
Towards the Clinical Application of Gene Therapy for Genetic Inner Ear Diseases
by
 , ,
, ,
Ghizlene Lahlou
1,2,
Charlotte Calvet
1,3 ,
,
Marie Giorgi
1,
Marie-José Lecomte
1 and
Saaid Safieddine
1,4,* 1
Institut Pasteur/Institut de l’Audition, Technologie et Thérapie Génique de la Surdité, Sorbonne Université, INSERM, Sorbonne Université, 75012 Paris, France
2
Département d’Oto-Rhino-Laryngologie, Unité Fonctionnelle Implants Auditifs, Groupe Hospitalo-Universitaire Pitié-Salpêtrière, APHP Sorbonne Université, 75013 Paris, France
3
Zurich Integrative Rodent Physiology (ZIRP), University of Zurich, CH-8057 Zurich, Switzerland
4
Centre National de la Recherche Scientifique, 75016 Paris, France
*
Author to whom correspondence should be addressed.
J. Clin. Med. 2023, 12(3), 1046; https://doi.org/10.3390/jcm12031046
Submission received: 22 December 2022
/
Revised: 20 January 2023
/
Accepted: 26 January 2023
/
Published: 29 January 2023
(This article belongs to the Special Issue Clinical and Translational Research in Auditory Processing Disorder)
Abstract
:Hearing loss, the most common human sensory defect worldwide, is a major public health problem. About 70% of congenital forms and 25% of adult-onset forms of deafness are of genetic origin. In total, 136 deafness genes have already been identified and there are thought to be several hundred more awaiting identification. However, there is currently no cure for sensorineural deafness. In recent years, translational research studies have shown gene therapy to be effective against inherited inner ear diseases, and the application of this technology to humans is now within reach. We provide here a comprehensive and practical overview of current advances in gene therapy for inherited deafness, with and without an associated vestibular defect. We focus on the different gene therapy approaches, considering their prospects, including the viral vector used, and the delivery route. We also discuss the clinical application of the various strategies, their strengths, weaknesses, and the challenges to be overcome.
1. Introduction
Deafness, with and without associated balance defects, is the most common sensory disorder in humans. It is a major public health issue, affecting people of all ages. According to the World Health Organization, 466 million people worldwide, including 34 million children, have disabling hearing impairment. The cause of hearing loss is genetic in 70% of congenital cases and 25% of adult-onset cases [1,2]. Congenital hearing loss has a negative impact on children’s lives, causing delays in speech and language acquisition, and social and emotional development problems [1,3]. In adults, hearing impairment is associated with social and psychological difficulties and with more severe cognitive decline in the elderly [4,5]. Balance disorders due to vestibular dysfunctions result in gaze instability, which worsens during movement, significantly decreasing participation in physical activities, and having major consequences for the personal and professional lives of those affected [6].
There is currently no specific cure for inner ear disorders. Patients with sensorineural hearing loss benefit from hearing rehabilitation with conventional hearing aids for milder cases, and cochlear implants for the most severe cases. Vestibular disorders are mostly managed by vestibular physiotherapy, which provides central compensation.
Improvements in our understanding of the genetics of hearing impairment over the last four decades have generated a growing interest in gene therapy for inner ear defects among researchers, clinicians, public and private funding organizations, industry, and patients. This keen interest is due to several preclinical studies on animal models of human genetic hearing loss showing promising results for a potential treatment of genetic causes of deafness [7,8,9,10,11,12,13]. These successes have laid the foundations for the future use of gene therapy to treat hearing loss and vestibular disorders of genetic origin. However, many challenges remain in terms of treatment efficacy, administration techniques, and safety.
We discuss here gene therapy approaches for alleviating and/or curing monogenic inherited hearing and balance impairments. We report recent trends in inner gene therapy, including the latest approaches being developed and improvements in therapeutic strategy that may be approved in the near future. We also discuss the additional challenges that must be overcome to ensure the safe and effective transfer of these therapies to humans.
2. Gene Therapy Strategies
The perspectives for gene therapy have broadened with respect to its initial definition: the treatment of a genetic disease by the replacement of a defective gene with a functional copy by gene transfer. Gene therapy is now considered to be the transfer of a nucleic acid, either DNA or RNA, to treat or prevent a disorder through various strategies, including gene replacement (or gene augmentation), gene silencing, and base and prime editing/in situ repair [14,15]. The challenges inherent to all these approaches are similar and include the specific, safe, and efficient delivery of the therapeutic material (DNA, RNA, oligonucleotides, siRNA, or the molecular elements of the CRISPR/Cas9 system) into target cells, mostly with the aid of a modified viral vector (as discussed below), or, more rarely, non-viral vectors, such as liposomes.
Various efforts have been made to treat or prevent hearing loss in mouse models of human deafness forms, with various vectors used to transfer the genetic material into the inner ear (see Table 1). We will focus here on the most recent and promising investigations, some of which have now progressed beyond the preclinical stage, and renewed interest in gene therapy for hearing loss, both in industry and in academic laboratories engaged in translational research.
2.1. Gene Replacement Prevents and Cures Congenital Deafness
The gene replacement strategy involves delivering a corrected copy of the gene responsible for deafness, to supplement the function of a non-functional mutant gene [36]. This is the strategy of choice for treating inherited disorders of the inner ear in which protein function is lost: autosomal recessive diseases (DFNB deafness and syndromic recessive deafness) and autosomal dominant diseases displaying haploinsufficiency (i.e., the product of the single functional allele is insufficient to ensure normal cell function). Various degrees of success have been achieved in preclinical investigations of gene therapy based on gene replacement strategies in mouse models of human deafness. These studies are summarized in Table 1. Outcomes are variable mostly due to the therapeutic time window and/or low transduction rates of target cells. The efficacy of gene therapy for hereditary deafness and balance disorders has been evaluated in murine models at different stages of inner ear maturation, from in utero to after the onset of hearing [12,16], but most studies to date were performed at early neonatal stages [7,10,20,22,26,27,31,32,33]. These studies have raised hopes for clinical trials in patients with hearing and balance disorders in the near future, despite the modest nature of some results obtained.
2.2. RNA Interference Therapy for Hearing Loss
RNA interference (RNAi) is a natural posttranscriptional process for the regulation of gene expression in eukaryote cells [37]. The discovery of RNAi has considerably extended the field of gene therapy to include RNA as targets [38]. RNA-based therapy involves the use of microRNAs, siRNAs, or antisense oligonucleotides (ASOs) designed on the basis of sequence complementarity to interfere with the posttranslational regulation process and silence harmful mRNAs. RNAi can be triggered by synthetic nucleotides and can, therefore, interfere with almost any gene of interest, including those difficult to target selectively with pharmaceutical approaches [39,40].
Almost 80% of autosomal-dominant forms of non-syndromic hearing loss in humans are caused by disruptive point mutations resulting in potential gain-of-function or dominant negative mutations, for which mRNA-based gene-silencing technology is highly suitable. This approach can be used to silence the dominant allele without altering the expression of the wild-type gene.
One proof-of-principle study by Maeda et al. [41] focused on DFNA3 hearing loss, which is caused by dominant mutations of the GJB2 gene [42,43] encoding the transmembrane protein connexin 26 (CX26) expressed by the cochlear supporting cells [44]. Maeda et al. showed that the use of siRNA in vivo in mice selectively suppressed expression of the mutant Gjb2R75W allele, which causes hearing loss through a dominant-negative effect, without significantly affecting expression of the wild-type Gjb2 allele [41]. This treatment prevented the hearing loss otherwise associated with the R75W dominant-negative variant of CX26.
An elegant RNAi approach based on an artificial microRNA (miRNA) was recently shown to rescue the progressive hearing-loss phenotype of the Beethoven (Bth) mouse, a model of the DFNA36 human autosomal-dominant non-syndromic form of deafness. This mouse carries a dominant gain-of-function mutation of Tmc1 (transmembrane channel-like 1) [22]. A specific siRNA sequence for silencing this gene was designed and embedded in an artificial miRNA scaffold for delivery to the cochlea of neonatal or adult Tmc1Bth/+ with an associated adenovirus (AAV) vector. This selectively suppressed the Tmc1 c.1235T > A (p.Met412Lys) dominant gain-of-function allele and prevented the development of profound hearing loss [22] (Table 1). These observations highlight the potential of allele-specific RNAi-based therapeutic approaches to mitigate sensorineural deafness caused by dominant-negative mutations, particularly as this type of auditory deficit is postlingual and progressive, providing a large therapeutic time window for intervention.
2.3. Gene-Editing Therapy for Hearing Loss
The advent of gene-editing technologies based on the clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 endonucleases is revolutionizing gene therapy by making it possible to perform manipulations highly efficiently at any endogenous locus, thereby facilitating gene manipulation in situ. This technology largely outperforms ex vivo homologous recombination and techniques based on TALENS and zinc finger nucleases [45,46]. The CRISPR/Cas9 system was first discovered in bacteria, in which it serves as an adaptive immune system against invasive viral genomes [47]. One of the principal reasons for the success and efficiency of CRISPR/Cas9 is its high specificity for the target sequence, similar to that of the RNAi approach [47,48,49]. Like RNAi approaches, it also uses synthetic RNAs specific to the target sequence (short-guide RNAs), which are delivered together with a Cas9 enzyme to mediate the creation of specific double-strand breaks in the target DNA sequence. The DNA repair pathway operates by frameshift and/or stop codon mutations [50]. Gene editing with CRISPR/Cas9 provides an interesting approach complementary to RNAi for the treatment of autosomal-dominant hearing loss due to disruptive point mutations. Gene editing was recently shown to prevent hearing loss in the Beethoven mouse model of DFNA36 [9,23] (Table 1), as previously reported for RNAi. A single injection of Cas9 plus the guide RNA mixture selectively suppressed the dominant gain-of-function mutation in Beethoven mice and prevented progressive deafness in the newborn mutant mice. Results for the correction of point mutations by CRISPR/Cas gene editing are promising, but the efficiency of homologous direct repair remains low and restricted to mitotic cells [51], and is generally surpassed by non-homologous end joining junction (NHEJ), which frequently leads to unwanted indel outcomes [52,53]. However, NHEJ occasionally leads to indels that coincide with the desired editing outcomes. This was the case for an in vitro frameshift repair using a strategy based on the gRNA-Cas9-induced precise cleavage and the NHEJ-mediated highly biased editing without a template [54,55,56]. Remarkably, using this strategy in vivo, Liu et al. were able to restore hearing and balance function in a mouse model of human deafness DFNB23, which is due to a spontaneous single nucleotide insertion causing a frameshift in the 7.9 kb Pcdh15 transcript [35]. The NHEJ-mediated frame restoration strategy is attractive for developing treatments for frameshift mutations. However, the frequency of indel byproducts remains high when considering a clinical application scenario. Further improvements are needed, including Cas9 and RNA guide optimization, delivery to the inner ear target cells, and precisely controlled indels. Several studies have shown that point mutations can be corrected, but they were often associated with aberrant editing within the target site itself, i.e., in which CRISPR-Cas is designed to initiate the repair process [57,58,59].
To overcome the major challenges and limitations of the CRISPR/Cas use for the treatment of human diseases, extensive CRISPR/Cas protein complex engineering led to the development of “base editing” and “prime editing” strategies [15]. The base editing approach allows the precise conversion of a single base into another in a genomic DNA with relatively high efficiencies and without generating double-strand breaks or exogenous donor DNA templates, minimizing the unwanted indels [60,61,62]. Remarkably, optimized cytosine base editors virally delivered in vivo to the inner ear corrected the Tmc1 C.A545G, a deafness point mutation, to wild-type sequence (c.A545A), leading to restored sensory transduction and improved auditory function in the treated mouse [21]. This discovery provides strong evidence in favor of the development of a base editing strategy to correct virtually all kinds of deafness mutation.
