
Synthesis, Structure and Bonding in Pentagonal Bipyramidal Cluster Compounds Containing a cyclo-Sn5 Ring, [(CO)3MSn5M(CO)3]4− (M = Cr, Mo)



Abstract
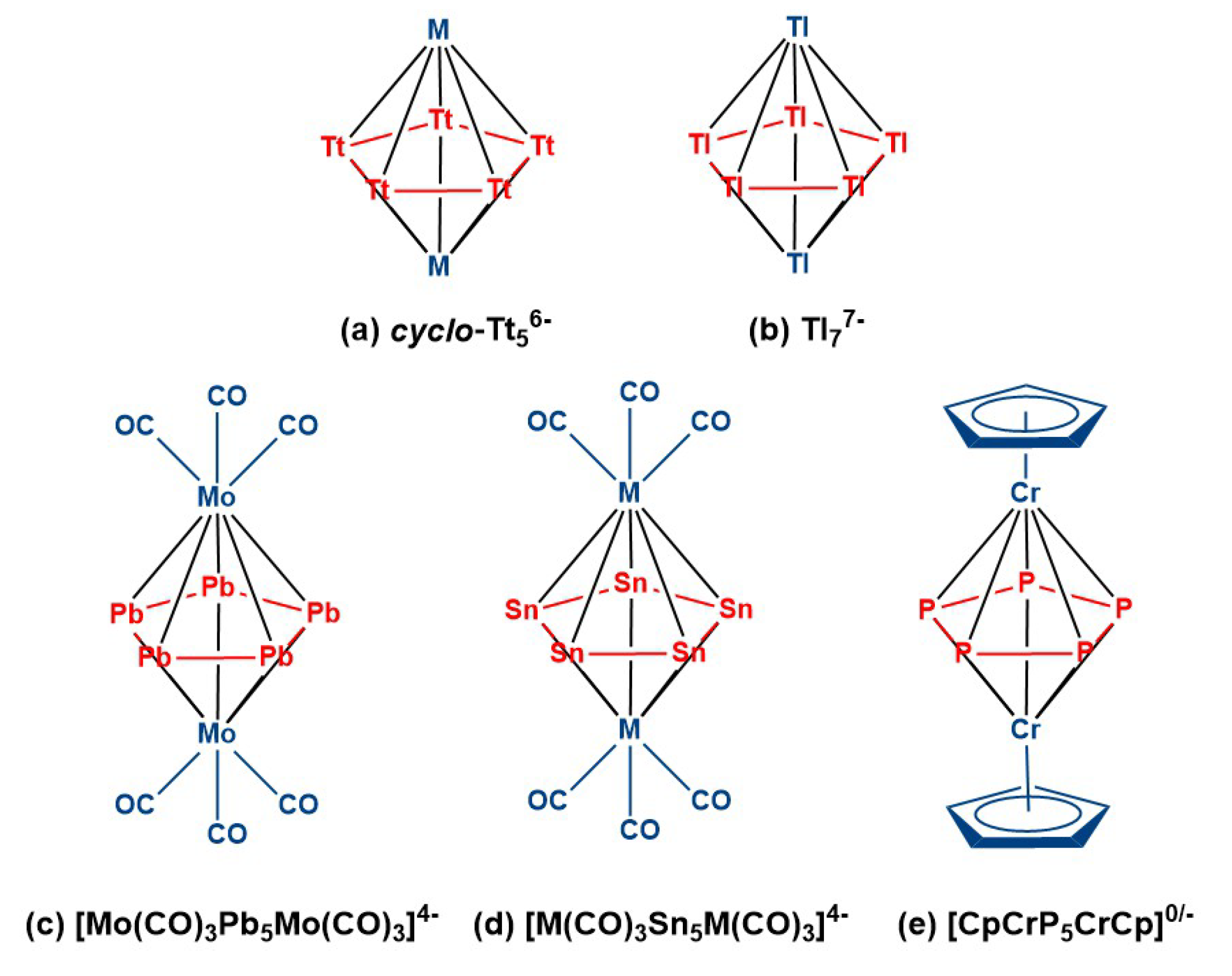
:1. Introduction
2. Materials and Methods
2.1. Synthesis of K2(en)3[K([2.2.2]-crypt)]2[Cr(CO)3Sn5Cr(CO)3].2en, (1)
2.2. Synthesis of K[K(18-crown-6)]3[Mo(CO)3Sn5Mo(CO)3] (2)
2.3. X-ray Diffraction
2.4. Electrospray Ionisation Mass Spectrometry (ESI-MS)
2.5. Energy Dispersive X-ray (EDX) Analysis
2.6. Computational Details
3. Results
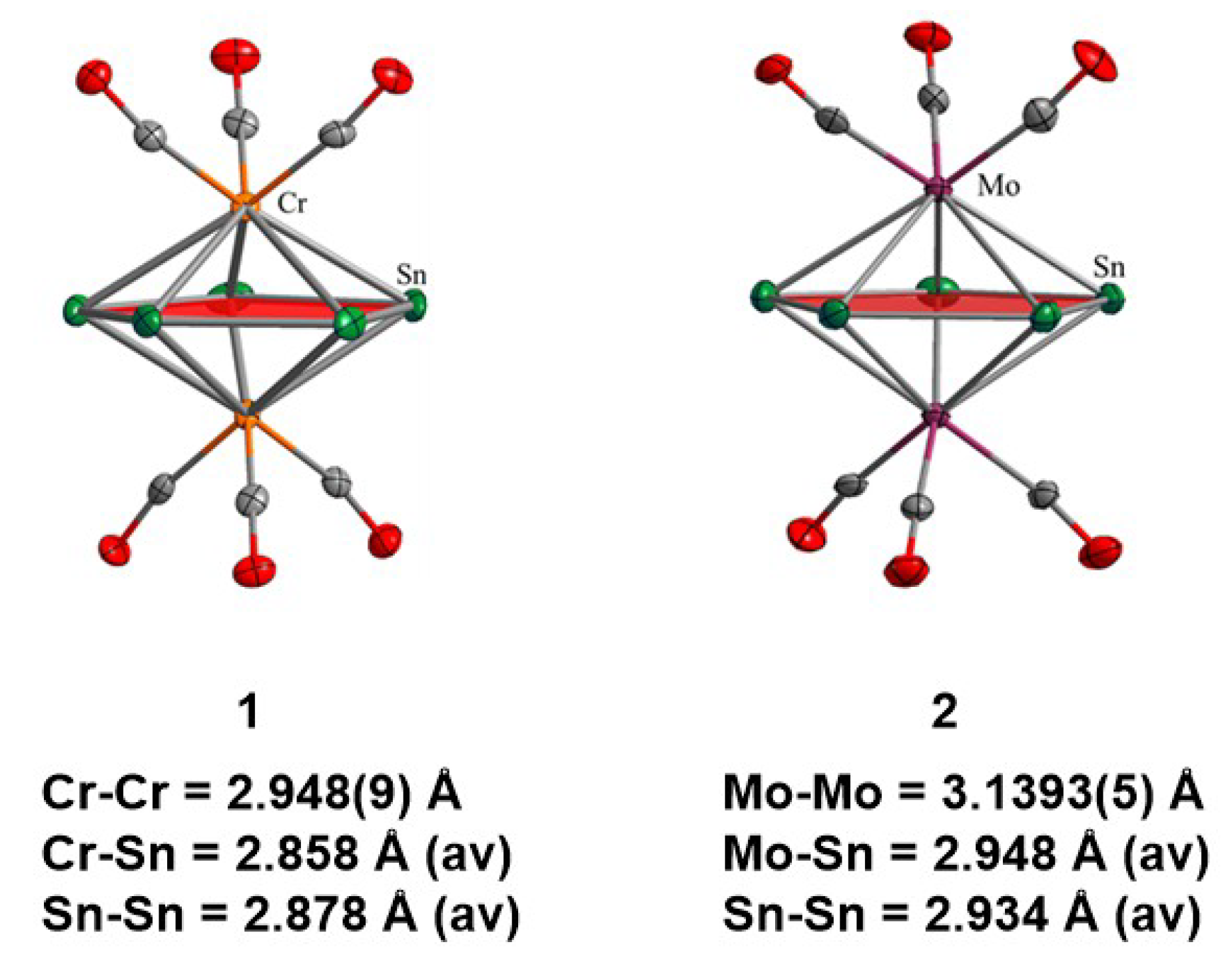
3.1. Synthesis and Structural Characterisation
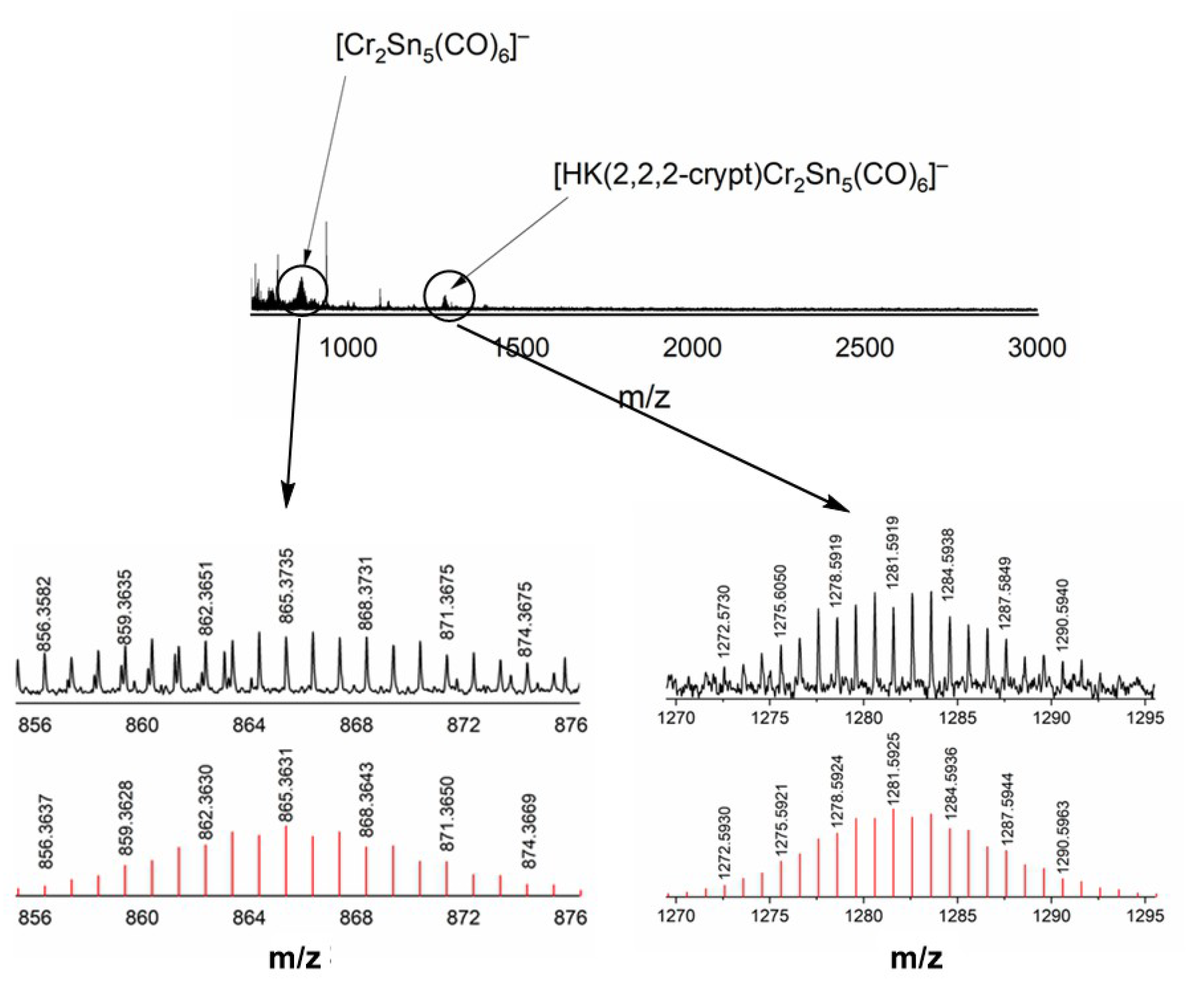
3.2. ESI-MS
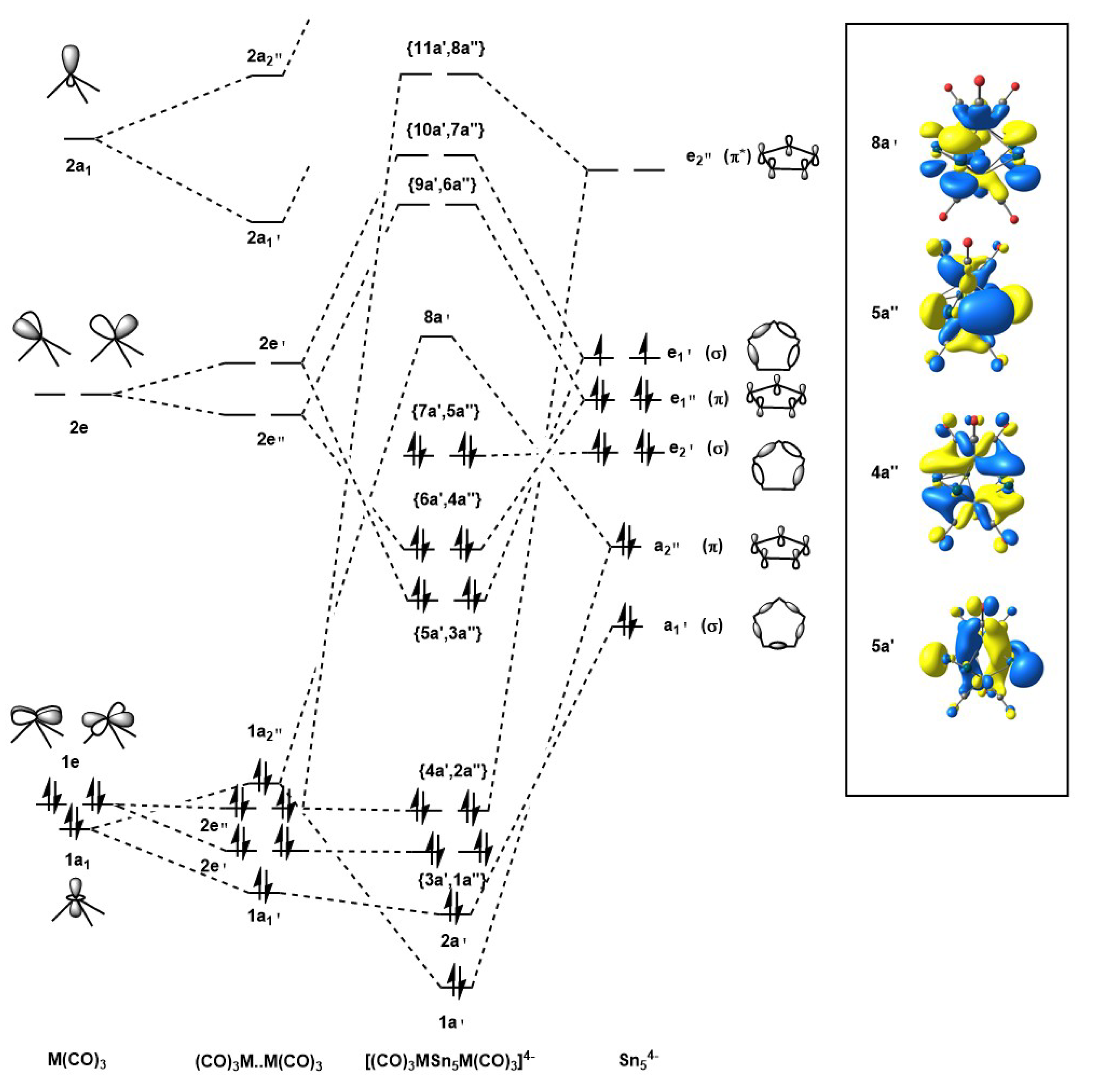
3.3. Electronic Structure Analysis
4. Discussion
Electron-Counting and Metal–Metal Bonding in Pentagonal Bipyramidal Clusters
