
Fetal Bradycardia Caused by Monogenic Disorders—A Review of the Literature
 , , and
, , and
Abstract
:1. Background
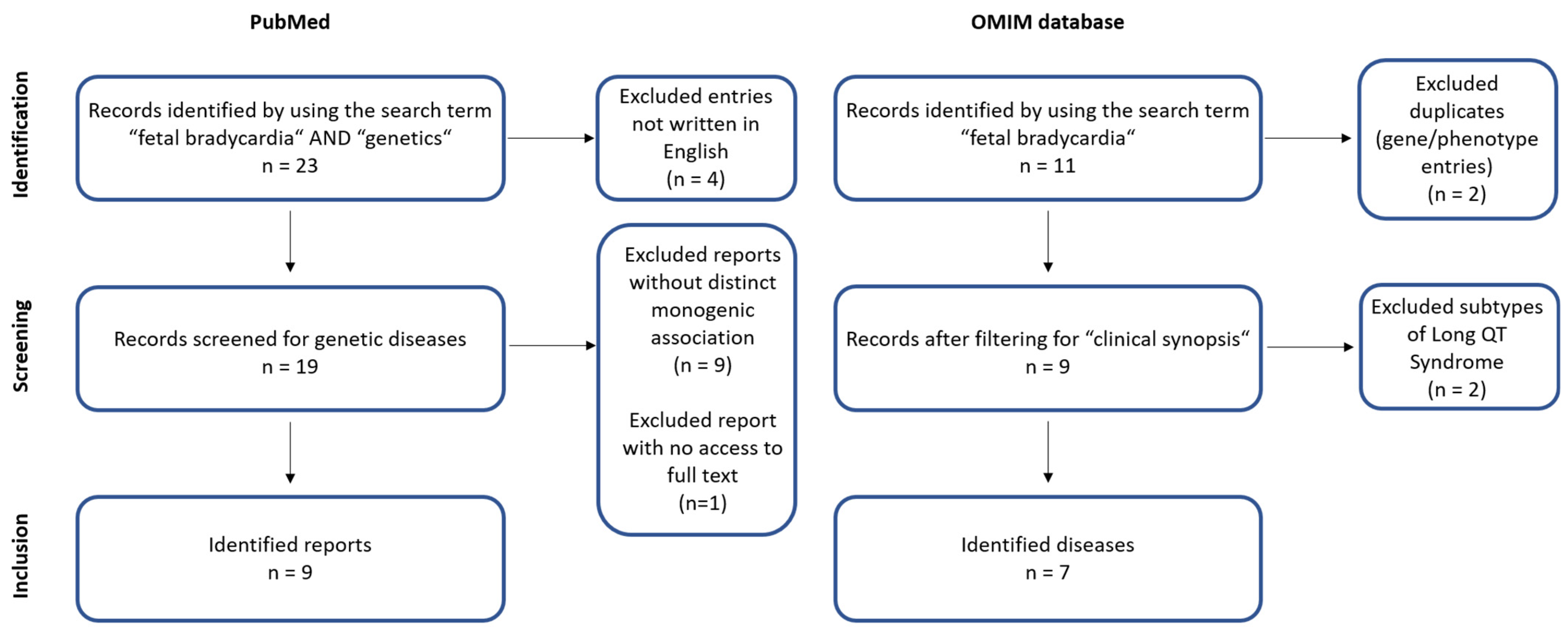
2. Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Macones, G.; Blackwell, S.; Moore, T.; Spong, C.; Hauth, J.; Hankins, G. Management of intrapartum fetal heart rate tracings: ACOG practice bulletin no. 116. Obstet. Gynecol. 2010, 116, 1232–1240. [Google Scholar]
- Widnes, C.; Flo, K.; Wilsgaard, T.; Kiserud, T.; Acharya, G. Sex differences in umbilical artery Doppler indices: A longitudinal study. Biol. Sex Differ. 2018, 9, 16. [Google Scholar] [CrossRef] [Green Version]
- Wacker-Gussmann, A.; Wakai, R.T.; Strasburger, J.F. Importance of Fetal Arrhythmias to the Neonatologist and Pediatrician. Neoreviews 2016, 17, e568–e578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, A.W.; Snijders, R.; Geerts, L.; Spencer, K.; Nicolaides, K.H. Fetal heart rate in chromosomally abnormal fetuses. Ultrasound. Obstet. Gynecol. 2000, 16, 610–613. [Google Scholar] [CrossRef]
- Wacker-Gussmann, A.; Strasburger, J.F.; Cuneo, B.F.; Wakai, R.T. Diagnosis and treatment of fetal arrhythmia. Am. J. Perinatol. 2014, 31, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Low, J.A.; Cox, M.J.; Karchmar, E.J.; McGrath, M.J.; Pancham, S.R.; Piercy, W.N. The prediction of intrapartum fetal metabolic acidosis by fetal heart rate monitoring. Am. J. Obstet. Gynecol. 1981, 139, 299–305. [Google Scholar] [CrossRef]
- Mitchell, J.L.; Cuneo, B.F.; Etheridge, S.P.; Horigome, H.; Weng, H.Y.; Benson, D.W. Fetal heart rate predictors of long QT syndrome. Circulation 2012, 126, 2688–2695. [Google Scholar] [CrossRef] [PubMed]
- Strand, S.; Strasburger, J.F.; Cuneo, B.F.; Wakai, R.T. Complex and Novel Arrhythmias Precede Stillbirth in Fetuses With De Novo Long QT Syndrome. Circ. Arrhythm. Electrophysiol. 2020, 13, e008082. [Google Scholar] [CrossRef] [PubMed]
- Lupoglazoff, J.M.; Denjoy, I.; Villain, E.; Fressart, V.; Simon, F.; Bozio, A.; Berthet, M.; Benammar, N.; Hainque, B.; Guicheney, P. Long QT syndrome in neonates: Conduction disorders associated with HERG mutations and sinus bradycardia with KCNQ1 mutations. J. Am. Coll. Cardiol. 2004, 43, 826–830. [Google Scholar] [CrossRef] [Green Version]
- Cuneo, B.F.; Strasburger, J.F.; Yu, S.; Horigome, H.; Hosono, T.; Kandori, A.; Wakai, R.T. In utero diagnosis of long QT syndrome by magnetocardiography. Circulation 2013, 128, 2183–2191. [Google Scholar] [CrossRef] [Green Version]