The prime editing strategy consists in directing the prime editing molecular machinery to the genomic region to be corrected and replacing it with the desired sequence. The prime editing molecular machinery involves a unique guide RNA, known as the prime editing gRNA (pegRNA), which guides a catalytically impaired Cas9 endonuclease fused to a reverse transcriptase enzyme (RT) [15]. Once the endonuclease notches the polynucleotide strand to be replaced, the RT generates in situ the correct complementary DNA using the pegRNA as a template. The proof of concept for the in vivo effectiveness of prime editing has recently been provided in a mouse model of type I tyrosinemia diseases [63,64]. To date, there are no in vivo data available concerning the field of genetic deafness; however, the prime editing is in remarkable and constant optimization, including precision, efficiency, specificity, and safety, which leaves the option of an eventual clinical application being conceivable for genetic deafness.
3. Viral Vectors
3.1. Adenovirus and Lentivirus
Despite the safety issues associated with adenoviruses (AdVs), such as toxicity and immune responses, these vectors are being tested in a large number (~20%) of clinical trials currently underway around the world [65,66]. The first phase I clinical trial for the treatment of deafness by gene therapy methods used AdVs (NCT02132130). However, a functional defect of the outer hair cells (OHC) was reported upon AdV delivery to the cochlea [67,68,69], making the use of these viruses as vectors for gene transfer to the inner ear challenging. However, recent advances in adenovirus engineering have significantly decreased their immunogenicity, extended transgene expression, and improved safety [70], which remains to be demonstrated for the inner ear.
Lentiviruses (LVs), which belong to a subclass of Retroviridae, have emerged as another possible vehicle for gene delivery applications. One of the principal obstacles to their use for in vivo gene therapy is the risk of insertional mutagenesis, which could disrupt gene function in the transduced cells [71]. Although several safer integrase-defective lentiviral vectors (IDLVs) have since been engineered [72,73], and the third generation of these vectors is now widely used [71]. Interestingly, LV vectors have been shown to deliver Myo7A cDNA efficiently and to mediate the correction of several abnormal retinal phenotypes in mouse models of Usher syndrome type 1B. These results represent a major step forward in the development of LV-mediated gene therapy for the treatment of Usher 1B blindness [74,75]. It remains unclear whether lentivirus-mediated gene therapy would be able to rescue the inner ear defect of Usher syndrome type 1B. Proofs-of-concept for the consistent transduction of inner ear tissues with LVs are currently lacking [76]. Attempts are currently being made to improve the specificity of LVs and their targeting of various cell types [77]. LV variants efficiently targeting the inner ear sensory cell will probably follow, together with the successful establishment of gene therapy to treat deafness with LV technology.
3.2. Adeno-Associated Virus
AAV, a single-stranded DNA virus, is one of the smallest known viruses and is among the potential gene therapy vehicles most actively investigated for therapeutic strategies [78,79]. AAV-based gene therapies have been used clinically in a number of situations, with remarkable therapeutic benefits and an excellent safety record [79]. These vectors are frequently chosen for gene therapy because they are highly stable, non-pathogenic, and capable of infecting diverse cells without undergoing site-specific integration into the chromosomes of the target cell [80,81]. To date, 13 naturally occurring AAV serotypes and over 108 AAV variants have been identified or engineered, and this number will undoubtedly continue to grow, as will the number of cell types that can be targeted [82]. Unsurprisingly, AAVs have emerged as the vector of choice for inner ear gene delivery in vivo. Indeed, it is becoming increasingly clear that the considerable diversity of the cells in the inner ear potentially affected during hearing loss will require the use of more than one vector.
Transduction rates and the cell types transduced in the inner ear can be influenced by several factors, including the AAV capsid/promoter combination, virus preparation and purification methods, the route and technique of administration, and the dose administered [81,83,84,85,86]. Furthermore, AAV cell tropism and transduction rate can vary between cell types, animal species, and inner ear development stages [84]. Many studies have investigated the tropism of rAAV in inner ear cells (mainly cochlear cells) in vivo in mice, and, more recently, in non-human primates (NHP). The results obtained in mice are summarized in Table 2.
A few recent studies have focused on the transduction rates and profiles obtained in NHP. Two modified AAV9 vectors (AAV9.PHP.B and AAV-S) robustly transduced both hair cells and supporting cells towards the cochlea after injection through the round window membrane (RWM) [33,87,88]. Following injection of the Anc80L65 vector through the RWM with oval window fenestration, Andres-Mateos et al. reported transduction rates of up to 90% for inner hair cells (IHCs), with a more variable percentage of the cells transduced in vestibular organs, in which transduction rates were much lower with AAV1 (up to 30% of IHCs in the apical region) [89]. Interestingly, a recent study found a transduction of IHC and spiral ganglion cells after cerebro-spinal fluid delivery of numerous AAV serotypes variants (AAV1, AAV2, and AAV9) to a Chorocebus aethiops (African green) monkey [90]. The common finding that emerges from these cell tropism studies is that most mature OHCs are refractory to transduction by existing AAV serotypes. It will therefore be essential to identify or design serotypes capable of targeting these cells to ensure the complete restoration of hearing.

{kind=link}
Table 2.
Results of viral transduction after the injection into the inner ear of several AAV serotypes driving the expression of eGFP under the control of a constitutive promoter.
Table 2.
Results of viral transduction after the injection into the inner ear of several AAV serotypes driving the expression of eGFP under the control of a constitutive promoter.
| Capsid | Injection Stage | Injection Route | Inner Ear Hair Cell Transduction (%) | Transduction of Other Inner Ear Cells | References | ||
|---|---|---|---|---|---|---|---|
| IHC | OHC | VHC | |||||
| AAV1 | Neonatal | RW | 0–67 | 0–14 | 0 | Inner phalangeal cells and Deiters’ cells | Askew et al., 2015 [19]; György et al., 2017 [24]; Landegger et al., 2017 [91]; Pan et al., 2017 [27]; Emptoz et al., 2017 [7] |
| CO | 36 | 17 | NR | Marginal cells | Chang et al., 2015 [26]; György et al., 2017 [24] | ||
| Mature | RW with PSCC fenestration | 10 | <5 | <10 | Stria vascularis cells | Omichi et al., 2020 [92] | |
| PSCC | 6 | 0 | NR | NR | Tao et al., 2018 [93] | ||
| AAV2 | Neonatal | RW | 0–78 | 0–50 | NR | NR | Emptoz et al., 2017 [7]; Askew et al., 2015 [19]; Landegger et al., 2017 [91]; Geng et al., 2017 [94] |
| PSCC | 44 | 54 | NR | Pillar cells | Isgrig et al., 2019 [95] | ||
| Mature | RW with PSCC fenestration | 95 | 80 | 0 | 0 | Omichi et al., 2020 [92] | |
| PSCC | 85 | 10 | 7 | NR | Tao et al., 2018 [93] | ||
| AAV5 | Neonatal | RW | 0 | 0 | 0 | Supporting and mesothelial cells | Emptoz et al., 2017 [7] |
| CO | 0 | 0 | 0 | Supporting, mesothelial, and Reissner’s membrane cells | Iizuka et al., 2015 [96] | ||
| AAV6 | Neonatal | RW | 15–20 | 5–10 | NR | NR | Askew et al., 2015 [19]; Landegger et al., 2017 [91] |
| Mature | PSCC | 5 | 0 | NR | NR | Tao et al., 2018 [93] | |
| AAV8 | Neonatal | RW | 10–90 | 5–28 | 90 | Spiral ganglion neurons | Askew et al., 2015 [19]; Chien et al., 2015 [97]; Emptoz et al., 2017 [7]; Landegger et al., 2017 [91]; Geng et al., 2017 [94]; Dulon et al., 2018 [32]; Xia et al., 2012 [98] |
| PSCC | 49–86 | 13–52 | 53 | Marginal, vestibular supporting, and pillar cells | Isgrig et al., 2019 [95]; Guo et al., 2017 [99] | ||
| Mature | RW with PSCC fenestration | 90 | <10 | 35 | Stria vascularis cells and spiral ganglion neurons | Omichi et al., 2020 [92] | |
| PSCC | 75 | 0 | 41 | NR | Tao et al., 2018 [93] | ||
| AAV9 | Neonatal | RW | 5 | 5 | NR | NR | Askew et al., 2015 [19] |
| CO | 57 | 15 | 12 | NR | Gu et al., 2019 [100] | ||
| Mature | RW + PSCC | 95 | <5 | good | NR | Yoshimura et al., 2018 [101] | |
| RW | 30 | 0 | 0 | NR | Yoshimura et al., 2018 [101] | ||
| RW with PSCC fenestration | 100 | 0 | 20 | Stria vascularis cells, and spiral ganglion neurons | Omichi et al., 2020 [92] | ||
| PSCC | 60 | 0 | 20 | NR | Tao et al., 2018 [93] | ||
| Anc80L65 | Neonatal | RW | 90–100 | 80–95 | 95 | Pillar and Deiters’ cells | Pan et al., 2017 [27]; Landegger et al., 2017 [91]; Lee et al., 2020 [86] |
| CO | 100 | 90 | NR | Supporting cells | Gu et al., 2019 [100] | ||
| PSCC | 94 | 67 | NR | Pillar cells | Isgrig et al., 2019 [95] | ||
| Utricle | 100 | 30–90 | robust | Pillar and Deiters’ cells | Lee et al., 2020 [86] | ||
| Mature | PSCC post | 95–100 | 40–50 | 40 | NR | Suzuki et al., 2017 [102]; Tao et al., 2018 [93] | |
| RW + PSCC | 90 | - | good | NR | Yoshimura et al., 2018 [101] | ||
| RW with PSCC fenestration | 100 | 50 | 35 | Stria vascularis cells, and spiral ganglion neurons | Omichi et al., 2020 [92] | ||
| Utricle | 100 | 0–20 | moderate | NR | Lee et al., 2020 [86] | ||
| AAV2 quadY-F | Mature | RW | 85 | NR | NR | NR | Akil et al., 2019 [13] |
| AAV2.7m8 | Neonatal | PSCC | 84 | 83 | NR | Pillar and internal phalangeal cells | Isgrig et al., 2019 [95] |
| Utricle | 40–100 | 40 | NR | NR | Lee et al., 2020 [86] | ||
| Mature | RW | 84 | 75 | NR | NR | Isgrig et al., 2019 [95] | |
| AAV8BP2 | Neonatal | PSCC | 56 | 44 | NR | NR | Isgrig et al., 2019 [95] |
| AAV9-PHP.B | Neonatal | RW | 70–100 | 35–70 | NR | NR | György et al., 2019 [33]; Lee et al., 2020 [86] |
| Utricle | 100 | 100 | robust | NR | Lee et al., 2020 [86] | ||
| Mature | PSCC | 100 | 0 | robust | NR | György et al., 2019 [33] | |
| Utricle | 100 | 20–80 | robust | NR | Lee et al., 2020 [86] | ||
| AAVrh.39 | Mature | PSCC | 55 | 0 | NR | NR | Tao et al., 2018 [93] |
| AAVrh.43 | Mature | PSCC | 95 | 0 | NR | NR | Tao et al., 2018 [93] |
| AAV-S | Neonatal | RW | 100 | 50–75 | robust | Interdental, inner and outer sulcus, Claudius cells, and spiral ganglion neurons | Ivanchenko et al., 2021 [88] |
| AAV-ie | Neonatal | RW | 100 | 60–100 | 100% | All cell types of supporting cells | Tan et al., 2019 [103] |
CO = cochleostomy; IHC = inner hair cells; NR = not reported; OHC = outer hair cells; PSCC = posterior semicircular canal; RW = round window; VHCs = vestibular hair cells.