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DFT | Density Functional Theory |
| QTAIM | Quantum Theory of Atoms in Molecules |
References
- Scherer, O.J. Complexes with Substituent-free Acyclic and Cyclic Phosphorus, Arsenic, Antimony, and Bismuth Ligands. Angew. Chem. Int. Ed. Engl. 1990, 29, 1104–1122. [Google Scholar] [CrossRef]
- Turbervill, R.S.P.; Goicoechea, J.M. From Clusters to Unorthodox Pnictogen Sources: Solution-Phase Reactivity of [E7]3– (E = P–Sb) Anions. Chem. Rev. 2014, 114, 10807–10828. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, W.; Sun, Z.; Meng, L.; Li, X. Construction of Pn10M Sandwich Compounds from Pn5− and Pn5M (Pn = N-Bi; M = Li, Na, K, Be, Mg, Ca, Fe, Co and Ni): A Theoretical Assessment. Comp. Theor. Chem. 2016, 1098, 50–55. [Google Scholar] [CrossRef]
- Dillon, K.B.; Mathey, F.; Nixon, J.F. Phosphorus: The Carbon Copy, 1st ed.; Wiley: New York, NY, USA, 1998. [Google Scholar]
- Simpson, M.C.; Protasiewicz, J.D. Phosphorus as a Carbon Copy and as a Photocopy: New Conjugated Materials Featuring Multiply Bonded Phosphorus. Pure App. Chem. 2013, 85, 801–815. [Google Scholar] [CrossRef]
- Heinl, C.; Peresypkina, E.; Balázs, G.; Mädl, E.; Virovets, A.V.; Scheer, M. The Missing Parent Compound [(C5H5)Fe(η5−P5)]: Synthesis, Characterization, Coordination Behavior and Encapsulation. Chem. Eur. J. 2021, 27, 7542–7548. [Google Scholar] [CrossRef] [PubMed]
- Todorov, I.; Sevov, S.C. Heavy-Metal Aromatic Rings: Cyclopentadienyl Anion Analogues Sn56− and Pb56− in the Zintl Phases Na8BaPb6, Na8BaSn6, and Na8EuSn6. Inorg. Chem. 2004, 43, 6490–6494. [Google Scholar] [CrossRef] [PubMed]
