET/CT in determining anti-inflammatory treatment for fibrotic pulmonary sarcoidosis. Future studies are needed to determine optimal utility of FDG-PET/CT imaging in fibrotic pulmonary sarcoidosis.
3.2. Pulmonary Function Testing
On pulmonary function testing (PFT), fibrotic sarcoidosis presents with varying degrees of gas-exchange, airflow-obstruction, ventilatory-restriction, and mixed defects [12]. One study found associations between HRCT anomalies and pulmonary function testing, revealing a connection between restrictive defects and reduced diffusion capacity with interstitial fibrosis and subpleural honeycombing, while airflow obstruction correlated with bronchial distortion. Linear opacities without septal changes were found to have the least functional impairment [55]. Patients with fibrotic disease were shown to have a higher prevalence of mixed ventilatory defects, lower diffusion capacity for carbon monoxide, and higher mortality in another study [67]. A recent study characterizing different pulmonary function phenotypes in sarcoidosis found that fibrocystic patterns on chest imaging (n = 22) were more commonly seen in Black individuals, and patients with fibrocystic patterns had a greater degree of restriction and mixed pulmonary function phenotypes than patients with nonfibrotic pulmonary sarcoidosis [68]. The findings of this study emphasize that fibrotic disease is linked to a higher prevalence of restrictive and mixed defects [68]. On 6-min walk tests, individuals with fibrotic sarcoidosis may have reduced walk distance, which has been associated with increased mortality, sarcoidosis-associated pulmonary hypertension, reduced forced vital capacity, and exertional hypoxia [69,70].
3.3. Serum Biomarkers
Inflammatory biomarkers have been proposed as a method to monitor disease activity and treatment response in pulmonary sarcoidosis. These biomarkers have not been studied in the setting of fibrotic disease, and larger prospective studies are needed to assess clinical utility. Nevertheless, research indicates promising results for a few of these biomarkers, such as serum angiotensin-converting enzyme (ACE), human chitotriosidase, C-reactive protein (CRP), and Krebs von den Lungen-6 (KL-6).
Serum ACE is a glycoprotein produced by alveolar macrophages that converts angiotensin I to angiotensin II in the renin-angiotensin pathway and degrades bradykinin. Granulomas express alveolar macrophages, and serum ACE levels may reflect granulomatous burden [71]. ACE levels are currently the most frequently used laboratory testing in sarcoidosis as a marker for disease activity, although they are neither sensitive nor specific [12,72,73,74]. High serum ACE levels may be seen in patients with greater HRCT abnormalities, including ground-glass opacities, interlobular septal thickening, nodularity, and consolidation [75]. They may be used to monitor treatment effects in sarcoidosis patients. An observational cohort study assessing treatment response with methotrexate by measuring serum ACE and soluble IL-2 receptor (sIL-2R), a marker of T-cell activation, found high baseline levels of ACE correlated with lung function improvement after treatment; and decreases in ACE and sIL-2R after treatment correlated with improved lung function, especially with change in DLCO [73]. In addition, T-helper type 1 cells secrete IL-2 and bind to IL-2R, stimulating T-cell proliferation [76]. sIL-2R is a marker of T-cell activation, whereas ACE reflects total body granulomas. In this study, ACE had a greater correlation with lung function change after methotrexate therapy than sIL-2R [73]. CRP is a proinflammatory acute phase reactant elevated in chronic sarcoidosis, and elevated baseline values may correlate with disease severity, physiologic progression of disease, and treatment response [77,78]. CRP may be useful in monitoring disease activity but requires validation. Further research of serum biomarkers is needed on the clinical utility of these in sarcoidosis in general as well as fibrotic pulmonary sarcoidosis.
4. Prognosis
The presence of fibrosis on high-resolution computed tomography (HRCT) scans indicates a poorer prognosis, disease progression, and an elevated risk of mortality [13,55,58,79]. One study proposed a clinicoradiological staging system using HRCT patterns and composite physiological indices (CPI, a weighted index of lung function variables) to determine prognosis in pulmonary sarcoidosis [58]. The staging system was composed of CPI, main pulmonary artery diameter to ascending aorta diameter ratio (MPAD/AAD), and fibrosis threshold of ≥20% [58]. The staging system was found to be straightforward yet reliable for identifying patients with increased risk of mortality [58]. The results further emphasized that CPI was the strongest predictor of mortality [58].
A prospective study in fibrotic pulmonary sarcoidosis evaluated the feasibility of employing percent fibrosis on HRCT, reduced DLCO, or increased CPI score to predict a clinal worsening event over an 18-month study period [80]. A clinical worsening event was defined as death, lung transplant, or greater than absolute 10% drop in percent predicted FVC [80]. Though the study was underpowered at 16 participants due to poor enrollment, it found that individuals with at least 20% fibrosis on HRCT and DLCO less than 30% predicted were more likely to experience a clinical worsening event [80].
In a recent study, HRCT features of fibrotic pulmonary sarcoidosis and its impact on pulmonary function and survival were assessed [81]. The study found that the presence of over 20% fibrosis and basal subpleural honeycombing were predictive of deteriorating pulmonary function and worse survival in fibrotic pulmonary disease [81]. Moreover, the researchers found that independent predictors of poor survival included basal subpleural honeycombing, DLCO < 40%, and White race [81]. This is the first study to assess patterns of fibrosis with mortality.
Associations between race and survival have been made by prior studies. As mentioned earlier, a United States population-based study found increased rates of pulmonary hypertension and pulmonary fibrosis in African Americans, and a significantly disproportionate increase in mortality amongst young African-American women compared with their Caucasian counterparts [6]. The recent finding of higher mortality in White race as noted above was shown after controlling for extent of fibrosis, fibrotic pattern on HRCT, presence or absence of sarcoidosis-associated pulmonary hypertension, age, and study location. The uncertainty surrounding the relationship between race, sex, and mortality in fibrotic pulmonary sarcoidosis underscores the need for additional research.
In a retrospective study conducted in France, individuals with fibrotic pulmonary sarcoidosis displayed a mortality rate of 11.3% over an average follow-up period of seven years [12]. Respiratory complications accounted for 75% of patient deaths, while 31.2% were attributed to pulmonary hypertension, and 25% were linked to chronic respiratory failure [12]. Other complications as contributory causes of death included extrapulmonary cardiac involvement, immunosuppressive therapy, and aspergilloma infection [12]. On univariate analysis, the authors found New York Heart Association [82] (NYHA) functional class, forced expiratory volume in 1 s (FEV1) below 63% predicted, forced vital capacity (FVC) below 72% predicted, total lung capacity (TLC) below 74% predicted, diffusion capacity of carbon monoxide (DLCO) below 58% predicted, room-air arterial oxygen tension (PaO2) below 81 mmHg, and the presence of pulmonary hypertension exhibited a significant association with increased risk of mortality [12].
5. Management
Management of fibrotic pulmonary sarcoidosis is challenging, largely due to the lack of standardized therapy and variability in presentation and evolution of the disease and needs long-term studies into treatment options. Treatment decisions are often guided by clinical experience and expert opinion. In general, a comprehensive approach involves the integration of various diagnostic tools, including serum biomarkers, PFT, 6MWT, imaging studies, and echocardiography. These assessments can be used to monitor disease progression, identify exacerbations and new complications such as pulmonary hypertension, and progressive respiratory failure. A personalized and multidisciplinary treatment strategy is necessary to address the complexities of the disease, manage comorbidities, deliver supportive care, and consider the possibility of lung transplantation (Figure 4). The next few paragraphs cover the basics of management of patients with fibrotic pulmonary sarcoidosis, but details can be found in corresponding sections of this series.
5.1. Anti-Inflammatory Therapy
Anti-inflammatory agents may preserve or improve lung function, aid in symptom management, and prevent progression of disease in certain patients with fibrotic pulmonary sarcoidosis [12,83]. However, identifying which patients would benefit from treatment remains uncertain. Due to a lack of evidence-based therapies in fibrotic pulmonary sarcoidosis, the Delphi consensus and the ERS clinical practice guidelines on treatment of pulmonary sarcoidosis have not focused on this subset [83,84]. Currently, anti-inflammatory therapy in the setting of fibrotic pulmonary sarcoidosis is done in a case-by-case scenario.
In patients with acute or chronic disease, the primary method for managing inflammation involves the use of glucocorticoids (prednisone 20–40 mg daily) as the initial treatment [83,84]. As clinical symptoms resolve, glucocorticoids are rapidly tapered to doses less than 10 mg or to the lowest effective dose. In cases where glucocorticoids are unable to be tapered, the addition of antimetabolites with methotrexate or azathioprine as second-line agents is considered [83,84,85]. In the context of progressive fibrotic disease, prior use of methotrexate may pose a challenge by raising concerns about pulmonary toxicity, necessitating a thorough evaluation of patients for potential adverse effects. If feasible and without financial barriers, FDG-PET/CT may be useful in identifying areas of inflammation. If unavailable, other disease-modifying agents may have to be chosen. Anti-TNF-alpha agents such as infliximab can also be considered and have been shown to improve or maintain FVC [86,87]. In certain situations with ongoing disease progression, repository corticotrophin injections, rituximab, and JAK inhibitors can be explored, although a consensus is yet to be established [83]. The potential for adverse effects induced by chronic glucocorticoids and chronic immunosuppressive agents requires frequent monitoring and vigilance in identifying drug-induced toxicities. Relapse after discontinuation of therapy can occur, and patients require clinical monitoring. Use of anti-inflammatory agents in the setting of fibrotic pulmonary sarcoidosis needs more evidence. While there are multiple agents being studied in the management of pulmonary sarcoidosis, most of these studies exclude patients with fibrosis >20%. It would be intriguing to see how these agents potentially impact the course of chronic pulmonary sarcoidosis and fibrotic pulmonary sarcoidosis.