- Vrtel, R.; Verhoef, S.; Bouman, K.; Maheshwar, M.M.; Nellist, M.; van Essen, A.J.; Bakker, P.L.; Hermans, C.J.; Bink-Boelkens, M.T.; van Elburg, R.M.; et al. Identification of a nonsense mutation at the 5’ end of the TSC2 gene in a family with a presumptive diagnosis of tuberous sclerosis complex. J. Med. Genet. 1996, 33, 47–51. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.A.; Fong, J.C.; Basson, C.T. Holt-Oram Syndrome. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Sun, H.; Liu, X.; Hao, X.; Wang, J.; Han, J.; Liang, M.; Zhang, H.; He, Y. Case Report: Biventricular Noncompaction Cardiomyopathy With Pulmonary Stenosis and Bradycardia in a Fetus With KCNH2 Mutation. Front. Genet. 2022, 24, 821226. [Google Scholar] [CrossRef] [PubMed]
- Wacker-Gussmann, A.; Oberhoffer-Fritz, R.; Westphal, D.S.; Hessling, G.; Wakai, R.T.; Strasburger, J.F. The missense variant p.(Gly482Arg) in HCN4 is responsible for fetal tachy-bradycardia syndrome. HeartRhythm Case Rep. 2020, 6, 352–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, L.; Wakai, R.; Tsao, S.; Strasburger, J.; Gotteiner, N.; Patel, A. Fetal diagnosis of KCNQ1-variant long QT syndrome using fetal echocardiography and magnetocardiography. Pacing Clin. Electrophysiol. 2020, 43, 430–433. [Google Scholar] [CrossRef] [PubMed]
- Gusic, M.; Schottmann, G.; Feichtinger, R.G.; Du, C.; Scholz, C.; Wagner, M.; Mayr, J.A.; Lee, C.Y.; Yepez, V.A.; Lorenz, N.; et al. Bi-Allelic UQCRFS1 Variants Are Associated with Mitochondrial Complex III Deficiency, Cardiomyopathy, and Alopecia Totalis. Am. J. Hum. Genet. 2020, 106, 102–111. [Google Scholar] [CrossRef]
- Sepp, R.; Hategan, L.; Bácsi, A.; Cseklye, J.; Környei, L.; Borbás, J.; Széll, M.; Forster, T.; Nagy, I.; Hegedus, Z. Timothy syndrome 1 genotype without syndactyly and major extracardiac manifestations. Am. J. Med. Genet. A 2017, 173, 784–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoosien, M.; Ahearn, M.E.; Myerburg, R.J.; Pham, T.V.; Miller, T.E.; Smets, M.J.; Baumbach-Reardon, L.; Young, M.-L.; Farooq, A.; Bishopric, N.H. Dysfunctional potassium channel subunit interaction as a novel mechanism of long QT syndrome. Heart Rhythm. 2013, 10, 728–737. [Google Scholar] [CrossRef] [Green Version]
- Schneider, U.; Haueisen, J.; Loeff, M.; Bondarenko, N.; Schleussner, E. Prenatal diagnosis of a long QT syndrome by fetal magnetocardiography in an unshielded bedside environment. Prenat. Diagn. 2005, 25, 704–708. [Google Scholar] [CrossRef]