One of the drawbacks of AAVs, limiting their utility for gene therapy for deafness, is their limited packaging capacity of 4.7 kb, below the size of most of the identified deafness genes. Several research groups have attempted to overcome this limitation by adopting an approach to double AAV packaging capacity by splitting a large transgene into two fragments, each packaged in a different AAV vector. The full-length expression cassette is reconstituted upon co-infection of the same cell with the two AAV vectors containing the fragments [104,105,106,107]. Such dual AAV gene therapy was recently adapted for use with the otoferlin gene, defects of which underlie DFNB9, one of the most frequent prelingual forms of non-syndromic human deafness [108,109,110,111]. The otoferlin cDNA, split between two AAVs delivered together to the cochlea of DFNB9 mice, restored hearing in these otoferlin-null mice [13].
4. Routes for Inner Ear Gene Delivery
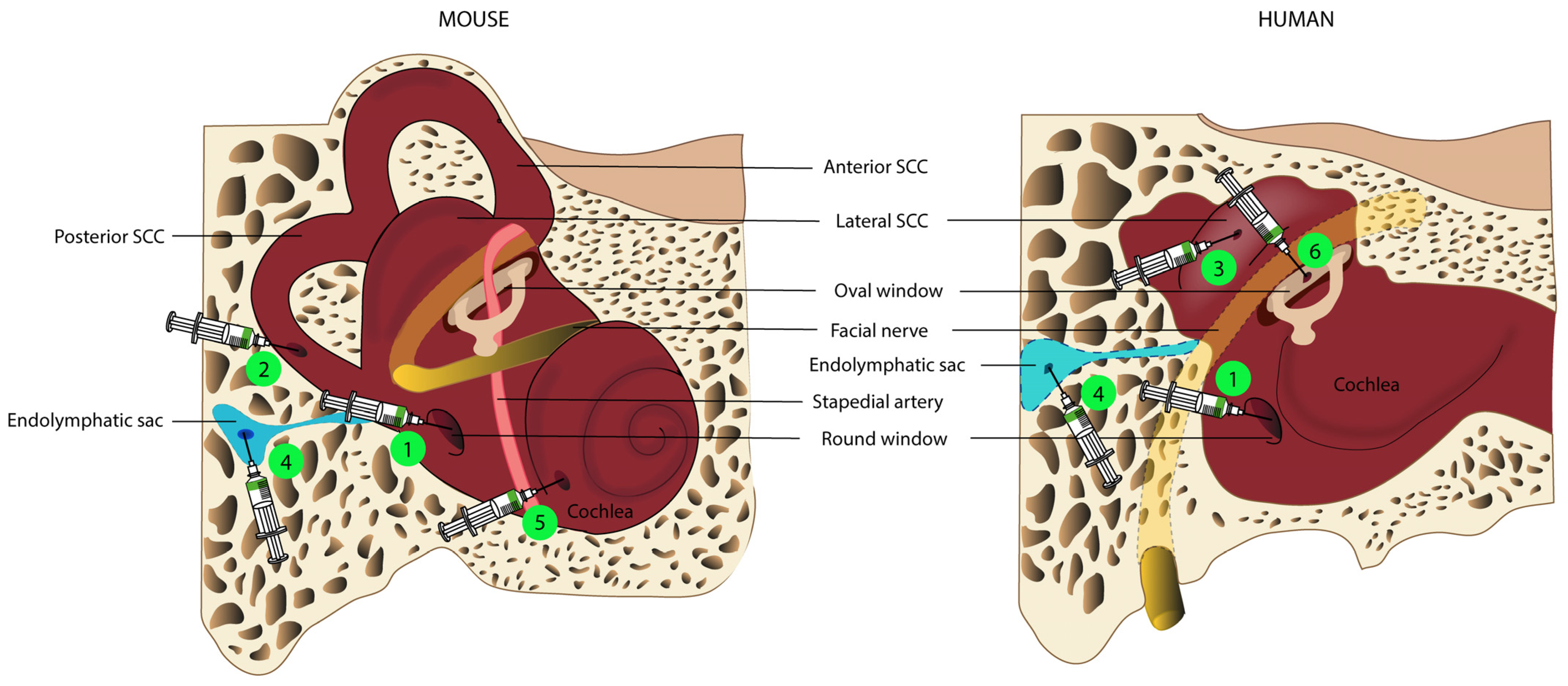
One of the major challenges for gene delivery to the inner ear is the anatomic, physiological, and cellular barriers present in this structure. The inner ear is a small but complex organ, with a highly regulated environment, encased in the temporal bone. The membranous labyrinth, filled with endolymph and located within the otic capsule, holds the cochlear and vestibular sensory organs: the two main targets of gene therapy for inner ear disorders. The space between the membranous labyrinth and the otic capsule is filled with perilymphatic fluid, which has a composition similar to that of cerebrospinal fluid. Additional obstacles are blood–perilymph and blood–strial barriers, a network of vascular endothelial cells equipped with tight junctions that greatly limits access to the inner ear following systemic administration [112]. One recent study showed that inner ear cells were transduced following the intravenous injection of the AAV2/9 viral vector only if high doses were administered via the superficial temporal vein at a neonatal stage [113]. Local routes of delivery within the inner ear, to the perilymphatic or endolymphatic space, appear to be necessary for inner ear gene therapy. Surgical delivery to the right place, without inflicting significant damage on the inner ear, is, therefore, a major challenge.
4.1. Delivery to the Endolymphatic Space: Cochleostomy, Endolymphatic Sac, and Utricle Administration
Delivery into the endolymphatic space, and through a cochleostomy into the scala media, has been tested in preclinical studies [30,114]. Nevertheless, this route is technically difficult and it can lead to a permanent increase in hearing thresholds in adult mice. [97], especially at high frequencies [114]. Another way to access the endolymphatic space is an injection into the endolymphatic sac, a closed sac located on the intracranial side of the petrous bone (Figure 1) and connected to the endolymphatic space via the vestibular aqueduct [115]. Few studies have investigated this route of administration [116,117,118], of which one carried out in a guinea pig showed that the injection of a recombinant AdV into the endolymphatic sac led to significant levels of transduction in the cochlear and vestibular sensory organs [116]. It may be possible to deliver therapeutic agents via the endolymphatic sac in humans, as this surgical approach is already well-established for the treatment of Ménière’s disease [119]. However, precise evaluations of the volume of therapeutic agent to be delivered, and of the pressure and administration rate, would be required to prevent endolymphatic hydrops, which could result in permanent damage to the cochlea or vestibule.
Lee et al. recently described injection into the utricle, to target the endolymphatic space, in neonatal mice [86]. Injections via this route, which is easily accessible in neonatal mice, led to a high rate of transduction in IHCs (almost 100%) and other inner ear cell types, with no damage to auditory or vestibular functions. However, the authors of this study stressed that it was difficult to ensure that the gene therapy agent was injected into the endolymph and not into the perilymphatic space. Furthermore, the endolymphatic utricular space is less easily accessible in humans, as it is almost completely covered by the facial nerve. (Figure 1).
4.2. Delivery to the Perilymphatic Space: Round Window, Posterior Semi-Circular Canal, and Oval Window Administration
Delivery to the perilymphatic space is the most widely used technique in preclinical studies, resulting in viral transduction rates and profiles similar to those achieved following delivery to the scala media [97]. The perilymphatic fluid can be assessed directly via two injection sites.
Injection through the RWM, which is readily accessible in both newborn and adult rodents after perforation of the tympanic bulla, results in the robust transduction of both vestibular and cochlear sensory hair cells [7,13,33,97,98]. Surgical access is more difficult in NHPs, requiring either mastoidectomy with posterior tympanotomy, a technique routinely practiced by ENT specialists during cochlear implantation [87], or trans-canal hypotympanotomy. The injection of a phosphate-buffered saline vehicle through the RWM of NHPs does not alter hearing or vestibular function [120], whereas modifications to these functions have been reported after the injection of an AAV recombinant vector via this route [33]. These findings probably reflect perilymph leakage after RWM puncture and changes in intracochlear pressure during the injection.
As a means of limiting the risk of leakage and inner ear defects, attempts have been made to administer the therapeutic gene, not by injection, but by diffusion through an intact RWM [121], potentially with the assistance of partial enzymatic digestion [98]. Although this approach results in only limited transduction of IHCs [98], and failed to restore hearing and balance in a mouse model of Usher syndrome type 1C (USH1C) [11]. Zhang et al. recently showed that ultrasound-microbubble cavitation facilitates viral transduction across an intact RWM [122]. Efforts are now being made to improve this technique, to facilitate the use of ultrasound-microbubble cavitation as an approach like any other, but with greater efficacy and fewer adverse effects, thereby optimizing the outcome of gene therapy. Furthermore, this technique would be easy to use in humans, with the possibility of administration via the external auditory canal under general or local anesthesia, for children and adults, respectively.
Another route of administration commonly used in preclinical studies is canalostomy, mostly through the posterior semicircular canal, in neonatal and adult mice [33,93,95,102]. This approach involves opening the bony canal and injecting the gene therapy agent into the perilymphatic space without damaging the membranous labyrinth. The semicircular canal is very small, rendering accurate injection difficult, and making it impossible to determine, with certainty, which compartment—perilymphatic or endolymphatic—actually received the therapeutic product. It would also be difficult to transpose the exact method to humans, due to the anatomical position of the posterior semicircular canal. Injection through the lateral semi-circular canal might be feasible in the human inner ear, provided this method had already been demonstrated to be safe in large-animal models.
By combining two techniques (RWM injection associated with canal fenestration to provide an exit hole), Yoshimura et al. greatly improved viral gene delivery to the mouse inner ear and the rates of transduction of both cochlear and vestibular hair cells, without affecting auditory function. This improvement resulted from the use of canal fenestration to create an exit path, facilitating a longitudinal flow of the injected viral preparation throughout the inner ear [101]. Interestingly, a similar delivery approach was also found to enhance virus-mediated gene delivery and inner ear hair cell transduction rates relative to RWM injection alone in NHPs. Longitudinal flow can easily be reproduced in humans, by injection through the RWM with the creation of an oval window opening, thereby significantly increasing transduction rates for inner ear hair cells, as already demonstrated in NHPs [89]. Nevertheless, it remains unclear whether this double opening of the cochlea and the resulting longitudinal flow pattern preserve vestibular and auditory function in NHPs [89].
Finally, another route of access to the perilymphatic compartment routinely used in otologic surgery is the opening of the oval window by platinotomy. This route is not used in preclinical studies in rodents because the oval window is covered by the stapedial artery in these animals, but this artery is not present in primates [123]. Only one phase I/II clinical trial for hearing loss to date has used this route of administration to deliver the viral vector, and the results of this study have yet to be reported (NCT02132130).
The perilymphatic space is likely to be the main target for clinical applications, but fluid may be exchanged between the cerebrospinal fluid and the perilymph across the cochlear aqueduct, in which case the therapeutic agent may diffuse outside the inner ear [115]. Improvements in our understanding and a clear identification of the routes of diffusion (in and out of the inner ear) of the therapeutic agent will undoubtedly help to optimize the targeting of particular cells and compartments.
4.3. Volume Injected and Its Flow Rate
The volume of the inner ear fluids is correlated with body mass index: the perilymph has a volume of about 0.62 µL in mice, 8.66 µL in guinea pigs [124], 26.7 µL in Macaca nigra [125], and 51 µL in humans [124].
In small mammals, relatively large volumes (1 to 2 µL), exceeding that of the perilymphatic compartment, are routinely locally injected in preclinical studies without generating hearing or vestibular impairment, probably because the overflow towards the CSF via the cochlear aqueduct remains open [126] preventing changes in intracochlear pressure during the injection. In macaques, there is no evidence of damage to the inner ear after administration of up to 30 µL, corresponding to more than 100% of the perilymph volume [120], suggesting clearance through the cochlear aqueduct or a leak around the injection point. NHPs, particularly subspecies from the Old-World group of monkeys, such as Macaca (Macaca fuscata, nigra, rhesus, or fascicularis), have a number of features in common with humans, including inner ear architecture and function [127], potentially allowing future extrapolations in terms of the pharmacokinetics and safety of inner ear injection [128].