5.2. Sarcoidosis-Associated Bronchiectasis
Patients with advanced pulmonary sarcoidosis can develop granulomatous infiltration of the airways, causing fibrotic changes with airway distortion and traction bronchiectasis. Airway abnormalities, along with chronic inflammatory treatments and poor mucociliary clearance, create optimal environments for infections and mycetomas [88,89,90].
5.3. Acute Pulmonary Exacerbations of Sarcoidosis
There is no consensus on the definition of acute exacerbations in sarcoidosis. Exacerbations have been described in the literature as new or worsening pulmonary symptoms with a decline in FVC or FEV1 for greater than one month, and exclusion of alternative causes [91,92]. Exacerbations may be related to bronchiectasis, infection, and impaired immune response, and require treatment with antibiotics and/or glucocorticoids [88]. In patients with fibrotic sarcoidosis, a small trial (n = 38) found that patients with greater than two exacerbations who were treated with roflumilast, a phosphodiesterase-4 inhibitor, had improved FEV1 in subsequent visits and quality of life than those treated with placebo [93]. A larger prospective trial confirming these results would provide valuable insights into the efficacy of roflumilast and could help establish a more standardized approach in identifying and managing acute exacerbations.
5.4. Infections
Infections such as aspergillus, mycobacteria, cryptococcus, nocardia, and histoplasma can complicate clinical course and increase morbidity and mortality in patients with sarcoidosis [94,95]. Mycetomas, particularly chronic pulmonary aspergillosis, have been reported in 3–12% of patients with APS [96]. Though frequently asymptomatic, they can cause life-threatening hemoptysis and may require long-term antifungal therapy, bronchial artery embolization, and surgical resection [97].
5.5. Sarcoidosis-Associated Pulmonary Hypertension
Sarcoidosis-associated pulmonary hypertension (SAPH) is classified into World Health Organization (WHO) group 5 and has been noted to be in 73.8% of patients with advanced pulmonary sarcoidosis awaiting lung transplantation [98]. It is a major cause of morbidity and mortality in APS and a predictor of lung transplant waitlist mortality [13,79,98,99]. Not all pulmonary vasodilator therapies may be appropriate, and treatment decisions regarding pulmonary vasodilator therapy should be made by experts with clinical experience in SAPH due to the multifactorial nature of SAPH and limited double-blind placebo-control trials in patients with precapillary SAPH.
5.6. Antifibrotic Therapy
There are no established guidelines for the use of antifibrotic therapies in the context of fibrotic pulmonary sarcoidosis. Targeted treatments are considered on an individualized basis and continue to be investigated. Insights from the INBUILD trial demonstrated that nintedanib decreased the rate of decline in FVC in progressive pulmonary fibrosis. However, the trial was underpowered for fibrotic sarcoidosis (n = 12), and nintedanib needs to be examined in a larger cohort [100]. Similarly, the RELIEF trial showed that pirfenidone had a slower decline in percent predicted FVC, but the study was terminated prematurely due to challenges related to slow recruitment in non-IPF progressive fibrotic lung disease [101]. Moreover, the study excluded patients with sarcoidosis, and the results limit the applicability to this cohort [101]. The PirFS trial, initially designed as a double-blind placebo controlled trial to assess the antifibrotic effect of pirfenidone on fibrotic pulmonary sarcoidosis, was subsequently converted to a phase-4 feasibility trial due to poor enrollment during the COVID pandemic [80]. Preliminary results suggested the potential use of DLCO < 40% predicted as an inclusion criterion for evaluating the efficacy of antifibrotic agents as these patients reached the defined time to clinical worsening [80]. Due to the inflammatory basis of fibrosis in pulmonary sarcoidosis and potential improvement in symptomology and physiologic parameters, the exact role of antifibrotic therapy in fibrotic pulmonary sarcoidosis is unclear at this time. Currently, there is no consensus on whether antifibrotic therapy should be used alone, in conjunction with, or after anti-inflammatory therapy has failed to slow the progression of fibrosis in patients with fibrotic pulmonary sarcoidosis. This needs to be decided on an individual basis.
5.7. Supportive Management
Supportive management, including pulmonary rehabilitation, preventative vaccinations, and supplemental oxygen therapy, may improve overall wellbeing for individuals affected by fibrotic pulmonary sarcoidosis. Pulmonary rehabilitation is a comprehensive program tailored to each patient’s needs, and involves personalized evaluations, exercise training, educational sessions, and behavioral modifications aimed at enhancing overall wellbeing [102]. Studies primarily conducted among patients diagnosed with chronic obstructive pulmonary disease (COPD) have demonstrated significant improvements in mortality, exercise capacity, overall quality of life, and efficient utilization of healthcare resources [103,104,105,106]. Emerging evidence suggests that individuals with interstitial lung disease, pulmonary hypertension, and those undergoing evaluation for lung transplantation derive benefit as well in exercise tolerance and decreased dyspnea [107,108,109]. According to the latest guidelines from the European Respiratory Society, pulmonary rehabilitation for a duration of 6–12 weeks is conditionally recommended for managing fatigue in patients with chronic sarcoidosis [84]. One observational pilot study evaluated the impact of a 12-week physical training program in 24 patients with IPF and fibrotic pulmonary sarcoidosis. Upon finishing the program, over 50% of patients had improvements in exercise capacity as assessed by 6-min walk distance, while others maintained their initial levels [110]. Another systematic review found that pulmonary rehabilitation may enhance exercise capacity and alleviate dyspnea in individuals with sarcoidosis, irrespective of stage of the disease [111]. These results highlight the potential in enhancing overall functional status among patients with sarcoidosis in general and with fibrotic pulmonary sarcoidosis.
Patients with sarcoidosis have dysregulated immune responses caused by underlying granulomatous inflammation and concurrent use of immunosuppressive agents, which can affect the efficacy of vaccinations [112]. Considering this, the timing of immunosuppressive treatments must be taken into account when administering inactivated and live vaccines. In the case of live vaccinations, the benefits should be carefully weighed against the associated risks, and therapy should be temporarily delayed before and after administration of live vaccinations [112]. Especially with B-cell depleting therapies such as rituximab, vaccination dosing and frequency require careful consideration of scheduling [112,113].
Though the ERS treatment guidelines on sarcoidosis do not make specific recommendations regarding oxygen supplementation, patients with chronic hypoxemic respiratory failure due to pulmonary sarcoidosis should be supported with supplemental oxygen therapy [114]. Non-invasive ventilation may be used as supportive therapy in cases of respiratory failure. Currently, there is limited evidence regarding the potential benefits or risks associated with the use of supplemental oxygenation in pulmonary sarcoidosis, particularly concerning aspects such as nocturnal hypoxemia, exertional hypoxemia, dyspnea, and exercise endurance. Further studies in this area are essential for a comprehensive understanding of the implications and appropriate management strategies for these patients with chronic hypoxemic respiratory failure due to APS.
5.8. Lung Transplantation
Lung transplantation serves as a final option for patients with fibrotic pulmonary sarcoidosis suffering from respiratory failure and pulmonary hypertension. According to International Society for Heart and Lung Transplantation (ISHLT) registry data, sarcoidosis accounts for 2.4% of all lung transplantations, and has a median survival of 6.1 years following transplantation [115,116]. There are no guidelines specifically tailored to sarcoidosis for lung transplantation; they currently follow those for ILD [117,118]. Following the implementation of the lung allocation score (LAS) in 2005, a greater percentage of sarcoidosis patients received lung allografts, leading to reduced waitlist mortality [119]. However, recent studies have found that, compared with patients with COPD and IPF, individuals with sarcoidosis continue to face disproportionately higher waitlist mortalities [120]. The several factors contributing to waitlist mortality were identified as pulmonary hypertension, poorer functional status, oxygen dependence, lower reduced output, and female sex [121,122]. Moreover, waitlisted patients’ percent predicted FVC was found to be significantly lower than the thresholds recommended by ISHLT lung transplant referrals, underscoring potential delays in referral for lung transplant [122]. Following transplantation, patients may experience increased perioperative morbidity and mortality attributed to higher rates of primary graft dysfunction, hemothorax, and prolonged dependence on ventilatory support [116,123,124,125]. Despite these initial risks, long-term survival rates appear to be comparable to those observed in other chronic lung conditions with a risk for disease recurrence [116,123,126]. Further research and development of more specific guidelines on selection and post-transplant management for patients with fibrotic pulmonary sarcoidosis are needed.
6. Conclusions
Patients suffering from fibrotic pulmonary sarcoidosis experience higher morbidity and mortality compared with those without chronic and/or advanced disease. This may be due to the progressive nature of the disease with variable complications. Factors such as age, imaging findings, respiratory failure, and pulmonary hypertension may assist in prognostication, but this needs refinement and validation. The variability in disease presentation and progression makes determining the best approach for management challenging, and the approach should be individualized for each patient. There is a critical need to evaluate management strategies and continue research efforts aimed at improving patient outcomes and quality of life.