- Chang, C.-C.; Acharfi, S.; Wu, M.-H.; Chiang, F.-T.; Wang, J.-K.; Sung, T.-C.; Chahine, M. A novel SCN5A mutation manifests as a malignant form of long QT syndrome with perinatal onset of tachycardia/bradycardia. Cardiovasc. Res. 2004, 64, 268–278. [Google Scholar] [CrossRef] [Green Version]
- Tester, D.J.; McCormack, J.; Ackerman, M.J. Prenatal molecular genetic diagnosis of congenital long QT syndrome by strategic genotyping. Am. J. Cardiol. 2004, 93, 788–791. [Google Scholar] [CrossRef]
- Alders, M.; Bikker, H.; Christiaans, I. Long QT Syndrome. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Krause, U.; Gravenhorst, V.; Kriebel, T.; Ruschewski, W.; Paul, T. A rare association of long QT syndrome and syndactyly: Timothy syndrome (LQT 8). Clin. Res. Cardiol. 2011, 100, 1123–1127. [Google Scholar] [CrossRef]
- Brackley, K.J.; Farndon, P.A.; Weaver, J.B.; Dow, D.J.; Chapman, S.; Kilby, M.D. Prenatal diagnosis of tuberous sclerosis with intracerebral signs at 14 weeks’ gestation. Prenat. Diagn. 1999, 19, 575–579. [Google Scholar] [CrossRef]
- Regalado, J.J.; Rodriguez, M.M.; Ferrer, P.L. Infantile hypertrophic cardiomyopathy of glycogenosis type IX: Isolated cardiac phosphorylase kinase deficiency. Pediatr. Cardiol. 1999, 20, 304–307. [Google Scholar] [CrossRef]
- Friederich, M.W.; Timal, S.; Powell, C.A.; Dallabona, C.; Kurolap, A.; Palacios-Zambrano, S.; Bratkovic, D.; Derks, T.G.J.; Bick, D.; Bouman, K.; et al. Pathogenic variants in glutamyl-tRNA(Gln) amidotransferase subunits cause a lethal mitochondrial cardiomyopathy disorder. Nat. Commun. 2018, 9, 4065. [Google Scholar] [CrossRef]
- Muru, K.; Kalev, I.; Teek, R.; Sonajalg, M.; Kuuse, K.; Reimand, T.; Ounap, K. A Boy with Holt-Oram Syndrome Caused by Novel Mutation c.1304delT in the TBX5 Gene. Mol. Syndromol. 2011, 1, 307–310. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Lahrouchi, N.; Tadros, R.; Crotti, L.; Mizusawa, Y.; Postema, P.G.; Beekman, L.; Walsh, R.; Hasegawa, K.; Barc, J.; Ernsting, M.; et al. Transethnic Genome-Wide Association Study Provides Insights in the Genetic Architecture and Heritability of Long QT Syndrome. Circulation 2020, 142, 324–338. [Google Scholar] [CrossRef]
- Xie, T.; Wang, B.; Nolte, I.M.; van der Most, P.J.; Oldehinkel, A.J.; Hartman, C.A.; Snieder, H. Genetic Risk Scores for Complex Disease Traits in Youth. Circ. Genom. Precis. Med. 2020, 13, e002775. [Google Scholar] [CrossRef] [PubMed]
- Jervell, A.; Lange-Nielsen, F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am. Heart J. 1957, 54, 59–68. [Google Scholar] [CrossRef]