5. Unresolved Issues
5.1. The Temporal Window for Therapeutic Intervention
Mouse models for human deafness have been widely and successfully used to investigate various aspects of auditory gene therapy, including delivery routes, vector specificity and efficiency, and spatiotemporally controlled gene expression. A number of preclinical investigations have been performed, which have established proof of concept for the feasibility and efficacy of gene therapy for the treatment of deafness (Table 1). However, there is still an absence of clinical trials with encouraging outcomes because the murine models for human deafness used are not ideal for predicting outcomes in patients. Indeed, mice are born deaf and do not begin to hear until the 12th postnatal day (P12); the cochlea is, thus, immature and continues to develop after birth [129]. By contrast, humans begin to hear after about four months in utero [130]. Most of the translational gene therapy studies performed in mouse models to date involved interventions performed from P1 to P9 (Table 1). The corresponding therapeutic window in humans would fall between 18 and 25 weeks of gestation, a period during which the risks of intervention are much higher, with potential safety issues relating to fetal intervention, delivery of the transgene, infection, premature delivery, and even the potential loss of the fetus. These multiple risks are the principal reason for the lack of planned trials in humans in the near future [131]. However, it should be borne in mind that patients with the most common causes of deafness potentially treatable by gene therapy are usually diagnosed during the neonatal period. The critical question is whether gene therapy shortly after birth can be effective in these patients. For any clinical application of gene therapy for deafness in humans to be considered in the near future, it will, therefore, be essential to identify the optimal therapeutic time window during which gene therapy can reverse existing deafness or prevent the progression of hearing impairment regardless of the stage already reached. A major step forward for gene therapy for deafness was taken with the study by Akil et al., 2019 [13], which addressed two of the most serious issues faced in inner ear gene therapy: the limited DNA packaging capacity of adeno-associated virus (AAV) vectors (about 5 kb, below the size of most known deafness genes), and the lack of evidence for gene therapy being able to reverse an existing deafness phenotype (cure as opposed to prevention). The authors focused on DFNB9 (MIM601071), which accounts for 2–8% of cases of prelingual hearing impairment [108,109] and is caused by biallelic mutations of the OTOF gene, encoding otoferlin [110,132]. Akil et al. adopted a dual AAV-vector strategy to transfer the Otof cDNA (~6 kb) with two different recombinant vectors, one containing the 5′ and the other the 3′ portion of the otoferlin cDNA. The authors demonstrated that local gene therapy in mutant mice not only prevents deafness when administered to immature hearing organs, but also durably restores hearing when administered at a mature stage, after hearing onset, which occurs several days after birth in mice. This finding, considered a major breakthrough, raises hopes for the possibility of future gene therapy trials in DFNB9 patients. In fact, several pharmaceutical companies are currently developing gene therapy products to treat DFNB9 patients.
5.2. Does the Inner Ear Have Immune Privilege?
The immune system is known to be an important determinant of gene therapy outcomes. Improvements in the design of the viral vectors most widely used in current gene therapy studies, including AdV, LV, and AAV vectors, have clearly decreased the risk of insertional mutagenesis. Nevertheless, the possibility of deleterious effects of in vivo gene therapy on the target cells or organs, including inflammation and/or immunotoxicity, remains a significant issue requiring careful consideration. There is currently insufficient evidence to support the assertion that the immune privilege of the inner ear is complete.
The eye, which is at the forefront of the field of gene therapy, has long been considered immune-privileged, but recent data have shown that viral gene transfer to the eye can trigger an adaptive immune response in both NHPs and patients [133,134,135]. A similar immunoreaction to the therapeutic vector cannot be excluded in the inner ear. The auditory hair cells are postmitotic at birth and are not subsequently regenerated. Comprehensive investigations are therefore required, to assess the risks associated with a possible immune response to inner ear gene therapy, such as damage to the treated tissue, for example, to maximize the safety and efficacy of the approach. These investigations will include evaluations of humoral and cellular immune responses, and studies of possible histological changes reflecting local immune responses in tissues of interest. Studies of these immune responses to different routes of cochlear administration will need to be carried out in both NHP and murine models.
Another issue concerns the high prevalence of neutralizing anti-AAV antibodies in humans. Seropositivity rates range from about 72% for AAV2 and 67% for AAV1 to about 47% for AAV9, 46% for AAV6, 40% for AAV5, and 30% for AAV8 [136,137]. This is a potentially serious problem, as these antibodies may completely prevent the transduction of a target tissue, rendering the treatment ineffective [138,139,140]. It will be essential to develop strategies for circumventing immune responses or preventing adaptive responses to capsid- or transgene-derived antigens through transient immunosuppression or immunomodulation, to ensure the long-term expression of the therapeutic gene.
6. Conclusions
Gene therapy has proved effective for preventing or treating several genetic causes of hearing impairment and/or vestibular defects in mouse models and may have potential as a curative treatment in humans. Various strategies could be adopted for the design of personalized gene therapy for patients, depending mainly on the time course of the pathophysiology of hearing loss and the presence or absence of an associated balance disorder. However, several factors must be considered before any application in humans. First, an evaluation of the therapy in mouse models, during a time window transposable to humans, is essential, because the chronological maturation of the inner ear differs between mice and humans. Second, an optimization of the viral vector, including capsid, promoter, and transgene engineering, will be required to improve transduction rates for the target inner ear cells in humans. This will involve an evaluation of inner ear cell tropism in large-animal models (especially NHPs), and in human inner ear cells. The safety of surgery, the gene delivery vector, and the therapeutic transgene will also need to be evaluated. Approaches based on injection through the RW and OW are commonly practiced by otological surgeons, but their safety must be evaluated for AAV vector-mediated cochlear gene therapy, to ensure the preservation of inner ear structure and function. Establishment of the appropriate volume of therapeutic agent and its rate of delivery is also a concern, given the small volume of the fluids present in the inner ear. Underdosage may limit AAV transduction efficiency and overdosage may result in avoidable toxicity. It is therefore essential to determine optimal dose windows for AAVs. Biodistribution studies will make it possible to evaluate the potential risk of adverse effects in cases of off-target transgene expression. Finally, the possible occurrence of an immune response to gene therapy must be considered before inner ear treatments are applied in humans. As for injections into the brain and eye, we expect the immune response to be weaker than that following systemic delivery, but preventive treatments may nevertheless be required when gene therapy agents are administered to the inner ear. The use of gene therapy to treat genetic causes of hearing impairment may become possible in the near future, paving the way for cures for several genetic causes of hearing loss.
Author Contributions
Literature search, conceptualization, and manuscript drafting G.L., C.C. and S.S.; Critical revision of the manuscript M.G. and M.-J.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Fondation pour l’Audition (FPA IDA08 to S.S.), RHU AUDINNOVE (ANR-18-RHUS-0007 to S.S.), and the Scholarship CRS Amplifon 2018.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Smith, R.J.H.; Bale, J.F.; White, K.R. Sensorineural Hearing Loss in Children. Lancet 2005, 365, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Haile, L.M.; Kamenov, K.; Briant, P.S.; Orji, A.U.; Steinmetz, J.D.; Abdoli, A.; Abdollahi, M.; Abu-Gharbieh, E.; Afshin, A.; Ahmed, H.; et al. Hearing Loss Prevalence and Years Lived with Disability, 1990–2019: Findings from the Global Burden of Disease Study 2019. Lancet 2021, 397, 996–1009. [Google Scholar] [CrossRef]
- François, M.; Boukhris, M.; Noel-Petroff, N. Schooling of Hearing-Impaired Children and Benefit of Early Diagnosis. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2015, 132, 251–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, F.R.; Yaffe, K.; Xia, J.; Xue, Q.L.; Harris, T.B.; Purchase-Helzner, E.; Satterfield, S.; Ayonayon, H.N.; Ferrucci, L.; Simonsick, E.M. Hearing Loss and Cognitive Decline in Older Adults. JAMA Intern. Med. 2013, 173, 293–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, L.L.; Tucci, D.L. Hearing Loss in Adults. N. Engl. J. Med. 2017, 377, 2465–2473. [Google Scholar] [CrossRef]
- Guinand, N.; Boselie, F.; Guyot, J.; Kingma, H. Quality of Life of Patients with Bilateral Vestibulopathy. Ann. Otol. Rhinol. Laryngol. 2012, 121, 471–477. [Google Scholar] [CrossRef]
- Emptoz, A.; Michel, V.; Lelli, A.; Akil, O.; de Monvel, J.B.; Lahlou, G.; Meyer, A.; Dupont, T.; Nouaille, S.; Ey, E.; et al. Local Gene Therapy Durably Restores Vestibular Function in a Mouse Model of Usher Syndrome Type 1G. Proc. Natl. Acad. Sci. USA 2017, 114, 9695–9700. [Google Scholar] [CrossRef] [Green Version]