Author Contributions
Writing—original draft preparation, J.S.K.; writing—review and editing, R.G.; visualization, J.S.K.; supervision, R.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hunninghake, G.W.; Costabel, U.; Ando, M.; Baughman, R.; Cordier, J.F.; Bois, R.D.; Eklund, A.; Kitaichi, M.; Lynch, J.; Rizzato, G.; et al. Ats/ers/wasog statement on sarcoidosis. American thoracic society/european respiratory society/world association of sarcoidosis and other granulomatous disorders. Sarcoidosis Vasc. Diffuse Lung Dis. 1999, 16, 149–173. [Google Scholar] [PubMed]
- Judson, M.A.; Thompson, B.W.; Rabin, D.L.; Steimel, J.; Knattereud, G.L.; Lackland, D.T.; Rose, C.; Rand, C.S.; Baughman, R.P.; Teirstein, A.S.; et al. The diagnostic pathway to sarcoidosis. Chest 2003, 123, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Crouser, E.D.; Maier, L.A.; Wilson, K.C.; Bonham, C.A.; Morgenthau, A.S.; Patterson, K.C.; Abston, E.; Bernstein, R.C.; Blankstein, R.; Chen, E.S.; et al. Diagnosis and detection of sarcoidosis. An official american thoracic society clinical practice guideline. Am. J. Respir. Crit. Care Med. 2020, 201, e26–e51. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.P.; Lower, E.E.; Bois, R.M.D. Sarcoidosis. Lancet 2003, 361, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Hena, K.M. Sarcoidosis epidemiology: Race matters. Front. Immunol. 2020, 11, 537382. [Google Scholar] [CrossRef] [PubMed]
- Mirsaeidi, M.; Machado, R.F.; Schraufnagel, D.; Sweiss, N.J.; Baughman, R.P. Racial difference in sarcoidosis mortality in the united states. Chest 2015, 147, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Scadding, J.G. Prognosis of intrathoracic sarcoidosis in england. A review of 136 cases after five years’ observation. Br. Med. J. 1961, 2, 1165–1172. [Google Scholar] [CrossRef]
- Judson, M.A.; Boan, A.D.; Lackland, D.T. The clinical course of sarcoidosis: Presentation, diagnosis, and treatment in a large white and black cohort in the united states. Sarcoidosis Vasc. Diffuse Lung Dis. 2012, 29, 119–127. [Google Scholar]
- Yeager, H.; Rossman, M.D.; Baughman, R.P.; Teirstein, A.S.; Judson, M.A.; Rabin, D.L.; Iannuzzi, M.C.; Rose, C.; Bresnitz, E.A.; DePalo, L.; et al. Pulmonary and psychosocial findings at enrollment in the access study. Sarcoidosis Vasc. Diffuse Lung Dis. 2005, 22, 147–153. [Google Scholar]
- Gupta, R.; Judson, M.A.; Baughman, R.P. Management of advanced pulmonary sarcoidosis. Am. J. Respir. Crit. Care Med. 2022, 205, 495–506. [Google Scholar] [CrossRef]
- Gupta, R.; Baughman, R.P. Advanced pulmonary sarcoidosis. Semin. Respir. Crit. Care Med. 2020, 41, 700–715. [Google Scholar] [CrossRef]
- Nardi, A.; Brillet, P.Y.; Letoumelin, P.; Girard, F.; Brauner, M.; Uzunhan, Y.; Naccache, J.M.; Valeyre, D.; Nunes, H. Stage iv sarcoidosis: Comparison of survival with the general population and causes of death. Eur. Respir. J. 2011, 38, 1368–1373. [Google Scholar] [CrossRef]
- Jeny, F.; Uzunhan, Y.; Lacroix, M.; Gille, T.; Brillet, P.Y.; Nardi, A.; Bouvry, D.; Planes, C.; Nunes, H.; Valeyre, D. Predictors of mortality in fibrosing pulmonary sarcoidosis. Respir. Med. 2020, 169, 105997. [Google Scholar] [CrossRef]
- Shlobin, O.A.; Kouranos, V.; Barnett, S.D.; Alhamad, E.H.; Culver, D.A.; Barney, J.; Cordova, F.C.; Carmona, E.M.; Scholand, M.B.; Wijsenbeek, M.; et al. Physiological predictors of survival in patients with sarcoidosis-associated pulmonary hypertension: Results from an international registry. Eur. Respir. J. 2020, 55, 1901747. [Google Scholar] [CrossRef]
- Shorr, A.F.; Davies, D.B.; Nathan, S.D. Outcomes for patients with sarcoidosis awaiting lung transplantation. Chest 2002, 122, 233–238. [Google Scholar] [CrossRef]
- Ronsmans, S.; De Ridder, J.; Vandebroek, E.; Keirsbilck, S.; Nemery, B.; Hoet, P.H.M.; Vanderschueren, S.; Wuyts, W.A.; Yserbyt, J. Associations between occupational and environmental exposures and organ involvement in sarcoidosis: A retrospective case-case analysis. Respir. Res. 2021, 22, 224. [Google Scholar] [CrossRef]
- Beijer, E.; Veltkamp, M. The emerging role of inorganic elements as potential antigens in sarcoidosis. Curr. Opin. Pulm. Med. 2021, 27, 430–438. [Google Scholar] [CrossRef]
- Beijer, E.; Meek, B.; Bossuyt, X.; Peters, S.; Vermeulen, R.C.H.; Kromhout, H.; Veltkamp, M. Immunoreactivity to metal and silica associates with sarcoidosis in dutch patients. Respir. Res. 2020, 21, 141. [Google Scholar] [CrossRef]
- Grunewald, J.; Grutters, J.C.; Arkema, E.V.; Saketkoo, L.A.; Moller, D.R.; Muller-Quernheim, J. Sarcoidosis. Nat. Rev. Dis. Primers 2019, 5, 45. [Google Scholar] [CrossRef]
- Iannuzzi, M.C.; Maliarik, M.J.; Poisson, L.M.; Rybicki, B.A. Sarcoidosis susceptibility and resistance hla-dqb1 alleles in african americans. Am. J. Respir. Crit. Care Med. 2003, 167, 1225–1231. [Google Scholar] [CrossRef]
- Iannuzzi, M.C.; Iyengar, S.K.; Gray-McGuire, C.; Elston, R.C.; Baughman, R.P.; Donohue, J.F.; Hirst, K.; Judson, M.A.; Kavuru, M.S.; Maliarik, M.J.; et al. Genome-wide search for sarcoidosis susceptibility genes in african americans. Genes Immun. 2005, 6, 509–518. [Google Scholar] [CrossRef]
- Gray-McGuire, C.; Sinha, R.; Iyengar, S.; Millard, C.; Rybicki, B.A.; Elston, R.C.; Iannuzzi, M.C.; Consortium, S.S. Genetic characterization and fine mapping of susceptibility loci for sarcoidosis in african americans on chromosome 5. Hum. Genet. 2006, 120, 420–430. [Google Scholar] [CrossRef]
- Rossman, M.D.; Thompson, B.; Frederick, M.; Maliarik, M.; Iannuzzi, M.C.; Rybicki, B.A.; Pandey, J.P.; Newman, L.S.; Magira, E.; Beznik-Cizman, B.; et al. Hla-drb1*1101: A significant risk factor for sarcoidosis in blacks and whites. Am. J. Hum. Genet. 2003, 73, 720–735. [Google Scholar] [CrossRef]
- Levin, A.M.; Iannuzzi, M.C.; Montgomery, C.G.; Trudeau, S.; Datta, I.; McKeigue, P.; Fischer, A.; Nebel, A.; Rybicki, B.A. Association of anxa11 genetic variation with sarcoidosis in african americans and european americans. Genes Immun. 2013, 14, 13–18. [Google Scholar] [CrossRef]
- Gazzerro, E.; Pereira, R.C.; Jorgetti, V.; Olson, S.; Economides, A.N.; Canalis, E. Skeletal overexpression of gremlin impairs bone formation and causes osteopenia. Endocrinology 2005, 146, 655–665. [Google Scholar] [CrossRef]
- Salez, F.; Gosset, P.; Copin, M.C.; Degros, C.L.; Tonnel, A.B.; Wallaert, B. Transforming growth factor-beta1 in sarcoidosis. Eur. Respir. J. 1998, 12, 913–919. [Google Scholar] [CrossRef]
- Heron, M.; van Moorsel, C.H.; Grutters, J.C.; Huizinga, T.W.; van der Helm-van Mil, A.H.; Nagtegaal, M.M.; Ruven, H.J.; van den Bosch, J.M. Genetic variation in grem1 is a risk factor for fibrosis in pulmonary sarcoidosis. Tissue Antigens 2011, 77, 112–117. [Google Scholar] [CrossRef]