- Bauer, R.; Timothy, K.W.; Golden, A. Update on the Molecular Genetics of Timothy Syndrome. Front. Pediatr. 2021, 9, 668546. [Google Scholar] [CrossRef]
- Schweizer, P.A.; Schroter, J.; Greiner, S.; Haas, J.; Yampolsky, P.; Mereles, D.; Buss, S.J.; Seyler, C.; Bruehl, C.; Draguhn, A.; et al. The symptom complex of familial sinus node dysfunction and myocardial noncompaction is associated with mutations in the HCN4 channel. J. Am. Coll. Cardiol. 2014, 64, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Milano, A.; Vermeer, A.M.; Lodder, E.M.; Barc, J.; Verkerk, A.O.; Postma, A.V.; van der Bilt, I.A.; Baars, M.J.; van Haelst, P.L.; Caliskan, K.; et al. HCN4 mutations in multiple families with bradycardia and left ventricular noncompaction cardiomyopathy. J. Am. Coll. Cardiol. 2014, 64, 745–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donofrio, M.T.; Moon-Grady, A.J.; Hornberger, L.K.; Copel, J.A.; Sklansky, M.S.; Abuhamad, A.; Cuneo, B.F.; Huhta, J.C.; Jonas, R.A.; Krishnan, A.; et al. Diagnosis and treatment of fetal cardiac disease: A scientific statement from the American Heart Association. Circulation 2014, 129, 2183–2242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strasburger, J.F.; Wakai, R.T. Fetal cardiac arrhythmia detection and in utero therapy. Nat. Rev. Cardiol. 2010, 7, 277–290. [Google Scholar] [CrossRef] [Green Version]
- Miyake, A.; Sakaguchi, H.; Miyazaki, A.; Miyoshi, T.; Aiba, T.; Shiraishi, I. Successful prenatal management of ventricular tachycardia and second-degree atrioventricular block in fetal long QT syndrome. HeartRhythm Case Rep. 2017, 3, 53–57. [Google Scholar] [CrossRef] [Green Version]
- Austin, S.L.; Chiou, A.; Sun, B.; Case, L.E.; Govendrageloo, K.; Hansen, P.; Kishnani, P.S. Alglucosidase alfa enzyme replacement therapy as a therapeutic approach for a patient presenting with a PRKAG2 mutation. Mol. Genet. Metab. 2017, 120, 96–100. [Google Scholar] [CrossRef]


{kind=link}
| Name | Type | Reference |
|---|---|---|
| Case Report: Biventricular Noncompaction Cardiomyopathy With Pulmonary Stenosis and Bradycardia in a Fetus With KCNH2 Mutation | Case report | [13] |
| The missense variant p.(Gly482Arg) in HCN4 is responsible for fetal tachy-bradycardia syndrome | Case report | [14] |
| Fetal diagnosis of KCNQ1-variant long QT syndrome using fetal echocardiography and magnetocardiography | Case report | [15] |
| Bi-Allelic UQCRFS1 Variants Are Associated with Mitochondrial Complex III Deficiency, Cardiomyopathy, and Alopecia Totalis | Case report | [16] |