- Akil, O.; Seal, R.P.; Burke, K.; Wang, C.; Alemi, A.; During, M.; Edwards, R.H.; Lustig, L.R. Restoration of Hearing in the VGLUT3 Knockout Mouse Using Virally-Mediated Gene Therapy. Neuron 2012, 75, 283–293. [Google Scholar] [CrossRef] [Green Version]
- György, B.; Nist-Lund, C.; Pan, B.; Asai, Y.; Karavitaki, K.D.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Solanes, P.; Spataro, S.; et al. Allele-Specific Gene Editing Prevents Deafness in a Model of Dominant Progressive Hearing Loss. Nat. Med. 2019, 25, 1123–1130. [Google Scholar] [CrossRef]
- Delmaghani, S.; Defourny, J.; Aghaie, A.; Beurg, M.; Dulon, D.; Thelen, N.; Perfettini, I.; Zelles, T.; Aller, M.; Meyer, A.; et al. Hypervulnerability to Sound Exposure through Impaired Adaptive Proliferation of Peroxisomes. Cell 2015, 163, 894–906. [Google Scholar] [CrossRef]
- Lentz, J.J.; Pan, B.; Ponnath, A.; Tran, C.M.; Nist-Lund, C.; Galvin, A.; Goldberg, H.; Robillard, K.N.; Jodelka, F.M.; Farris, H.E.; et al. Direct Delivery of Antisense Oligonucleotides to the Middle and Inner Ear Improves Hearing and Balance in Usher Mice. Mol. Ther. 2020, 28, 2662–2676. [Google Scholar] [CrossRef]
- Kim, M.A.; Cho, H.J.; Bae, S.H.; Lee, B.; Oh, S.K.; Kwon, T.J.; Ryoo, Z.Y.; Kim, H.Y.; Cho, J.H.; Kim, U.K.; et al. Methionine Sulfoxide Reductase B3-Targeted in Utero Gene Therapy Rescues Hearing Function in a Mouse Model of Congenital Sensorineural Hearing Loss. Antioxid. Redox Signal. 2015, 24, 590–602. [Google Scholar] [CrossRef] [Green Version]
- Akil, O.; Dyka, F.; Calvet, C.; Emptoz, A.; Lahlou, G.; Nouaille, S.; de Monvel, J.B.; Hardelin, J.-P.; Hauswirth, W.W.; Avan, P.; et al. Dual AAV-Mediated Gene Therapy Restores Hearing in a DFNB9 Mouse Model. Proc. Natl. Acad. Sci. USA 2019, 116, 4496–4501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Yang, Z.; Tang, X. Chemical Modifications of Nucleic Acid Drugs and Their Delivery Systems for Gene-Based Therapy. Med. Res. Rev. 2018, 38, 829–869. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Bauer, D.E.; Chiarle, R. Assessing and Advancing the Safety of CRISPR-Cas Tools: From DNA to RNA Editing. Nat. Commun. 2023, 14, 212. [Google Scholar] [CrossRef] [PubMed]
- Miwa, T.; Minoda, R.; Ise, M.; Yamada, T.; Yumoto, E. Mouse Otocyst Transuterine Gene Transfer Restores Hearing in Mice with Connexin 30 Deletion-Associated Hearing Loss. Mol. Ther. 2013, 21, 1142–1150. [Google Scholar] [CrossRef] [Green Version]
- Crispino, G.; Galindo Ramirez, F.; Campioni, M.; Zorzi, V.; Praetorius, M.; di Pasquale, G.; Chiorini, J.A.; Mammano, F. In Vivo Genetic Manipulation of Inner Ear Connexin Expression by Bovine Adeno-Associated Viral Vectors. Sci. Rep. 2017, 7, 6567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Wang, Y.; Chang, Q.; Wang, J.; Gong, S.; Li, H.; Lin, X. Virally Expressed Connexin26 Restores Gap Junction Function in the Cochlea of Conditional Gjb2 Knockout Mice. Gene Ther. 2014, 21, 71–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Askew, C.; Rochat, C.; Pan, B.; Asai, Y.; Ahmed, H.; Child, E.; Schneider, B.L.; Aebischer, P.; Holt, J.R. Tmc Gene Therapy Restores Auditory Function in Deaf Mice. Sci. Transl. Med. 2015, 7, 295ra108. [Google Scholar] [CrossRef] [PubMed]
- Nist-Lund, C.A.; Pan, B.; Patterson, A.; Asai, Y.; Chen, T.; Zhou, W.; Zhu, H.; Romero, S.; Resnik, J.; Polley, D.B.; et al. Improved TMC1 Gene Therapy Restores Hearing and Balance in Mice with Genetic Inner Ear Disorders. Nat. Commun. 2019, 10, 236. [Google Scholar] [CrossRef]
- Yeh, W.H.; Shubina-Oleinik, O.; Levy, J.M.; Pan, B.; Newby, G.A.; Wornow, M.; Burt, R.; Chen, J.C.; Holt, J.R.; Liu, D.R. In Vivo Base Editing Restores Sensory Transduction and Transiently Improves Auditory Function in a Mouse Model of Recessive Deafness. Sci. Transl. Med. 2020, 12, eaay9101. [Google Scholar] [CrossRef]
- Yoshimura, H.; Shibata, S.B.; Ranum, P.T.; Moteki, H.; Smith, R.J.H. Targeted Allele Suppression Prevents Progressive Hearing Loss in the Mature Murine Model of Human TMC1 Deafness. Mol. Ther. 2019, 27, 681–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Tao, Y.; Lamas, V.; Huang, M.; Yeh, W.; Pan, B.; Hu, Y.; Hu, J.H.; Thompson, D.B.; Shu, Y.; et al. Treatment of Autosomal Dominant Hearing Loss by in Vivo Delivery of Genome Editing Agents. Nature 2018, 553, 217–221. [Google Scholar] [CrossRef] [PubMed]
- György, B.; Sage, C.; Indzhykulian, A.A.; Scheffer, D.I.; Brisson, A.R.; Tan, S.; Wu, X.; Volak, A.; Mu, D.; Tamvakologos, P.I.; et al. Rescue of Hearing by Gene Delivery to Inner-Ear Hair Cells Using Exosome-Associated AAV. Mol. Ther. 2017, 25, 379–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Moyed, H.; Cepeda, A.P.; Jung, S.; Moser, T.; Kügler, S.; Reisinger, E. A Dual-AAV Approach Restores Fast Exocytosis and Partially Rescues Auditory Function in Deaf Otoferlin Knock-out Mice. EMBO Mol. Med. 2019, 11, e9396. [Google Scholar] [CrossRef]
- Chang, Q.; Wang, J.; Li, Q.; Kim, Y.; Zhou, B.; Wang, Y.; Li, H.; Lin, X. Virally Mediated Kcnq 1 Gene Replacement Therapy in the Immature Scala Media Restores Hearing in a Mouse Model of Human Jervell and Lange-Nielsen Deafness Syndrome. EMBO J. 2015, 7, 1077–1086. [Google Scholar]
- Pan, B.; Askew, C.; Galvin, A.; Heman-Ackah, S.; Asai, Y.; Indzhykulian, A.A.; Jodelka, F.M.; Hastings, M.L.; Lentz, J.J.; Vandenberghe, L.H.; et al. Gene Therapy Restores Auditory and Vestibular Function in a Mouse Model of Usher Syndrome Type 1c. Nat. Biotechnol. 2017, 35, 264–272. [Google Scholar] [CrossRef]
- Lentz, J.J.; Jodelka, F.M.; Hinrich, A.J.; Mccaffrey, K.E.; Farris, H.E.; Spalitta, M.J.; Bazan, N.G.; Duelli, D.M.; Rigo, F.; Hastings, M.L. Rescue of Hearing and Vestibular Function by Antisense Oligonucleotides in a Mouse Model of Human Deafness. Nat. Med. 2013, 19, 345–350. [Google Scholar] [CrossRef] [Green Version]
- Vijayakumar, S.; Depreux, F.F.; Jodelka, F.M.; Lentz, J.J.; Rigo, F.; Jones, T.A.; Hastings, M.L. Rescue of Peripheral Vestibular Function in Usher Syndrome Mice Using a Splice-Switching Antisense Oligonucleotide. Hum. Mol. Genet. 2017, 26, 3482–3494. [Google Scholar] [CrossRef] [Green Version]
- Chien, W.W.; Isgrig, K.; Roy, S.; Belyantseva, I.A.; Drummond, M.C.; May, L.A.; Fitzgerald, T.S.; Friedman, T.B.; Cunningham, L.L. Gene Therapy Restores Hair Cell Stereocilia Morphology in Inner Ears of Deaf Whirler Mice. Mol. Ther. 2015, 24, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Isgrig, K.; Shteamer, J.W.; Belyantseva, I.A.; Drummond, M.C.; Fitzgerald, T.S.; Vijayakumar, S.; Jones, S.M.; Griffith, A.J.; Friedman, T.B.; Cunningham, L.L.; et al. Gene Therapy Restores Balance and Auditory Functions in a Mouse Model of Usher Syndrome. Mol. Ther. 2017, 25, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Dulon, D.; Papal, S.; Patni, P.; Cortese, M.; Vincent, P.F.Y.; Tertrais, M.; Emptoz, A.; Tlili, A.; Bouleau, Y.; Michel, V.; et al. Clarin-1 Gene Transfer Rescues Auditory Synaptopathy in Model of Usher Syndrome. J. Clin. Investig. 2018, 128, 3382–3401. [Google Scholar] [CrossRef] [PubMed]
- György, B.; Meijer, E.J.; Ivanchenko, M.V.; Tenneson, K.; Emond, F.; Hanlon, K.S.; Indzhykulian, A.A.; Volak, A.; Karavitaki, K.D.; Tamvakologos, P.I.; et al. Gene Transfer with AAV9-PHP.B Rescues Hearing in a Mouse Model of Usher Syndrome 3A and Transduces Hair Cells in a Non-Human Primate. Mol. Ther. Methods Clin. Dev. 2019, 13, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Shubina-Oleinik, O.; Nist-Lund, C.; French, C.; Rockowitz, S.; Shearer, A.E.; Holt, J.R. Dual-Vector Gene Therapy Restores Cochlear Amplification and Auditory Sensitivity in a Mouse Model of DFNB16 Hearing Loss. Sci. Adv. 2021, 7, 7629. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zou, L.; Li, K.; Hou, H.; Hu, Q.; Liu, S.; Li, J.; Song, C.; Chen, J.; Wang, S.; et al. Template-Independent Genome Editing in the Pcdh15av−3j Mouse, a Model of Human DFNB23 Nonsyndromic Deafness. Cell Rep. 2022, 40, 111061. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Shubina-Oleinik, O.; Holt, J.R. Emerging Gene Therapies for Genetic Hearing Loss. JARO J. Assoc. Res. Otolaryngol. 2017, 18, 649–670. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Martinez, J.; Patkaniowska, A.; Lendeckel, W.; Tuschl, T. Functional Anatomy of SiRNAs for Mediating Efficient RNAi in Drosophila Melanogaster Embryo Lysate. EMBO J. 2001, 20, 6877–6888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowdy, S.F. Overcoming Cellular Barriers for RNA Therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef]
- Hannon, G.J.; Rossi, J.J. Unlocking the Potential of the Humangenome with RNA Interference. Nature 2004, 431, 371–378. [Google Scholar] [CrossRef]
- Nikitenko, N.A.; Speiseder, T.; Lam, E.; Rubtsov, P.M.; Tonaeva, K.D.; Borzenok, S.A.; Dobner, T.; Prassolov, V.S. Regulation of Human Adenovirus Replication by RNA Interference. Acta Nat. 2015, 7, 2015. [Google Scholar] [CrossRef] [Green Version]
- Maeda, Y.; Fukushima, K.; Nishizaki, K.; Smith, R.J.H. In Vitro and in Vivo Suppression of GJB2 Expression by RNA Interference. Hum. Mol. Genet. 2005, 14, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.; Smith, L.E.; Bailey, R.A.; Compton, J.G.; Bale, S.J.; White, T.W.; Paul, D.L.; Richard, G.; White, T.W.; Smith, L.E.; et al. RAPID COMMUNICATION Functional Defects of Cx26 Resulting from a Heterozygous Missense Mutation in a Family with Dominant Deaf-Mutism and Palmoplantar Keratoderma. Hum. Genet. 1998, 103, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Denoyelle, F.; Lina-Granade, G.; Plauchu, H.; Bruzzone, R.; Chaib, H.; Levi-Acobas, F.; Weil, D.; Petit, C. Connexin 26 Gene Linked to a Dominant Deafness. Nature 1998, 393, 319–320. [Google Scholar] [CrossRef] [PubMed]
- Kelsell, D.P.; Dunlop, J.; Stevens, H.P.; Lench, N.J.; Liang, J.N.; Parryll, G.; Mueller, R.F.; Leigh, I.M. Connexin 26 Mutations in Hereditary Non-Syndromic Sensorineural Deafness. Nature 1997, 387, 80–83. [Google Scholar] [CrossRef]