- Kruit, A.; Grutters, J.C.; Ruven, H.J.; van Moorsel, C.H.; Weiskirchen, R.; Mengsteab, S.; van den Bosch, J.M. Transforming growth factor-beta gene polymorphisms in sarcoidosis patients with and without fibrosis. Chest 2006, 129, 1584–1591. [Google Scholar] [CrossRef]
- Sato, H.; Williams, H.R.; Spagnolo, P.; Abdallah, A.; Ahmad, T.; Orchard, T.R.; Copley, S.J.; Desai, S.R.; Wells, A.U.; Bois, R.M.D.; et al. Card15/nod2 polymorphisms are associated with severe pulmonary sarcoidosis. Eur. Respir. J. 2010, 35, 324–330. [Google Scholar] [CrossRef]
- Hill, M.R.; Papafili, A.; Booth, H.; Lawson, P.; Hubner, M.; Beynon, H.; Read, C.; Lindahl, G.; Marshall, R.P.; McAnulty, R.J.; et al. Functional prostaglandin-endoperoxide synthase 2 polymorphism predicts poor outcome in sarcoidosis. Am. J. Respir. Crit. Care Med. 2006, 174, 915–922. [Google Scholar] [CrossRef]
- Gupta, R.; Kim, J.S.; Baughman, R.P. An expert overview of pulmonary fibrosis in sarcoidosis. Expert Rev. Respir. Med. 2023, 17, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Lepzien, R.; Liu, S.; Czarnewski, P.; Nie, M.; Osterberg, B.; Baharom, F.; Pourazar, J.; Rankin, G.; Eklund, A.; Bottai, M.; et al. Monocytes in sarcoidosis are potent tumour necrosis factor producers and predict disease outcome. Eur. Respir. J. 2021, 58, 2003468. [Google Scholar] [CrossRef] [PubMed]
- Weeratunga, P.; Moller, D.R.; Ho, L.P. Immune mechanisms in fibrotic pulmonary sarcoidosis. Eur. Respir. Rev. 2022, 31, 220178. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, D.N.; Armstrong, M.E.; Trujillo, G.; Cooke, G.; Keane, M.P.; Fallon, P.G.; Simpson, A.J.; Millar, A.B.; McGrath, E.E.; Whyte, M.K.; et al. The toll-like receptor 3 l412f polymorphism and disease progression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2013, 188, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Prasse, A.; Probst, C.; Bargagli, E.; Zissel, G.; Toews, G.B.; Flaherty, K.R.; Olschewski, M.; Rottoli, P.; Muller-Quernheim, J. Serum cc-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2009, 179, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Pechkovsky, D.V.; Prasse, A.; Kollert, F.; Engel, K.M.; Dentler, J.; Luttmann, W.; Friedrich, K.; Muller-Quernheim, J.; Zissel, G. Alternatively activated alveolar macrophages in pulmonary fibrosis-mediator production and intracellular signal transduction. Clin. Immunol. 2010, 137, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, R.; Kozhaya, L.; McKevitt, K.; Djuretic, I.M.; Carlson, T.J.; Quintero, M.A.; McCauley, J.L.; Abreu, M.T.; Unutmaz, D.; Sundrud, M.S. Pro-inflammatory human th17 cells selectively express p-glycoprotein and are refractory to glucocorticoids. J. Exp. Med. 2014, 211, 89–104. [Google Scholar] [CrossRef]
- Broos, C.E.; Koth, L.L.; van Nimwegen, M.; In, J.; Paulissen, S.M.J.; van Hamburg, J.P.; Annema, J.T.; Heller-Baan, R.; Kleinjan, A.; Hoogsteden, H.C.; et al. Increased t-helper 17.1 cells in sarcoidosis mediastinal lymph nodes. Eur. Respir. J. 2018, 51, 1701124. [Google Scholar] [CrossRef]
- Sano, T.; Huang, W.; Hall, J.A.; Yang, Y.; Chen, A.; Gavzy, S.J.; Lee, J.Y.; Ziel, J.W.; Miraldi, E.R.; Domingos, A.I.; et al. An il-23r/il-22 circuit regulates epithelial serum amyloid a to promote local effector th17 responses. Cell 2015, 163, 381–393. [Google Scholar] [CrossRef]
- Cho, W.C.; Yip, T.T.; Cheng, W.W.; Au, J.S. Serum amyloid a is elevated in the serum of lung cancer patients with poor prognosis. Br. J. Cancer 2010, 102, 1731–1735. [Google Scholar] [CrossRef]
- Beijer, E.; Roodenburg-Benschop, C.; Schimmelpennink, M.C.; Grutters, J.C.; Meek, B.; Veltkamp, M. Elevated serum amyloid a levels are not specific for sarcoidosis but associate with a fibrotic pulmonary phenotype. Cells 2021, 10, 585. [Google Scholar] [CrossRef]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory t cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef]
- Miyara, M.; Amoura, Z.; Parizot, C.; Badoual, C.; Dorgham, K.; Trad, S.; Kambouchner, M.; Valeyre, D.; Chapelon-Abric, C.; Debre, P.; et al. The immune paradox of sarcoidosis and regulatory t cells. J. Exp. Med. 2006, 203, 359–370. [Google Scholar] [CrossRef]
- Oswald-Richter, K.A.; Richmond, B.W.; Braun, N.A.; Isom, J.; Abraham, S.; Taylor, T.R.; Drake, J.M.; Culver, D.A.; Wilkes, D.S.; Drake, W.P. Reversal of global cd4+ subset dysfunction is associated with spontaneous clinical resolution of pulmonary sarcoidosis. J. Immunol. 2013, 190, 5446–5453. [Google Scholar] [CrossRef]
- Sakthivel, P.; Grunewald, J.; Eklund, A.; Bruder, D.; Wahlstrom, J. Pulmonary sarcoidosis is associated with high-level inducible co-stimulator (icos) expression on lung regulatory t cells—Possible implications for the icos/icos-ligand axis in disease course and resolution. Clin. Exp. Immunol. 2016, 183, 294–306. [Google Scholar] [CrossRef]
- Zhang, H.; Jiang, D.; Zhu, L.; Zhou, G.; Xie, B.; Cui, Y.; Costabel, U.; Dai, H. Imbalanced distribution of regulatory t cells and th17.1 cells in the peripheral blood and balf of sarcoidosis patients: Relationship to disease activity and the fibrotic radiographic phenotype. Front. Immunol. 2023, 14, 1185443. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. Mtor signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Pacheco, Y.; CLim, X.; Weichhart, T.; Valeyre, D.; Bentaher, A.; Calender, A. Sarcoidosis and the mtor, rac1, and autophagy triad. Trends Immunol. 2020, 41, 286–299. [Google Scholar] [CrossRef]
- Vukmirovic, M.; Yan, X.; Gibson, K.F.; Gulati, M.; Schupp, J.C.; DeIuliis, G.; Adams, T.S.; Hu, B.; Mihaljinec, A.; Woolard, T.N.; et al. Transcriptomics of bronchoalveolar lavage cells identifies new molecular endotypes of sarcoidosis. Eur. Respir. J. 2021, 58, 2002950. [Google Scholar] [CrossRef]
- Linke, M.; Pham, H.T.; Katholnig, K.; Schnoller, T.; Miller, A.; Demel, F.; Schutz, B.; Rosner, M.; Kovacic, B.; Sukhbaatar, N.; et al. Chronic signaling via the metabolic checkpoint kinase mtorc1 induces macrophage granuloma formation and marks sarcoidosis progression. Nat. Immunol. 2017, 18, 293–302. [Google Scholar] [CrossRef]
- Jeny, F.; Bernaudin, J.F.; Valeyre, D.; Kambouchner, M.; Pretolani, M.; Nunes, H.; Planes, C.; Besnard, V. Hypoxia promotes a mixed inflammatory-fibrotic macrophages phenotype in active sarcoidosis. Front. Immunol. 2021, 12, 719009. [Google Scholar] [CrossRef]
- Baughman, R.P.; Teirstein, A.S.; Judson, M.A.; Rossman, M.D.; Yeager, H., Jr.; Bresnitz, E.A.; DePalo, L.; Hunninghake, G.; Iannuzzi, M.C.; Johns, C.J.; et al. Clinical characteristics of patients in a case control study of sarcoidosis. Am. J. Respir. Crit. Care Med. 2001, 164, 1885–1889. [Google Scholar] [CrossRef]
- Iannuzzi, M.C.; Rybicki, B.A.; Teirstein, A.S. Sarcoidosis. N. Engl. J. Med. 2007, 357, 2153–2165. [Google Scholar] [CrossRef]