| Timothy syndrome 1 genotype without syndactyly and major extracardiac manifestations | Review | [17] |
| Dysfunctional potassium channel subunit interaction as a novel mechanism of long QT syndrome | Original article | [18] |
| Prenatal diagnosis of a long QT syndrome by fetal magnetocardiography in an unshielded bedside environment | Case report | [19] |
| A novel SCN5A mutation manifests as a malignant form of long QT syndrome with perinatal onset of tachycardia/bradycardia | Case report | [20] |
| Prenatal molecular genetic diagnosis of congenital long QT syndrome by strategic genotyping | Case report | [21] |
| Primary/Secondary Bradycardia | Associated Disease | Gene(s) | Inheritance | Further Prenatal Manifestations |
|---|---|---|---|---|
| Primary | Long QT Syndrome | KCNQ1, KCNH2, SCN5A * | AD, AR | AV block, prolonged QTc [22], syndactyly in Timothy Syndrome [23] |
| Sick Sinus Syndrome | HCN4, SCN5A | AD, AR | atrial flutter, prolonged QTc [14] | |
| Short QT Syndrome | KCNQ1, KCNH2, KCNJ2 | AD | not reported | |
| Holt Oram Syndrome | TBX5 | AD | structural heart defects (e.g., VSD), skeletal abnormalities (e.g., upper-limb malformations) [12] | |
| Tuberous sclerosis | TSC1, TSC2 | AD | neuronal migration disorder [24], cardiac rhabdomyosarcoma [11] | |
| Secondary | Lethal congenital glycogen storage disease of heart | PRKAG2 | AD | hypertrophic cardiomyopathy [25] |
| Combined oxidative phosphorylation deficiency, type 41 | GATB | AR | cardiomegaly, fetal hydrops [26] | |
| Familial erythrocytosis, type 2 | VHL | AR | not reported | |
| Nuclear mitochondrial complex III deficiency, type 10 | UQCRFS1 | AR | IUGR [16] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Westphal, D.S.; Hauser, M.; Beckmann, B.-M.; Wolf, C.M.; Hessling, G.; Oberhoffer-Fritz, R.; Wacker-Gussmann, A. Fetal Bradycardia Caused by Monogenic Disorders—A Review of the Literature. J. Clin. Med. 2022, 11, 6880. https://doi.org/10.3390/jcm11236880
Westphal DS, Hauser M, Beckmann B-M, Wolf CM, Hessling G, Oberhoffer-Fritz R, Wacker-Gussmann A. Fetal Bradycardia Caused by Monogenic Disorders—A Review of the Literature. Journal of Clinical Medicine. 2022; 11(23):6880. https://doi.org/10.3390/jcm11236880
Chicago/Turabian StyleWestphal, Dominik S., Michael Hauser, Britt-Maria Beckmann, Cordula M. Wolf, Gabriele Hessling, Renate Oberhoffer-Fritz, and Annette Wacker-Gussmann. 2022. "Fetal Bradycardia Caused by Monogenic Disorders—A Review of the Literature" Journal of Clinical Medicine 11, no. 23: 6880. https://doi.org/10.3390/jcm11236880