- Sontheimer, E.J.; Barrangou, R. The Bacterial Origins of the CRISPR Genome-Editing Revolution. Hum. Gene Ther. 2015, 26, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Hille, F.; Richter, H.; Wong, S.P.; Bratovič, M.; Ressel, S.; Charpentier, E. The Biology of CRISPR-Cas: Backward and Forward. Cell 2018, 172, 1239–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolotin, A.; Quinquis, B.; Sorokin, A.; Dusko Ehrlich, S. Clustered Regularly Interspaced Short Palindrome Repeats (CRISPRs) Have Spacers of Extrachromosomal Origin. Microbiology 2005, 151, 2551–2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrangou, R.; Doudna, J.A. Applications of CRISPR Technologies in Research and Beyond. Nat. Biotechnol. 2016, 34, 933–941. [Google Scholar] [CrossRef]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-CrRNA Ribonucleoprotein Complex Mediates Specific DNA Cleavage for Adaptive Immunity in Bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [Green Version]
- Mali, P.; Esvelt, K.M.; Church, G.M. Cas9 as a Versatile Tool for Engineering Biology. Nat. Methods 2013, 10, 957–963. [Google Scholar] [CrossRef] [Green Version]
- Di Stazio, M.; Foschi, N.; Athanasakis, E.; Gasparini, P.; d’Adamo, A.P. Systematic Analysis of Factors That Improve Homologous Direct Repair (HDR) Efficiency in CRISPR/Cas9 Technique. PLoS ONE 2021, 16, e0247603. [Google Scholar] [CrossRef] [PubMed]
- Paquet, D.; Kwart, D.; Chen, A.; Sproul, A.; Jacob, S.; Teo, S.; Olsen, K.M.; Gregg, A.; Noggle, S.; Tessier-Lavigne, M. Efficient Introduction of Specific Homozygous and Heterozygous Mutations Using CRISPR/Cas9. Nature 2016, 533, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Badran, A.H.; Liu, D.R. CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 2017, 168, 20–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.W.; Arbab, M.; Hsu, J.Y.; Worstell, D.; Culbertson, S.J.; Krabbe, O.; Cassa, C.A.; Liu, D.R.; Gifford, D.K.; Sherwood, R.I. Predictable and Precise Template-Free CRISPR Editing of Pathogenic Variants. Nature 2018, 563, 646–651. [Google Scholar] [CrossRef]
- Leenay, R.T.; Aghazadeh, A.; Hiatt, J.; Tse, D.; Roth, T.L.; Apathy, R.; Shifrut, E.; Hultquist, J.F.; Krogan, N.; Wu, Z.; et al. Large Dataset Enables Prediction of Repair after CRISPR–Cas9 Editing in Primary T Cells. Nat. Biotechnol. 2019, 37, 1034–1037. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.; Crepaldi, L.; Alsinet, C.; Strong, A.J.; Kleshchevnikov, V.; de Angeli, P.; Páleníková, P.; Khodak, A.; Kiselev, V.; Kosicki, M.; et al. Predicting the Mutations Generated by Repair of Cas9-Induced Double-Strand Breaks. Nat. Biotechnol. 2019, 37, 64–82. [Google Scholar] [CrossRef]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of Double-Strand Breaks Induced by CRISPR–Cas9 Leads to Large Deletions and Complex Rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Lessard, S.; Francioli, L.; Alfoldi, J.; Tardif, J.C.; Ellinor, P.T.; MacArthur, D.G.; Lettre, G.; Orkin, S.H.; Canver, M.C. Human Genetic Variation Alters CRISPR-Cas9 on- and off-Targeting Specificity at Therapeutically Implicated Loci. Proc. Natl. Acad. Sci. USA 2017, 114, E11257–E11266. [Google Scholar] [CrossRef] [Green Version]
- Modarai, S.R.; Kanda, S.; Bloh, K.; Opdenaker, L.M.; Kmiec, E.B. Precise and Error-Prone CRISPR-Directed Gene Editing Activity in Human CD34+ Cells Varies Widely among Patient Samples. Gene Ther. 2021, 28, 105–113. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable Editing of a Target Base in Genomic DNA without Double-Stranded DNA Cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Komor, A.C.; Zhao, K.T.; Packer, M.S.; Gaudelli, N.M.; Waterbury, A.L.; Koblan, L.W.; Kim, Y.B.; Badran, A.H.; Liu, D.R. Improved Base Excision Repair Inhibition and Bacteriophage Mu Gam Protein Yields C:G-to-T:A Base Editors with Higher Efficiency and Product Purity. Sci. Adv. 2017, 3, eaao4774. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Arazoe, T.; Yachie, N.; Banno, S.; Kakimoto, M.; Tabata, M.; Mochizuki, M.; Miyabe, A.; Araki, M.; Hara, K.Y.; et al. Targeted Nucleotide Editing Using Hybrid Prokaryotic and Vertebrate Adaptive Immune Systems. Science 2016, 353, aaf8729. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Jo, D.H.; Cho, C.S.; Shin, J.H.; Seo, J.H.; Yu, G.; Gopalappa, R.; Kim, D.; Cho, S.R.; Kim, J.H.; et al. Application of Prime Editing to the Correction of Mutations and Phenotypes in Adult Mice with Liver and Eye Diseases. Nat. Biomed. Eng. 2022, 6, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Dong, X.; Cheng, H.; Zheng, C.; Chen, Z.; Rodríguez, T.C.; Liang, S.Q.; Xue, W.; Sontheimer, E.J. A Split Prime Editor with Untethered Reverse Transcriptase and Circular RNA Template. Nat. Biotechnol. 2022, 40, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Ginn, S.; Alexander, I.E.; Edelstein, M.L.; Abedi, M.R.; Wixon, J. Gene Therapy Clinical Trials Worldwide to 2012–An Update. J. Gene Med. 2013, 15, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Zabner, J.; Couture, L.A.; Gregory, R.J.; Graham, S.M.; Smith, A.E.; Welsh, M.J. Adenovirus-Mediated Gene Transfer Transiently Corrects the Chloride Transport Defect in Nasal Epithelia of Patients with Cystic Fibrosis. Cell 1993, 75, 207–216. [Google Scholar] [CrossRef]
- Kawamoto, K.; Oh, S.H.; Kanzaki, S.; Brown, N.; Raphael, Y. The Functional and Structural Outcome of Inner Ear Gene Transfer via the Vestibular and Cochlear Fluids in Mice. Mol. Ther. 2001, 4, 575–585. [Google Scholar] [CrossRef]
- Luebke, A.E.; Foster, P.K.; Muller, C.D.; Peel, A.L. Cochlear Function and Transgene Expression in the Guinea Pig Cochlea, Using Adenovirus- and Adeno-Associated Virus-Directed Gene Transfer. Hum. Gene Ther. 2001, 12, 773–781. [Google Scholar] [CrossRef]
- Praetorius, M.; Baker, K.; Weich, C.; Plinkert, P.; Staecker, H. Hearing Preservation after Inner Ear Gene Therapy: The Effect of Vector and Surgical Approach. ORL J. Otorhinolaryngol. Relat. Spec. 2003, 65, 211–214. [Google Scholar] [CrossRef]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and Problems with the Use of Viral Vectors for Gene Therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef]
- Milone, M.C.; O’Doherty, U. Clinical Use of Lentiviral Vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Apolonia, L.; Waddington, S.N.; Fernandes, C.; Ward, N.J.; Bouma, G.; Blundell, M.P.; Thrasher, A.J.; Collins, M.K.; Philpott, N.J. Stable Gene Transfer to Muscle Using Non-Integrating Lentiviral Vectors. Mol. Ther. 2007, 15, 1947–1954. [Google Scholar] [CrossRef]
- Vannucci, L.; Lai, M.; Chiuppesi, F.; Ceccherini-Nelli, L.; Pistello, M. Viral Vectors: A Look Back and Ahead on Gene Transfer Technology. New Microbiol. 2013, 36, 1–22. [Google Scholar] [PubMed]
- Hashimoto, T.; Gibbs, D.; Lillo, C.; Azarian, S.M.; Legacki, E.; Zhang, X.M.; Yang, X.J.; Williams, D.S. Lentiviral Gene Replacement Therapy of Retinas in a Mouse Model for Usher Syndrome Type 1B. Gene Ther. 2007, 14, 584–594. [Google Scholar] [CrossRef] [Green Version]
- Zallocchi, M.; Binley, K.; Lad, Y.; Ellis, S.; Widdowson, P.; Iqball, S.; Scripps, V.; Kelleher, M.; Loader, J.; Miskin, J.; et al. EIAV-Based Retinal Gene Therapy in the Shaker1 Mouse Model for Usher Syndrome Type 1B: Development of UshStat. PLoS ONE 2014, 9, e94272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.J.; Mhatre, A.N.; Wareing, M.; Pettis, R.; Gao, W.-Q.; Zufferey, R.N.; Trono, D.; Lalwani, A.K. Transgene Expression in the Guinea Pig Cochlea Mediated by a Lentivirus-Derived Gene Transfer Vector. Hum. Gene Ther. 1999, 10, 1867–1873. [Google Scholar] [CrossRef]
- Parker, C.L.; Jacobs, T.M.; Huckaby, J.T.; Harit, D.; Lai, S.K. Efficient and Highly Specific Gene Transfer Using Mutated Lentiviral Vectors Redirected with Bispecific Antibodies. mBio 2020, 11, e02990-19. [Google Scholar] [CrossRef] [Green Version]
- Hastie, E.; Samulski, R.J. Adeno-Associated Virus at 50: A Golden Anniversary of Discovery, Research, and Gene Therapy Success–A Personal Perspective. Hum. Gene Ther. 2015, 26, 257–265. [Google Scholar]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L.; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef] [Green Version]
- Penaud-Budloo, M.; le Guiner, C.; Nowrouzi, A.; Toromanoff, A.; Chérel, Y.; Chenuaud, P.; Schmidt, M.; von Kalle, C.; Rolling, F.; Moullier, P.; et al. Adeno-Associated Virus Vector Genomes Persist as Episomal Chromatin in Primate Muscle. J. Virol. 2008, 82, 7875–7885. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Asokan, A.; Samulski, R.J. Adeno-Associated Virus Serotypes: Vector Toolkit for Human Gene Therapy. Mol. Ther. 2006, 14, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.E.; Rolling, F.; Li, C.; Conrath, H.; Xiao, W.; Xiao, X.; Samulski, R.J. Cross-Packaging of a Single Adeno-Associated Virus (AAV) Type 2 Vector Genome into Multiple AAV Serotypes Enables Transduction with Broad Specificity. J. Virol. 2002, 76, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Stieger, K.; Lheriteau, E.; Moullier, P.; Rolling, F. AAV-Mediated Gene Therapy for Retinal Disorders in Large Animal Models. ILAR J. 2009, 50, 206–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hordeaux, J.; Wang, Q.; Katz, N.; Buza, E.L.; Bell, P.; Wilson, J.M. The Neurotropic Properties of AAV-PHP.B Are Limited to C57BL/6J Mice. Mol. Ther. 2018, 26, 664–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, P.S.; Lee, V.; Wei, Z.; Song, J.Y.; Casal, G.; Cronin, T.; Willett, K.; Huckfeldt, R.; Morgan, J.I.W.; Aleman, T.S.; et al. Evaluation of Dose and Safety of AAV7m8 and AAV8BP2 in the Non-Human Primate Retina. Hum. Gene Ther. 2017, 28, 154–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Nist-Lund, C.; Solanes, P.; Goldberg, H.; Wu, J.; Pan, B.; Schneider, B.L.; Holt, J.R. Efficient Viral Transduction in Mouse Inner Ear Hair Cells with Utricle Injection and AAV9-PHP.B. Hear Res. 2020, 394, 107882. [Google Scholar] [CrossRef] [PubMed]
- Ivanchenko, M.V.; Hanlon, K.S.; Devine, M.K.; Tenneson, K.; Emond, F.; Lafond, J.F.; Kenna, M.A.; Corey, D.P.; Maguire, C.A. Preclinical Testing of AAV9-PHP.B for Transgene Expression in the Non-Human Primate Cochlea. Hear Res. 2020, 394, 107930. [Google Scholar] [CrossRef]
- Ivanchenko, M.V.; Hanlon, K.S.; Hathaway, D.M.; Klein, A.J.; Peters, C.W.; Li, Y.; Tamvakologos, P.I.; Nammour, J.; Maguire, C.A.; Corey, D.P. AAV-S: A Versatile Capsid Variant for Transduction of Mouse and Primate Inner Ear. Mol. Ther. Methods Clin. Dev. 2021, 21, 382–398. [Google Scholar] [CrossRef]