- Lofgren, R.; Meholic, A.; Ketai, L. Fundamentals of Chest Radiology; WB Saunders: Philadelphia, PA, USA, 1996; pp. 120–121. [Google Scholar]
- Abehsera, M.; Valeyre, D.; Grenier, P.; Jaillet, H.; Battesti, J.P.; Brauner, M.W. Sarcoidosis with pulmonary fibrosis: Ct patterns and correlation with pulmonary function. AJR Am. J. Roentgenol. 2000, 174, 1751–1757. [Google Scholar] [CrossRef]
- Udwadia, Z.F.; Pilling, J.R.; Jenkins, P.F.; Harrison, B.D. Bronchoscopic and bronchographic findings in 12 patients with sarcoidosis and severe or progressive airways obstruction. Thorax 1990, 45, 272–275. [Google Scholar] [CrossRef]
- Criado, E.; Sanchez, M.; Ramirez, J.; Arguis, P.; de Caralt, T.M.; Perea, R.J.; Xaubet, A. Pulmonary sarcoidosis: Typical and atypical manifestations at high-resolution ct with pathologic correlation. Radiographics 2010, 30, 1567–1586. [Google Scholar] [CrossRef]
- Walsh, S.L.; Wells, A.U.; Sverzellati, N.; Keir, G.J.; Calandriello, L.; Antoniou, K.M.; Copley, S.J.; Devaraj, A.; Maher, T.M.; Renzoni, E.; et al. An integrated clinicoradiological staging system for pulmonary sarcoidosis: A case-cohort study. Lancet Respir. Med. 2014, 2, 123–130. [Google Scholar] [CrossRef]
- Regis, C.; Benali, K.; Rouzet, F. Fdg pet/ct imaging of sarcoidosis. Semin. Nucl. Med. 2023, 53, 258–272. [Google Scholar] [CrossRef]
- Keijsers, R.G.; Grutters, J.C.; Thomeer, M.; Bois, R.M.D.; Van Buul, M.M.; Lavalaye, J.; Van Den Bosch, J.M.; Verzijlbergen, F.J. Imaging the inflammatory activity of sarcoidosis: Sensitivity and inter observer agreement of 67ga imaging and 18f-fdg pet. Q. J. Nucl. Med. Mol. Imaging 2011, 55, 66–71. [Google Scholar]
- Braun, J.J.; Kessler, R.; Constantinesco, A.; Imperiale, A. 18f-fdg pet/ct in sarcoidosis management: Review and report of 20 cases. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 1537–1543. [Google Scholar] [CrossRef]
- Vender, R.J.; Aldahham, H.; Gupta, R. The role of pet in the management of sarcoidosis. Curr. Opin. Pulm. Med. 2022, 28, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Mostard, R.L.; Verschakelen, J.A.; van Kroonenburgh, M.J.; Nelemans, P.J.; Wijnen, P.A.; Voo, S.; Drent, M. Severity of pulmonary involvement and 18f-fdg pet activity in sarcoidosis. Respir. Med. 2013, 107, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, V.; Zompatori, M.; Fasano, L.; Nanni, C.; Nava, S.; Rubello, D.; Fanti, S. 18f-fdg pet/ct for the assessment of disease extension and activity in patients with sarcoidosis: Results of a preliminary prospective study. Clin. Nucl. Med. 2013, 38, e171–e177. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Jin, R.; Wang, Y.; Li, L.; Li, K.; He, Y. The utility of 18f-fdg pet/ct for monitoring response and predicting prognosis after glucocorticoids therapy for sarcoidosis. BioMed Res. Int. 2018, 2018, 1823710. [Google Scholar] [CrossRef] [PubMed]
- Maturu, V.N.; Rayamajhi, S.J.; Agarwal, R.; Aggarwal, A.N.; Gupta, D.; Mittal, B.R. Role of serial f-18 fdg pet/ct scans in assessing treatment response and predicting relapses in patients with symptomatic sarcoidosis. Sarcoidosis Vasc. Diffuse Lung Dis. 2016, 33, 372–380. [Google Scholar] [PubMed]
- Kouranos, V.; Ward, S.; Kokosi, M.A.; Castillo, D.; Chua, F.; Judge, E.P.; Thomas, S.; Van Tonder, F.; Devaraj, A.; Nicholson, A.G.; et al. Mixed ventilatory defects in pulmonary sarcoidosis: Prevalence and clinical features. Chest 2020, 158, 2007–2014. [Google Scholar] [CrossRef] [PubMed]
- Sharp, M.; Psoter, K.J.; Balasubramanian, A.; Pulapaka, A.V.; Chen, E.S.; Brown, S.W.; Mathai, S.C.; Gilotra, N.A.; Chrispin, J.; Bascom, R.; et al. Heterogeneity of lung function phenotypes in sarcoidosis: Role of race and sex differences. Ann. Am. Thorac. Soc. 2023, 20, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.P.; Sparkman, B.K.; Lower, E.E. Six-minute walk test and health status assessment in sarcoidosis. Chest 2007, 132, 207–213. [Google Scholar] [CrossRef]
- Gupta, R.; Baughman, R.P.; Nathan, S.D.; Wells, A.U.; Kouranos, V.; Alhamad, E.H.; Culver, D.A.; Barney, J.; Carmona, E.M.; Cordova, F.C.; et al. The six-minute walk test in sarcoidosis associated pulmonary hypertension: Results from an international registry. Respir. Med. 2022, 196, 106801. [Google Scholar] [CrossRef]
- Rosen, Y. Pathology of sarcoidosis. Semin. Respir. Crit. Care Med. 2007, 28, 36–52. [Google Scholar] [CrossRef]
- Mathur, R.S.; Kurdikar, V.L.; Shah, J.R. Serum angiotensin converting enzyme in the diagnosis of pulmonary sarcoidosis. J. Assoc. Physicians India 1990, 38, 474–475. [Google Scholar] [PubMed]
- Vorselaars, A.D.; van Moorsel, C.H.; Zanen, P.; Ruven, H.J.; Claessen, A.M.; van Velzen-Blad, H.; Grutters, J.C. Ace and sil-2r correlate with lung function improvement in sarcoidosis during methotrexate therapy. Respir. Med. 2015, 109, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Popevic, S.; Sumarac, Z.; Jovanovic, D.; Babic, D.; Stjepanovic, M.; Jovicic, S.; Sobic-Saranovic, D.; Filipovic, S.; Gvozdenovic, B.; Omcikus, M.; et al. Verifying sarcoidosis activity: Chitotriosidase versus ace in sarcoidosis—A case-control study. J. Med. Biochem. 2016, 35, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Xu, Y.; Zhu, H.; Zhang, H.; Sun, S.; Sun, H.; Wang, W.; Xie, S. Relationship between ct activity score with lung function and the serum angiotensin converting enzyme in pulmonary sarcoidosis on chest hrct. Medicine 2018, 97, e12205. [Google Scholar] [CrossRef] [PubMed]
- Hunninghake, G.W.; Bedell, G.N.; Zavala, D.C.; Monick, M.; Brady, M. Role of interleukin-2 release by lung t-cells in active pulmonary sarcoidosis. Am. Rev. Respir. Dis. 1983, 128, 634–638. [Google Scholar] [PubMed]
- Sweiss, N.J.; Barnathan, E.S.; Lo, K.; A Judson, M.; Baughman, R. C-reactive protein predicts response to infliximab in patients with chronic sarcoidosis. Sarcoidosis Vasc. Diffuse Lung Dis. 2010, 27, 49–56. [Google Scholar] [PubMed]
- McDonnell, M.J.; Saleem, M.I.; Wall, D.; Gilmartin, J.J.; Rutherford, R.M.; O’Regan, A. Predictive value of c-reactive protein and clinically relevant baseline variables in sarcoidosis. Sarcoidosis Vasc. Diffuse Lung Dis. 2016, 33, 331–340. [Google Scholar]
- Kirkil, G.; Lower, E.E.; Baughman, R.P. Predictors of mortality in pulmonary sarcoidosis. Chest 2018, 153, 105–113. [Google Scholar] [CrossRef]
- Baughman, R.P.; Gupta, R.; Judson, M.A.; Lower, E.E.; Birring, S.S.; Stewart, J.; Reeves, R.; Wells, A.U. Value of pulmonary function testing identifying progressive pulmonary disease in fibrotic sarcoidosis: Results of a prospective feasibility study. Sarcoidosis Vasc. Diffuse Lung Dis. 2022, 39, e2022011. [Google Scholar] [CrossRef]