- Andres-Mateos, E.; Landegger, L.D.; Unzu, C.; Phillips, J.; Lin, B.M.; Dewyer, N.A.; Sanmiguel, J.; Nicolaou, F.; Valero, M.D.; Bourdeu, K.I.; et al. Choice of Vector and Surgical Approach Enables Efficient Cochlear Gene Transfer in Nonhuman Primate. Nat. Commun. 2022, 13, 1359. [Google Scholar] [CrossRef]
- Ranum, P.T.; Tecedor, L.; Keiser, M.S.; Chen, Y.H.; Leib, D.E.; Liu, X.; Davidson, B.L. Cochlear Transduction via Cerebrospinal Fluid Delivery of AAV in Non-Human Primates. Mol. Ther. 2023. [Google Scholar] [CrossRef]
- Landegger, L.D.; Pan, B.; Askew, C.; Wassmer, S.J.; Gluck, S.D.; Galvin, A.; Taylor, R.; Forge, A.; Stankovic, K.M.; Holt, J.R.; et al. A Synthetic AAV Vector Enables Safe and Efficient Gene Transfer to the Mammalian Inner Ear. Nat. Biotechnol. 2017, 35, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omichi, R.; Yoshimura, H.; Shibata, S.B.; Vandenberghe, L.H.; Smith, R.J.H. Hair Cell Transduction Efficiency of Single- and Dual-AAV Serotypes in Adult Murine Cochleae. Mol. Methods Clin. Dev. 2020, 17, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Huang, M.; Shu, Y.; Ruprecht, A.; Wang, H.; Tang, Y.; Vandenberghe, L.H.; Wang, Q.; Gao, G.; Kong, W.J.; et al. Delivery of Adeno-Associated Virus Vectors in Adult Mammalian Inner-Ear Cell Subtypes Without Auditory Dysfunction. Hum. Gene Ther. 2018, 29, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Geng, R.; Omar, A.; Gopal, S.R.; Chen, D.H.C.; Stepanyan, R.; Basch, M.L.; Dinculescu, A.; Furness, D.N.; Saperstein, D.; Hauswirth, W.; et al. Modeling and Preventing Progressive Hearing Loss in Usher Syndrome III. Sci. Rep. 2017, 7, 13480. [Google Scholar] [CrossRef] [Green Version]
- Isgrig, K.; McDougald, D.S.; Zhu, J.; Wang, H.J.; Bennett, J.; Chien, W.W. AAV2.7m8 Is a Powerful Viral Vector for Inner Ear Gene Therapy. Nat. Commun. 2019, 10, 427. [Google Scholar] [CrossRef] [Green Version]
- Iizuka, T.; Kamiya, K.; Gotoh, S.; Sugitani, Y.; Suzuki, M.; Noda, T.; Minowa, O.; Ikeda, K. Perinatal GJB2 Gene Transfer Rescues Hearing in a Mouse Model of Hereditary Deafness. Hum. Mol. Genet. 2015, 24, 3651–3661. [Google Scholar] [CrossRef] [Green Version]
- Chien, W.W.; McDougald, D.S.; Roy, S.; Fitzgerald, T.S.; Cunningham, L.L. Cochlear Gene Transfer Mediated by Adeno-Associated Virus: Comparison of Two Surgical Approaches. Laryngoscope 2015, 125, 2557–2564. [Google Scholar] [CrossRef]
- Xia, L.; Yin, S.; Wang, J. Inner Ear Gene Transfection in Neonatal Mice Using Adeno-Associated Viral Vector: A Comparison of Two Approaches. PLoS ONE 2012, 7, e43218. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Liu, Y.; Qu, T.; Peng, Z.; Xie, J.; Wang, G.; Gong, S. Cochleovestibular Gene Transfer in Neonatal Mice by Canalostomy. Neuroreport 2017, 28, 682–688. [Google Scholar] [CrossRef]
- Gu, X.; Chai, R.; Guo, L.; Dong, B.; Li, W.; Shu, Y.; Huang, X.; Li, H. Transduction of Adeno-Associated Virus Vectors Targeting Hair Cells and Supporting Cells in the Neonatal Mouse Cochlea. Front. Cell. Neurosci. 2019, 13, 1–16. [Google Scholar] [CrossRef]
- Yoshimura, H.; Shibata, S.B.; Ranum, P.T.; Smith, R.J.H. Enhanced Viral-Mediated Cochlear Gene Delivery in Adult Mice by Combining Canal Fenestration with Round Window Membrane Inoculation. Sci. Rep. 2018, 8, 2980. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Hashimoto, K.; Xiao, R.; Vandenberghe, L.H.; Liberman, M.C. Cochlear Gene Therapy with Ancestral AAV in Adult Mice: Complete Transduction of Inner Hair Cells without Cochlear Dysfunction. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tan, F.; Chu, C.; Qi, J.; Li, W.; You, D.; Li, K.; Chen, X.; Zhao, W.; Cheng, C.; Liu, X.; et al. AAV-Ie Enables Safe and Efficient Gene Transfer to Inner Ear Cells. Nat. Commun. 2019, 10, 3733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyka, F.M.; Boye, S.L.; Chiodo, V.A.; Hauswirth, W.W.; Boye, S.E. Dual Adeno-Associated Virus Vectors Result in Efficient in Vitro and in Vivo Expression of an Oversized Gene, MYO7A. Hum. Gene Ther. Methods 2014, 25, 166–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapani, I.; Colella, P.; Sommella, A.; Iodice, C.; Cesi, G.; de Simone, S.; Marrocco, E.; Rossi, S.; Giunti, M.; Palfi, A.; et al. Effective Delivery of Large Genes to the Retina by Dual AAV Vectors. EMBO Mol. Med. 2014, 6, 194–211. [Google Scholar] [CrossRef] [PubMed]
- Mcclements, M.E.; Maclaren, R.E. Adeno-Associated Virus (AAV) Dual Vector Strategies for Gene Therapy Encoding Large Transgenes. Yale J. Biol. Med. 2017, 90, 611–623. [Google Scholar]
- Koo, T.; Popplewell, L.; Athanasopoulos, T.; Dickson, G. Triple Trans-Splicing Adeno-Associated Virus Vectors Capable of Transferring the Coding Sequence for Full-Length Dystrophin Protein into Dystrophic Mice. Hum. Gene Ther. 2014, 25, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Ahmed, Z.M.; Riazuddin, S.; Bhinder, M.A.; Shahzad, M.; Husnain, T.; Riazuddin, S.; Griffith, A.J.; Friedman, T.B. Identities and Frequencies of Mutations of the Otoferlin Gene (OTOF) Causing DFNB9 Deafness in Pakistan. Clin. Genet. 2009, 75, 237–243. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Ballesteros, M.; Reynoso, R.; Olarte, M.; Villamar, M.; Morera, C.; Santarelli, R.; Arslan, E.; Medá, C.; Curet, C.; Völter, C.; et al. A Multicenter Study on the Prevalence and Spectrum of Mutations in the Otoferlin Gene (OTOF) in Subjects with Nonsyndromic Hearing Impairment and Auditory Neuropathy. Hum. Mutat. 2008, 29, 823–831. [Google Scholar] [CrossRef]
- Yasunaga, S.; Grati, M.; Cohen-Salmon, M.; El-Amraoui, A.; Mustapha, M.; Salem, N.; El-Zir, E.; Loiselet, J.; Petit, C. A Mutation in OTOF, Encoding Otoferlin, a FER-1-like Protein, Causes DFNB9, a Nonsyndromic Form of Deafness. Nat. Genet. 1999, 21, 363–369. [Google Scholar] [CrossRef]
- Yasunaga, S.; Grati, M.; Chardenoux, S.; Smith, T.N.; Friedman, T.B.; Lalwani, A.K.; Wilcox, E.R.; Petit, C. OTOF Encodes Multiple Long and Short Isoforms: Genetic Evidence That the Long Ones Underlie Recessive Deafness DFNB9. Am. J. Hum. Genet. 2000, 67, 591–600. [Google Scholar] [CrossRef] [Green Version]
- Shi, X. Pathophysiology of the Cochlear Intrastrial Fluid-Blood Barrier (Review). Hear Res. 2016, 338, 52–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, S.B.; Yoshimura, H.; Ranum, P.T.; Goodwin, A.T.; Smith, R.J.H. Intravenous RAAV2/9 Injection for Murine Cochlear Gene Delivery. Sci. Rep. 2017, 7, 9609. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, L.A.; Li, Q.; Yang, J.; Goddard, J.C.; Fekete, D.M.; Lang, H. Adeno-Associated Virus-Mediated Gene Delivery into the Scala Media of the Normal and Deafened Adult Mouse Ear. Gene Ther. 2011, 18, 569–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salt, A.N.; Hirose, K. Communication Pathways to and from the Inner Ear and Their Contributions to Drug Delivery. Hear Res. 2018, 362, 25–37. [Google Scholar] [CrossRef]
- Yamasoba, T.; Yagi, M.; Roessler, B.J.; Miller, J.M.; Raphael, Y. Inner Ear Transgene Expression after Adenoviral Vector Inoculation in the Endolymphatic Sac. Hum. Gene Ther. 1999, 10, 769–774. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, R.; Dai, C.; Steyger, P.S.; Yongfu, Y.Y. Comparison of Gentamicin Distribution in the Inner Ear Following Administration via the Endolymphatic Sac or Round Window. Laryngoscope 2010, 120, 2054–2060. [Google Scholar] [CrossRef] [Green Version]
- Colletti, V.; Mandalà, M.; Carner, M.; Barillari, M.; Cerini, R.; Pozzi Mucelli, R.; Colletti, L. Evidence of Gadolinium Distribution from the Endolymphatic Sac to the Endolymphatic Compartments of the Human Inner Ear. Audiol. Neurotol. 2010, 15, 353–363. [Google Scholar] [CrossRef]
- García, M.D.L.F.; Segura, C.D.L.L.; Lesser, J.C.C.; Pianese, C.P. Endolymphatic Sac Surgery for Ménière’s Disease–Current Opinion and Literature Review. Int. Arch. Otorhinolaryngol. 2017, 21, 179–183. [Google Scholar]
- Dai, C.; Lehar, M.; Sun, D.Q.; Rvt, L.S.; Carey, J.P.; MacLachlan, T.; Brough, D.; Staecker, H.; della Santina, A.M.; Hullar, T.E.; et al. Rhesus Cochlear and Vestibular Functions Are Preserved After Inner Ear Injection of Saline Volume Sufficient for Gene Therapy Delivery. JARO J. Assoc. Res. Otolaryngol. 2017, 18, 601–617. [Google Scholar] [CrossRef]
- Jero, J.; Mhatre, A.N.; Tseng, C.J.; Stern, R.E.; Coling, D.E.; Goldstein, J.A.; Hong, K.; Zheng, W.W.; Hoque, A.T.M.S.; Lalwani, A.K. Cochlear Gene Delivery through an Intact Round Window Membrane in Mouse. Hum. Gene Ther. 2001, 12, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, Z.; Fan, L.; Landry, T.; Brown, J.; Yu, Z.; Yin, S.; Wang, J. Ultrasound-Microbubble Cavitation Facilitates Adeno-Associated Virus Mediated Cochlear Gene Transfection across the Round-Window Membrane. Bioeng. Transl. Med. 2021, 6, e10189. [Google Scholar] [CrossRef] [PubMed]
- Hitier, M.; Zhang, M.; Labrousse, M.; Barbier, C.; Patron, V.; Moreau, S. Persistent Stapedial Arteries in Human: From Phylogeny to Surgical Consequences. Surg. Radiol. Anat. 2013, 35, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Thorne, M.; Salt, A.N.; DeMott, J.E.; Henson, M.M.; Henson, O.W.; Gewalt, S.L. Cochlear Fluid Space Dimensions for Six Species Derived from Reconstructions of Three-Dimensional Magnetic Resonance Images. Laryngoscope 1999, 109, 1661–1668. [Google Scholar] [CrossRef] [PubMed]
- Kirk, E.C.; Gosselin-Ildari, A.D. Cochlear Labyrinth Volume and Hearing Abilities in Primates. Anat. Rec. 2009, 292, 765–776. [Google Scholar] [CrossRef]
- Stöver, T.; Yagi, M.; Raphael, Y. Transduction of the Contralateral Ear after Adenovirus-Mediated Cochlear Gene Transfer. Gene Ther. 2000, 7, 377–383. [Google Scholar] [CrossRef]
- Burton, J.A.; Valero, M.D.; Hackett, T.A.; Ramachandran, R. The Use of Nonhuman Primates in Studies of Noise Injury and Treatment. J. Acoust. Soc. Am. 2019, 146, 3770–3789. [Google Scholar] [CrossRef] [Green Version]
- Manrique-Huarte, R.; de Linera-Alperi, M.A.; Parilli, D.; Rodriguez, J.A.; Borro, D.; Dueck, W.F.; Smyth, D.; Salt, A.; Manrique, M. Inner Ear Drug Delivery through a Cochlear Implant: Pharmacokinetics in a Macaque Experimental Model. Hear Res. 2021, 404, 108228. [Google Scholar] [CrossRef]
- Litovsky, R. Development of the Auditory System, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 129. [Google Scholar]
- Lim, R.; Brichta, A.M. Anatomical and Physiological Development of the Human Inner Ear. Hear Res. 2016, 338, 9–21. [Google Scholar] [CrossRef]
- Pedreira, D.A.L. Advances in Fetal Surgery. Einstein 2016, 14, 110–112. [Google Scholar] [CrossRef] [Green Version]
- Michalski, N.; Goutman, J.D.; Auclair, S.M.; de Monvel, J.B.; Tertrais, M.; Emptoz, A.; Parrin, A.; Nouaille, S.; Guillon, M.; Sachse, M.; et al. Otoferlin Acts as a Ca2+ Sensor for Vesicle Fusion and Vesicle Pool Replenishment at Auditory Hair Cell Ribbon Synapses. Elife 2017, 6, e31013. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Cideciyan, A.V.; Roman, A.J.; Sumaroka, A.; Schwartz, S.B.; Heon, E.; Hauswirth, W.W. Improvement and Decline in Vision with Gene Therapy in Childhood Blindness. N. Engl. J. Med. 2015, 372, 1920–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bainbridge, J.W.B.; Mehat, M.S.; Sundaram, V.; Robbie, S.J.; Barker, S.E.; Ripamonti, C.; Georgiadis, A.; Mowat, F.M.; Beattie, S.G.; Gardner, P.J.; et al. Long-Term Effect of Gene Therapy on Leber’s Congenital Amaurosis. N. Engl. J. Med. 2015, 372, 1887–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichel, F.F.; Dauletbekov, D.L.; Klein, R.; Peters, T.; Ochakovski, G.A.; Seitz, I.P.; Wilhelm, B.; Ueffing, M.; Biel, M.; Wissinger, B.; et al. AAV8 Can Induce Innate and Adaptive Immune Response in the Primate Eye. Mol. Ther. 2017, 25, 2648–2660. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, A.C.; Tuddenham, E.G.D.; Rangarajan, S.; Rosales, C.; McIntosh, J.; Linch, D.C.; Chowdary, P.; Riddell, A.; Pie, A.J.; Harrington, C.; et al. Adenovirus-Associated Virus Vector–Mediated Gene Transfer in Hemophilia, B. N. Engl. J. Med. 2011, 365, 2357–2365. [Google Scholar] [CrossRef]
- Hareendran, S.; Balakrishnan, B.; Sen, D.; Kumar, S.; Srivastava, A.; Jayandharan, G.R. Adeno-Associated Virus (AAV) Vectors in Gene Therapy: Immune Challenges and Strategies to Circumvent Them. Rev. Med. Virol. 2013, 23, 399–413. [Google Scholar] [CrossRef]
- Manno, C.S.; Arruda, V.R.; Pierce, G.F.; Glader, B.; Ragni, M.; Rasko, J.; Ozelo, M.C.; Hoots, K.; Blatt, P.; Konkle, B.; et al. Successful Transduction of Liver in Hemophilia by AAV-Factor IX and Limitations Imposed by the Host Immune Response. Nat. Med. 2006, 12, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; High, K.A. Immune Responses to AAV Vectors: Overcoming Barriers to Successful Gene Therapy. Blood 2013, 122, 23–36. [Google Scholar] [CrossRef]
- Masat, E.; Pavani, G.; Mingozzi, F. Humoral Immunity to AAV Vectors in Gene Therapy: Challenges and Potential Solutions. Discov. Med. 2013, 15, 379–389. [Google Scholar]
Figure 1.
Route of administration for inner ear gene therapy. Routes of administration evaluated in mouse models (left panel), and potential delivery routes in humans (surgical view through the middle ear cavities, right panel). The most widely used delivery route is injection through the round window membrane (1), which is possible in both mice and humans, followed by the posterior semicircular canal (SCC, 2) in mice, which is potentially equivalent to a lateral SCC injection (3) in humans. Other techniques that have been evaluated in mice include injection into the endolymphatic sac (4) and cochleostomy (5). Injection through the oval window (6) is possible in humans, but not in mice, due to the persistence of the stapedial artery in rodents.
Figure 1.
Route of administration for inner ear gene therapy. Routes of administration evaluated in mouse models (left panel), and potential delivery routes in humans (surgical view through the middle ear cavities, right panel). The most widely used delivery route is injection through the round window membrane (1), which is possible in both mice and humans, followed by the posterior semicircular canal (SCC, 2) in mice, which is potentially equivalent to a lateral SCC injection (3) in humans. Other techniques that have been evaluated in mice include injection into the endolymphatic sac (4) and cochleostomy (5). Injection through the oval window (6) is possible in humans, but not in mice, due to the persistence of the stapedial artery in rodents.

Table 1.
Preclinical studies of gene therapy in mouse models of genetic hearing impairment.
| Gene (Deafness) | Mouse Model | Stage | Approach | Vector | Strategy | Results | References |
|---|---|---|---|---|---|---|---|
| VGLUT3 (DFNA25) | Vglut3-/- | Mature | RW | AAV2/1 | Replacement | Improvement in hearing to near-normal ABR thresholds | Akil et al., 2012 [8] |
| Neonatal | RW/Co | AAV2/1 | Replacement | ||||
| GJB6 (DFNB1) | Gjb6-/- | In utero | Otocyst | - | Replacement | Improvement of hearing (thresholds: 50 dB) | Miwa et al., 2013 [16] |
| Gjb6-/- | Neonatal | PSCC | BAAV | Replacement | Protein production without hearing improvement | Crispino et al., 2017 [17] | |
| GJB2 (DFNB1) | Foxg1-cCx26KO | Neonatal | Co | AAV2/1 | Replacement | Protein production without hearing improvement | Yu et al., 2014 [18] |
| MSRB3 (DFNB74) | MsrB3-/- | In utero | Otocyst | AAV2/1 | Replacement | Improvement in hearing to near-normal ABR thresholds | M.-A. Kim et al., 2015 [12] |
| TMC1 (DFNB7/11) | Tmc1Δ/Δ | Neonatal | RW | AAV2/1 | Replacement | Partial improvement of hearing (thresholds: 90 dB) | Askew et al., 2015 [19] |
| Tmc1Δ/Δ | AAV2/Anc80L65 | Replacement | Partial improvement of hearing (thresholds: 60 dB) Improvement of auditory cortex responses | Nist-Lund et al., 2019 [20] | |||
| Tmc1Y182C/Y182C | Neonatal | NR | AAV2/Anc80L65 | Base editing | Partial and transient improvement of hearing (thresholds: 90 dB) | Yeh et al., 2020 [21] | |
| TMC1 (DFNA36) | Tmc1Bth/+ | Mature | RW + PSCC fenestration | AAV2/9 | Regulation (miRNA) | Prevention of the progression of deafness | Yoshimura et al., 2019 [22] |
| Tmc1Bth/+ | Neonatal | Co | Liposome | Gene editing (CRISPR-Cas9) | Prevention of the progression of deafness | Gao et al., 2017 [23] | |
| Tmc1Bth/+ | Neonatal | Intracochlear | AAV2/Anc80L65 | Gene editing (CRISPR-Cas9) | Prevention of the progression of deafness up to one year after treatment | György et al., 2019 [9] | |
| PJVK (DFNB59) | Pjvk-/- | Neonatal | RW | AAV2/8 | Replacement | Improvement in hearing to near-normal thresholds | Delmaghani et al., 2015 [10] |
| LHFPL5 (DFNB67) | Lhflp5-/- | Neonatal | RW | Exo-AAV2/1 | Replacement | Partial improvement of hearing (ABR thresholds: 80 dB) Partial improvement of vestibular function | György et al., 2017 [24] |
| OTOF (DFNB9) | Otof-/- | P6-P7 | RW | AAV2/6 | Replacement | Partial improvement of hearing (thresholds: 70 to 90 dB) | Al Moyed et al., 2019 [25] |
| Otof-/- | P10-P30 | RW | AAV2quadY-F | Replacement | Improvement in hearing to near-normal thresholds | Akil et al., 2019 [13] | |
| KCNQ1 (Jervell Lange-Nielsen) | Kcnq1-/- | Neonatal | RW | AAV2/1 | Replacement | Prevention of cochlear morphological abnormalities Improvement in hearing to near-normal thresholds | Chang et al., 2015 [26] |
| USH1C (Usher type 1C) | Ush1c c.216G > A | Neonatal | RW | Anc80L65 | Replacement | Complete restoration of balance Partial improvement of hearing (thresholds: 50 dB) | Pan et al., 2017 [27] |
| Neonatal | IP | - | Regulation (ASO) | Partial improvement of hearing (thresholds: 50 dB) | Lentz et al., 2013 [28] | ||
| Complete restoration of balance | Vijayakumar et al., 2017 [29] | ||||||
| Mature | Partial restoration of balance | ||||||
| Neonatal | RW/ITI | - | Regulation (ASO) | Complete restoration of balance Improvement in hearing to near-normal thresholds | Lentz et al., 2020 [11] | ||
| Mature | ITI | Partial improvement in hearing Significant improvement in balance function | |||||
| USH1G (Usher type 1G) | Ush1g-/- mice | Neonatal | RW | AAV2/8 | Replacement | Complete restoration of balance Partial improvement of hearing (thresholds: 50 dB) | Emptoz et al., 2017 [7] |
| WHRN (Usher type 2D) | Whrnwi/wi | Neonatal | RW | AAV2/8 | Replacement | Restoration of stereocilium structure and prevention of cell degeneration without improvement of hearing | Chien et al., 2015 [30] |
| Neonatal | PSCC | AAV2/8 | Replacement | Improvement of balance and hearing (thresholds: 90 dB) | Isgrig et al., 2017 [31] | ||
| CLRN (Usher type 3) | cClrn1KO | Neonatal | RW | AAV2/8 | Replacement | Preservation of synaptic morphology with a slight improvement in hearing | Dulon et al., 2018 [32] |
| Clrn-/- | Neonatal | PSCC | AAV9.PHP.B | Replacement | Partial improvement of hearing (thresholds: 40 dB for low frequencies) | György et al., 2019 [33] | |
| STRC (DFNB16) | Strc-/- | Neonatal | Utricle | AAV9.PHP.B | Replacement | Partial improvement of hearing (thresholds: 40 db) with restoration of DPOAE | Shubina-Oleinik et al., 2021 [34] |
| PCDH15 (DFNB23) | Pcdh15av3j | Neonatal | Co | AAV2/9 | Gene editing (CRISPR-Cas9) | Almost complete restoration of balance Partial improvement of hearing (thresholds: 90 dB) | Liu et al., 2022 [35] |
ASO = antisense oligonucleotides; Co = cochleostomy; RW = round window; PSCC = posterior semicircular canal; IP = intraperitoneal; ITI = intratympanic injection; NR = not reported.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lahlou, G.; Calvet, C.; Giorgi, M.; Lecomte, M.-J.; Safieddine, S. Towards the Clinical Application of Gene Therapy for Genetic Inner Ear Diseases. J. Clin. Med. 2023, 12, 1046. https://doi.org/10.3390/jcm12031046
AMA Style
Lahlou G, Calvet C, Giorgi M, Lecomte M-J, Safieddine S. Towards the Clinical Application of Gene Therapy for Genetic Inner Ear Diseases. Journal of Clinical Medicine. 2023; 12(3):1046. https://doi.org/10.3390/jcm12031046
Chicago/Turabian StyleLahlou, Ghizlene, Charlotte Calvet, Marie Giorgi, Marie-José Lecomte, and Saaid Safieddine. 2023. "Towards the Clinical Application of Gene Therapy for Genetic Inner Ear Diseases" Journal of Clinical Medicine 12, no. 3: 1046. https://doi.org/10.3390/jcm12031046
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.