- Obi, O.N.; Alqalyoobi, S.; Maddipati, V.; Lower, E.E.; Baughman, R.P. Hrct fibrotic patterns in stage 4 pulmonary sarcoidosis: Impact on pulmonary function and survival. Chest 2023, 23, S0012-3692(23)05666-0. [Google Scholar] [CrossRef]
- Nunes, H.; Uzunhan, Y.; Gille, T.; Lamberto, C.; Valeyre, D.; Brillet, P.Y. Imaging of sarcoidosis of the airways and lung parenchyma and correlation with lung function. Eur. Respir. J. 2012, 40, 750–765. [Google Scholar] [CrossRef] [PubMed]
- Rahaghi, F.F.; Baughman, R.P.; Saketkoo, L.A.; Sweiss, N.J.; Barney, J.B.; Birring, S.S.; Costabel, U.; Crouser, E.D.; Drent, M.; Gerke, A.K.; et al. Delphi consensus recommendations for a treatment algorithm in pulmonary sarcoidosis. Eur. Respir. Rev. 2020, 29, 190146. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.P.; Valeyre, D.; Korsten, P.; Mathioudakis, A.G.; Wuyts, W.A.; Wells, A.; Rottoli, P.; Nunes, H.; Lower, E.E.; Judson, M.A.; et al. Ers clinical practice guidelines on treatment of sarcoidosis. Eur. Respir. J. 2021, 58, 2004079. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.P.; Winget, D.B.; Lower, E.E. Methotrexate is steroid sparing in acute sarcoidosis: Results of a double blind, randomized trial. Sarcoidosis Vasc. Diffuse Lung Dis. 2000, 17, 60–66. [Google Scholar] [PubMed]
- Baughman, R.P.; Drent, M.; Kavuru, M.; Judson, M.A.; Costabel, U.; Bois, R.D.; Albera, C.; Brutsche, M.; Davis, G.; Donohue, J.F.; et al. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am. J. Respir. Crit. Care Med. 2006, 174, 795–802. [Google Scholar] [CrossRef]
- Rossman, M.D.; Newman, L.S.; Baughman, R.P.; Teirstein, A.; Weinberger, S.E.; Miller, W., Jr.; Sands, B.E. A double-blinded, randomized, placebo-controlled trial of infliximab in subjects with active pulmonary sarcoidosis. Sarcoidosis Vasc. Diffuse Lung Dis. 2006, 23, 201–208. [Google Scholar]
- Baughman, R.P.; Lower, E.E. Frequency of acute worsening events in fibrotic pulmonary sarcoidosis patients. Respir. Med. 2013, 107, 2009–2013. [Google Scholar] [CrossRef]
- Sawahata, M.; Shijubo, N.; Johkoh, T.; Hagiwara, K.; Konno, S.; Yamaguchi, T. Honeycomb lung-like structures resulting from clustering of traction bronchiectasis distally in sarcoidosis. Respirol. Case Rep. 2020, 8, e00539. [Google Scholar] [CrossRef]
- Lewis, M.M.; Mortelliti, M.P.; Yeager, H., Jr.; Tsou, E. Clinical bronchiectasis complicating pulmonary sarcoidosis: Case series of seven patients. Sarcoidosis Vasc. Diffuse Lung Dis. 2002, 19, 154–159. [Google Scholar]
- McKinzie, B.P.; Bullington, W.M.; Mazur, J.E.; Judson, M.A. Efficacy of short-course, low-dose corticosteroid therapy for acute pulmonary sarcoidosis exacerbations. Am. J. Med. Sci. 2010, 339, 1–4. [Google Scholar] [CrossRef]
- Panselinas, E.; Judson, M.A. Acute pulmonary exacerbations of sarcoidosis. Chest 2012, 142, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.P.; Judson, M.A.; Culver, D.A.; Birring, S.S.; Parambil, J.; Zeigler, J.; Lower, E.E. Roflumilast (daliresp(r)) to reduce acute pulmonary events in fibrotic sarcoidosis: A multi-center, double blind, placebo controlled, randomized clinical trial. Sarcoidosis Vasc. Diffuse Lung Dis. 2021, 38, e2021035. [Google Scholar] [CrossRef] [PubMed]
- Girard, C.; Hot, A.; Etienne-Mastroianni, B.; Chidiac, C.; Cordier, J.F. Opportunistic infections and sarcoidosis. Rev. Mal. Respir. 2004, 21, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Jamilloux, Y.; Maucort-Boulch, D.; Kerever, S.; Gerfaud-Valentin, M.; Broussolle, C.; Eb, M.; Valeyre, D.; Seve, P. Sarcoidosis-related mortality in france: A multiple-cause-of-death analysis. Eur. Respir. J. 2016, 48, 1700–1709. [Google Scholar] [CrossRef] [PubMed]
- Uzunhan, Y.; Nunes, H.; Jeny, F.; Lacroix, M.; Brun, S.; Brillet, P.Y.; Martinod, E.; Carette, M.F.; Bouvry, D.; Charlier, C.; et al. Chronic pulmonary aspergillosis complicating sarcoidosis. Eur. Respir. J. 2017, 49, 1602396. [Google Scholar] [CrossRef] [PubMed]
- Rafferty, P.; Biggs, B.A.; Crompton, G.K.; Grant, I.W. What happens to patients with pulmonary aspergilloma? Analysis of 23 cases. Thorax 1983, 38, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Shorr, A.F.; Helman, D.L.; Davies, D.B.; Nathan, S.D. Pulmonary hypertension in advanced sarcoidosis: Epidemiology and clinical characteristics. Eur. Respir. J. 2005, 25, 783–788. [Google Scholar] [CrossRef]
- Shorr, A.F.; Davies, D.B.; Nathan, S.D. Predicting mortality in patients with sarcoidosis awaiting lung transplantation. Chest 2003, 124, 922–928. [Google Scholar] [CrossRef]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.F.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in progressive fibrosing interstitial lung diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef]
- Behr, J.; Prasse, A.; Kreuter, M.; Johow, J.; Rabe, K.F.; Bonella, F.; Bonnet, R.; Grohe, C.; Held, M.; Wilkens, H.; et al. Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (relief): A double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2021, 9, 476–486. [Google Scholar] [CrossRef]
- Holland, A.E.; Cox, N.S.; Houchen-Wolloff, L.; Rochester, C.L.; Garvey, C.; ZuWallack, R.; Nici, L.; Limberg, T.; Lareau, S.C.; Yawn, B.P.; et al. Defining modern pulmonary rehabilitation. An official american thoracic society workshop report. Ann. Am. Thorac. Soc. 2021, 18, e12–e29. [Google Scholar] [CrossRef] [PubMed]
- Puhan, M.A.; Gimeno-Santos, E.; Cates, C.J.; Troosters, T. Pulmonary rehabilitation following exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst. Rev. 2016, 12, CD005305. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, B.; Casey, D.; Devane, D.; Murphy, K.; Murphy, E.; Lacasse, Y. Pulmonary rehabilitation for chronic obstructive pulmonary disease. Cochrane Database Syst. Rev. 2015, 2015, CD003793. [Google Scholar] [CrossRef] [PubMed]
- Hudson, L.D.; Tyler, M.L.; Petty, T.L. Hospitalization needs during an outpatient rehabilitation program for severe chronic airway obstruction. Chest 1976, 70, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Seymour, J.M.; Moore, L.; Jolley, C.J.; Ward, K.; Creasey, J.; Steier, J.S.; Yung, B.; Man, W.D.; Hart, N.; Polkey, M.I.; et al. Outpatient pulmonary rehabilitation following acute exacerbations of copd. Thorax 2010, 65, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Morisset, J.; Dube, B.P.; Garvey, C.; Bourbeau, J.; Collard, H.R.; Swigris, J.J.; Lee, J.S. The unmet educational needs of patients with interstitial lung disease. Setting the stage for tailored pulmonary rehabilitation. Ann. Am. Thorac. Soc. 2016, 13, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Morris, N.R.; Kermeen, F.D.; Holland, A.E. Exercise-based rehabilitation programmes for pulmonary hypertension. Cochrane Database Syst. Rev. 2017, 1, CD011285. [Google Scholar] [CrossRef]
- Li, M.; Mathur, S.; Chowdhury, N.A.; Helm, D.; Singer, L.G. Pulmonary rehabilitation in lung transplant candidates. J. Heart Lung Transplant. 2013, 32, 626–632. [Google Scholar] [CrossRef]
- Strookappe, B.; Swigris, J.; De Vries, J.; Elfferich, M.; Knevel, T.; Drent, M. Benefits of physical training in sarcoidosis. Lung 2015, 193, 701–708. [Google Scholar] [CrossRef]
- Alsina-Restoy, X.; Torres-Castro, R.; Caballeria, E.; Gimeno-Santos, E.; Solis-Navarro, L.; Francesqui, J.; Hernandez-Gonzalez, F.; Ramos-Casals, M.; Blanco, I.; Sellares, J. Pulmonary rehabilitation in sarcoidosis: A systematic review and meta-analysis. Respir. Med. 2023, 219, 107432. [Google Scholar] [CrossRef]
- Syed, H.; Ascoli, C.; Linssen, C.F.; Vagts, C.; Iden, T.; Syed, A.; Kron, J.; Polly, K.; Perkins, D.; Finn, P.W.; et al. Infection prevention in sarcoidosis: Proposal for vaccination and prophylactic therapy. Sarcoidosis Vasc. Diffuse Lung Dis. 2020, 37, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Haraoui, B.; Kumar, D.; Marshall, J.K.; Bissonnette, R.; Bitton, A.; Bressler, B.; Gooderham, M.; Ho, V.; Jamal, S.; et al. Vaccination guidelines for patients with immune-mediated disorders on immunosuppressive therapies. J. Cutan. Med. Surg. 2019, 23, 50–74. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.S.; Krishnan, J.A.; Lederer, D.J.; Ghazipura, M.; Hossain, T.; Tan, A.M.; Carlin, B.; Drummond, M.B.; Ekstrom, M.; Garvey, C.; et al. Home oxygen therapy for adults with chronic lung disease. An official american thoracic society clinical practice guideline. Am. J. Respir. Crit. Care Med. 2020, 202, e121–e141. [Google Scholar] [CrossRef] [PubMed]
- Valapour, M.; Lehr, C.J.; Skeans, M.A.; Smith, J.M.; Uccellini, K.; Lehman, R.; Robinson, A.; Israni, A.K.; Snyder, J.J.; Kasiske, B.L. Optn/srtr 2017 annual data report: Lung. Am. J. Transplant. 2019, 2, 404–484. [Google Scholar] [CrossRef] [PubMed]
- Yusen, R.D.; Edwards, L.B.; Kucheryavaya, A.Y.; Benden, C.; Dipchand, A.I.; Goldfarb, S.B.; Levvey, B.J.; Lund, L.H.; Meiser, B.; Rossano, J.W.; et al. The registry of the international society for heart and lung transplantation: Thirty-second official adult lung and heart-lung transplantation report—2015; focus theme: Early graft failure. J. Heart Lung Transplant. 2015, 34, 1264–1277. [Google Scholar] [CrossRef] [PubMed]
- Weill, D.; Benden, C.; Corris, P.A.; Dark, J.H.; Davis, R.D.; Keshavjee, S.; Lederer, D.J.; Mulligan, M.J.; Patterson, G.A.; Singer, L.G.; et al. A consensus document for the selection of lung transplant candidates: 2014—An update from the pulmonary transplantation council of the international society for heart and lung transplantation. J. Heart Lung Transplant. 2015, 34, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Gupta, R. Lung transplantation in pulmonary sarcoidosis. J. Autoimmun. 2023, 103135. [Google Scholar] [CrossRef]
- Egan, T.M.; Edwards, L.B. Effect of the lung allocation score on lung transplantation in the united states. J. Heart Lung Transplant. 2016, 35, 433–439. [Google Scholar] [CrossRef]
- Fleitas Sosa, D.C.; Gayen, S.; Zheng, M.; Gangemi, A.J.; Zhao, H.; Kim, V.; Sehgal, S.; Criner, G.J.; Gupta, R.; Mamary, A.J. Sarcoidosis lung transplantation waitlist mortality, a national registry database study. ERJ Open Res 2023, 9, 00738–2022. [Google Scholar] [CrossRef]
- Gayen, S.; Sosa, D.F.; Zheng, M.; Gangemi, A.; Sehgal, S.; Zhao, H.; Marchetti, N.; Criner, G.J.; Gupta, R.; Mamary, A.J. Lung transplantation waitlist mortality among sarcoidosis patients by lung allocation score grouping. Transplant. Proc. 2023, 55, 440–445. [Google Scholar] [CrossRef]
- Gangemi, A.J.; Myers, C.N.; Zheng, M.; Brown, J.; Butler-LeBair, M.; Cordova, F.; Marchetti, N.; Criner, G.J.; Gupta, R.; Mamary, A.J. Mortality for sarcoidosis patients on the transplant wait list in the lung allocation score era: Experience from a high volume center. Respir. Med. 2019, 157, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Le Pavec, J.; Valeyre, D.; Gazengel, P.; Holm, A.M.; Schultz, H.H.; Perch, M.; Le Borgne, A.; Reynaud-Gaubert, M.; Knoop, C.; Godinas, L.; et al. Lung transplantation for sarcoidosis: Outcome and prognostic factors. Eur. Respir. J. 2021, 58, 2003358. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.C. Lung transplantation for pulmonary sarcoidosis. Sarcoidosis Vasc. Diffuse Lung Dis. 2019, 36, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Shorr, A.F.; Helman, D.L.; Davies, D.B.; Nathan, S.D. Sarcoidosis, race, and short-term outcomes following lung transplantation. Chest 2004, 125, 990–996. [Google Scholar] [CrossRef]
- Taimeh, Z.; Hertz, M.I.; Shumway, S.; Pritzker, M. Lung transplantation for pulmonary sarcoidosis. Twenty-five years of experience in the USA. Thorax 2016, 71, 378–379. [Google Scholar] [CrossRef]
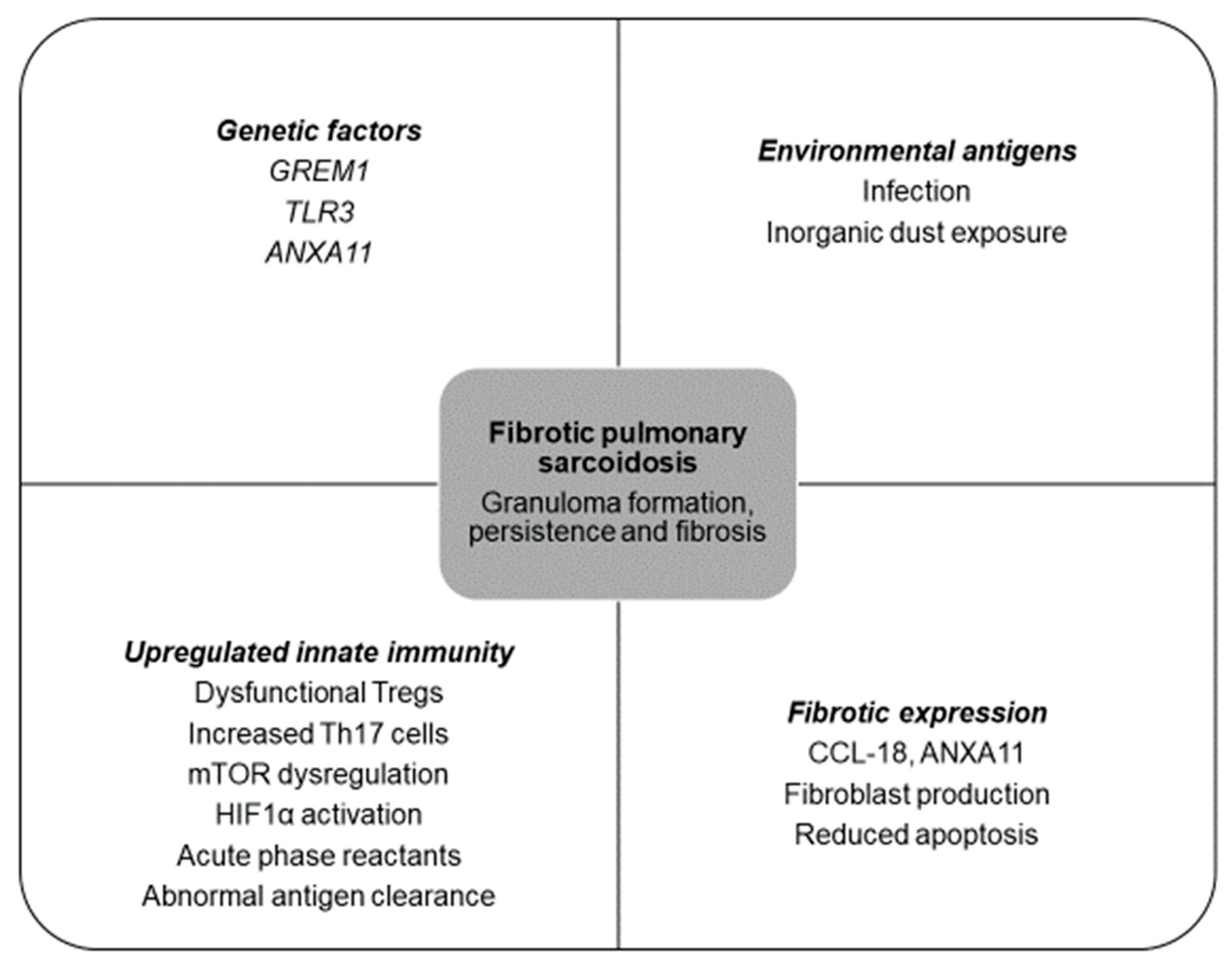
Figure 1.
Drivers that may be involved in fibrotic pulmonary sarcoidosis. GREM1, gremlin 1; TLR3, toll-like receptor 3; ANXA11, annexin 11; Tregs, T-regulatory cells; Th17, T-helper 17 cells; mTOR, mammalian target of rapamycin complex 1; HIF1α, hypoxia inducible factor 1; CCL-18, C-C motif chemokine ligand 18.
Figure 1.
Drivers that may be involved in fibrotic pulmonary sarcoidosis. GREM1, gremlin 1; TLR3, toll-like receptor 3; ANXA11, annexin 11; Tregs, T-regulatory cells; Th17, T-helper 17 cells; mTOR, mammalian target of rapamycin complex 1; HIF1α, hypoxia inducible factor 1; CCL-18, C-C motif chemokine ligand 18.

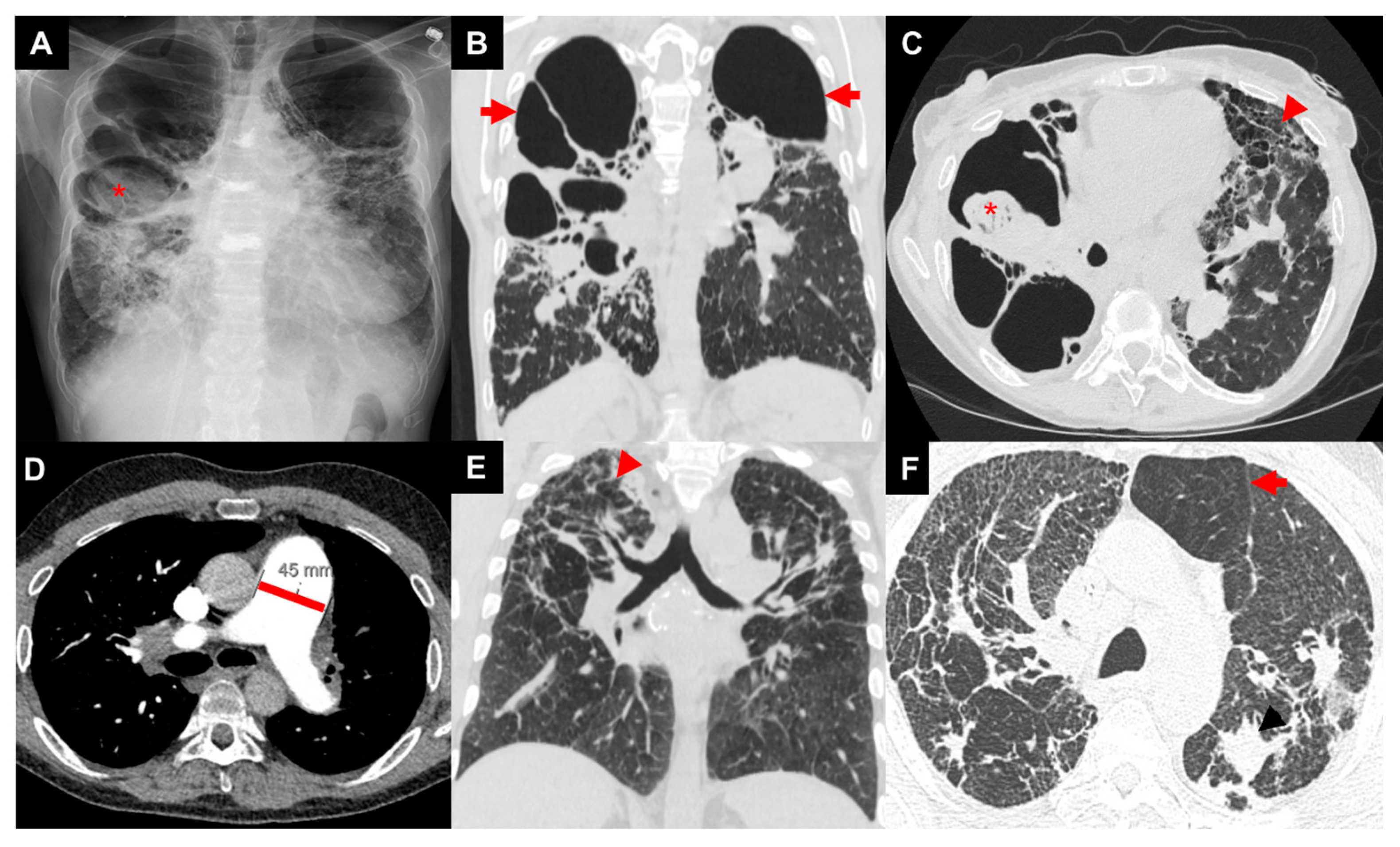
Figure 2.
Images of three patients with advanced pulmonary sarcoidosis: patient 1 with biapical cavities and mycetoma (asterisk) on chest X-ray (A), large biapical bronchiectatic cavities (arrows) on coronal image of CT chest (B), right apical mycetoma (asterisk) and extensive left-sided upper-zone predominant fibrosis (arrow head) of anterior lung on axial image of CT chest (C); patient 2 with enlarged pulmonary artery diameter (45 mm) on axial image of CT chest (D); patient 3 with bilateral irregular reticular and nodular fibrosis (arrow head) on coronal image of CT chest (E) with air trapping (arrow) on axial image of CT chest (F).
Figure 2.
Images of three patients with advanced pulmonary sarcoidosis: patient 1 with biapical cavities and mycetoma (asterisk) on chest X-ray (A), large biapical bronchiectatic cavities (arrows) on coronal image of CT chest (B), right apical mycetoma (asterisk) and extensive left-sided upper-zone predominant fibrosis (arrow head) of anterior lung on axial image of CT chest (C); patient 2 with enlarged pulmonary artery diameter (45 mm) on axial image of CT chest (D); patient 3 with bilateral irregular reticular and nodular fibrosis (arrow head) on coronal image of CT chest (E) with air trapping (arrow) on axial image of CT chest (F).

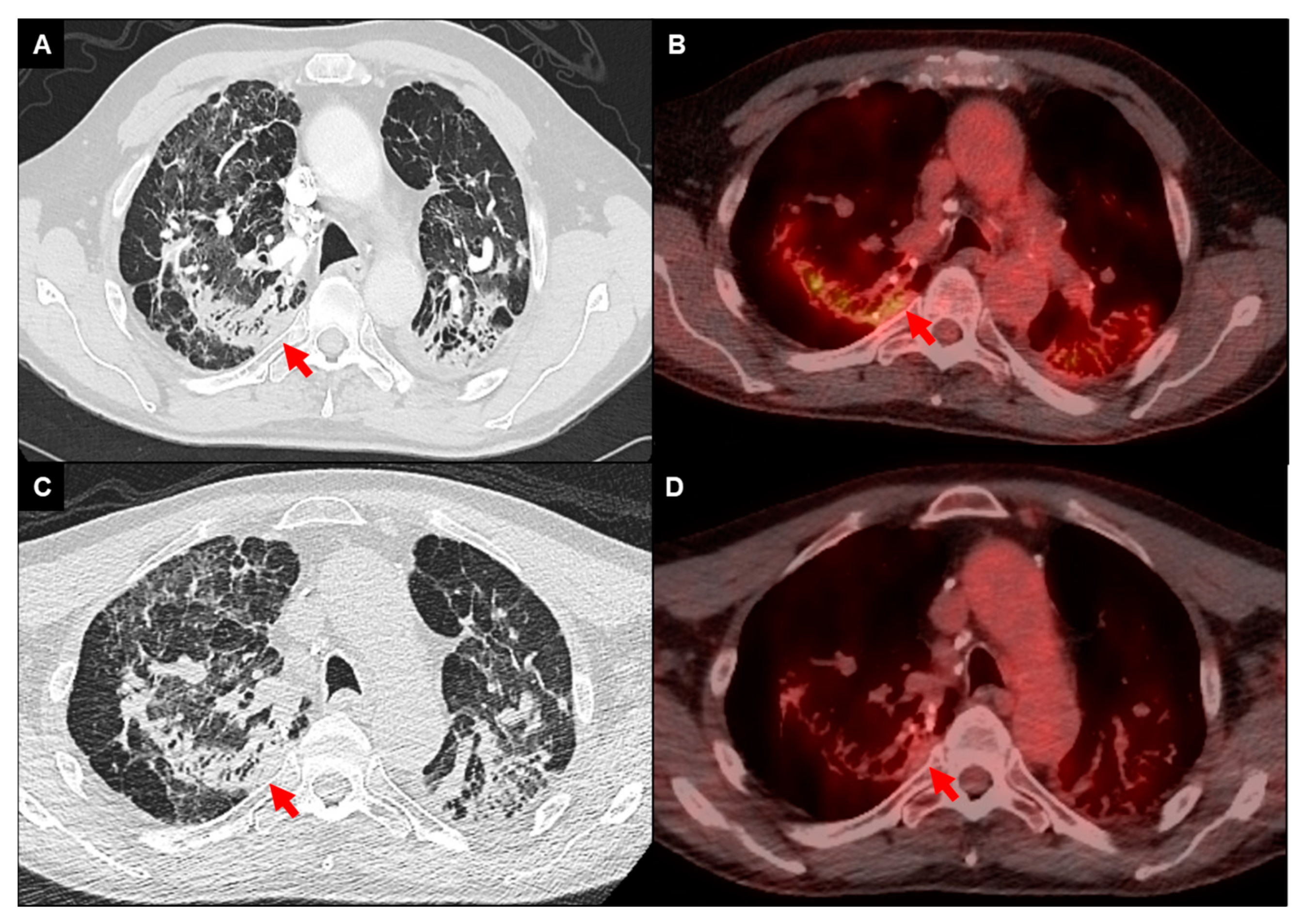
Figure 3.
FDG-PET/CT imaging of a 68-year-old male with fibrotic pulmonary sarcoidosis on chronic methotrexate with metabolically active infiltrates (arrows) in the right upper lobe and left upper lobe extending into the pleura (A,B); reduced density and activity on follow-up imaging 12 months after the addition of anti-TNF-alpha inhibitor (C,D).
Figure 3.
FDG-PET/CT imaging of a 68-year-old male with fibrotic pulmonary sarcoidosis on chronic methotrexate with metabolically active infiltrates (arrows) in the right upper lobe and left upper lobe extending into the pleura (A,B); reduced density and activity on follow-up imaging 12 months after the addition of anti-TNF-alpha inhibitor (C,D).

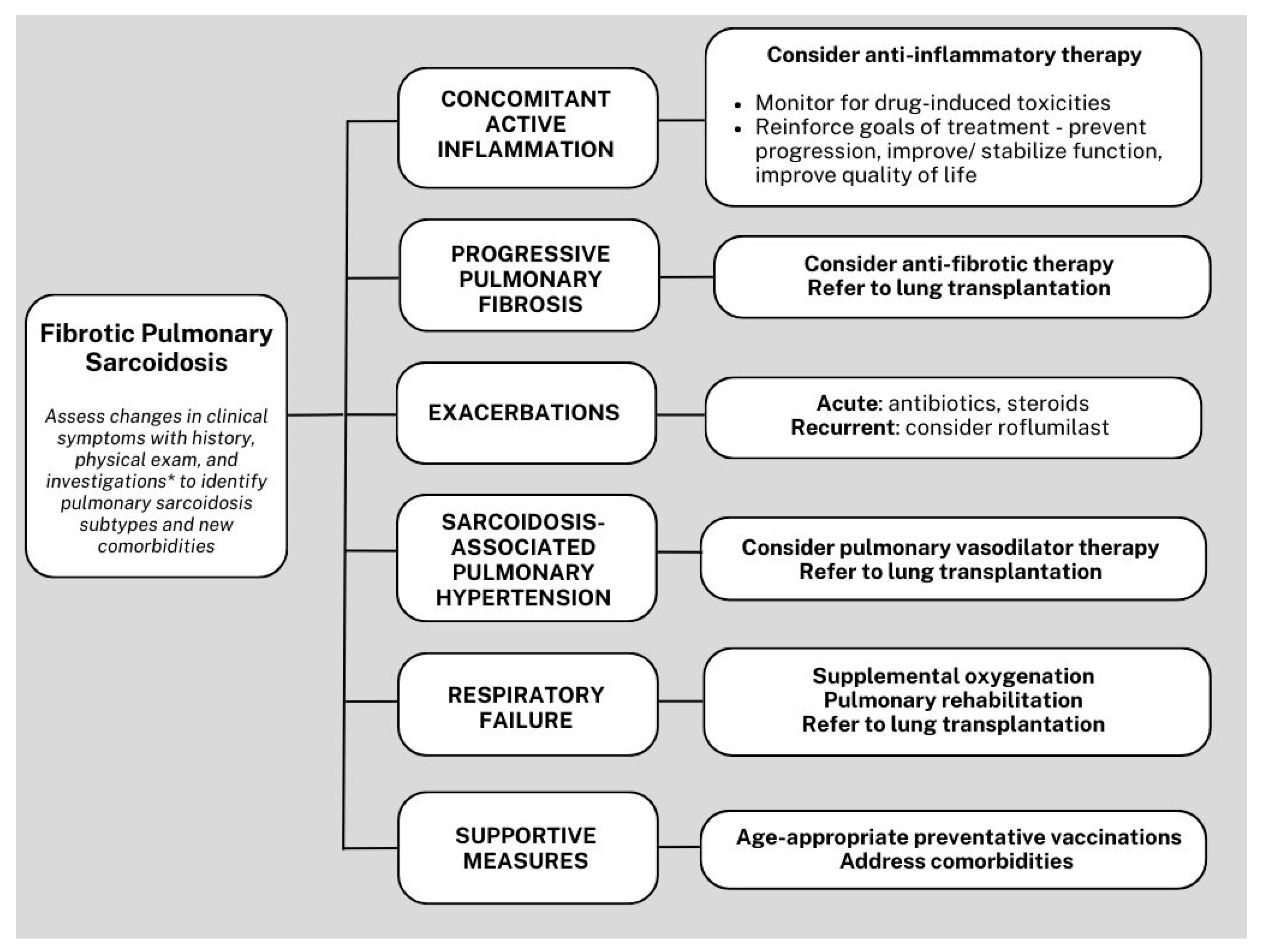
Figure 4.
Simplified algorithm for the management of fibrotic pulmonary sarcoidosis. *Investigations with pulmonary function test, 6-min walk test, high-resolution computed tomography, echocardiogram, and fluorodeoxyglucose positron emission tomography integrated with computed tomography.
Figure 4.
Simplified algorithm for the management of fibrotic pulmonary sarcoidosis. *Investigations with pulmonary function test, 6-min walk test, high-resolution computed tomography, echocardiogram, and fluorodeoxyglucose positron emission tomography integrated with computed tomography.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kim, J.S.; Gupta, R. Clinical Manifestations and Management of Fibrotic Pulmonary Sarcoidosis. J. Clin. Med. 2024, 13, 241. https://doi.org/10.3390/jcm13010241
AMA Style
Kim JS, Gupta R. Clinical Manifestations and Management of Fibrotic Pulmonary Sarcoidosis. Journal of Clinical Medicine. 2024; 13(1):241. https://doi.org/10.3390/jcm13010241
Chicago/Turabian StyleKim, Jin Sun, and Rohit Gupta. 2024. "Clinical Manifestations and Management of Fibrotic Pulmonary Sarcoidosis" Journal of Clinical Medicine 13, no. 1: 241. https://doi.org/10.3390/jcm13010241
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.