- von Schnering, H.G.; Nesper, R.; Curda, J.; Tebbe, K.F. Li12Si7, a Compound Having a Trigonal Planar Si4 Cluster and Planar Si5 Rings. Angew. Chem. Int. Ed. Engl. 1980, 19, 1033–1034. [Google Scholar] [CrossRef]
- Nesper, R.; Curda, J.; Von Schnering, H. Li8MgSi6, a Novel Zintl Compound Containing Quasi-Aromatic Si5 rings. J. Solid State Chem. 1986, 62, 199–206. [Google Scholar] [CrossRef]
- Frank, U.; Müller, W. Li11Ge6—A Phase with Isolated, Plane, Five-Membered Ge-Rings. Z. Naturf. B 1975, 30, 313–315. [Google Scholar] [CrossRef] [Green Version]
- Wade, K. Structural Significance of Number of Skeletal Bonding Electron-Pairs in Carboranes, Higher Boranes and Borane Anions, and Various Transition-Metal Carbonyl Cluster Compounds. J. Chem. Soc. D Chem. Comm. 1971, 15, 792–793. [Google Scholar] [CrossRef]
- Kaskel, S.; Corbett, J.D. Synthesis and Structure of K10Tl7: The First Binary Trielide Containing Naked Pentagonal Bipyramidal Tl7 Clusters. Inorg. Chem. 2000, 39, 778–782. [Google Scholar] [CrossRef]
- Yong, L.; Hoffmann, S.D.; Fässler, T.F.; Riedel, S.; Kaupp, M. [Pb5[Mo(CO)3]2]4−: A complex containing a planar Pb5 unit. Ang. Chem. Int. Ed. Engl. 2005, 44, 2092–2096. [Google Scholar] [CrossRef]
- Scherer, O.J.; Schwalb, J.; Wolmershäuser, G.; Kaim, W.; Gross, R. cyclo-P5 as Complex Ligand—The Phosphorus Analogue of the Cyclopentadienyl Ligand. Angew. Chem. Int. Ed. Engl. 1986, 25, 363–364. [Google Scholar] [CrossRef]
- Armstrong, R.S.; Aroney, M.J.; Barnes, C.M.; Nugent, K.W. Infrared and Raman Spectra of (η6−mesitylene)M(CO)3 Complexes (M = Cr, Mo or W): An Insight Into Metal-Arene Bonding. App. Organomet. Chem. 1990, 4, 569–580. [Google Scholar] [CrossRef]
- Gholiee, Y.; Salehzadeh, S.; Khodaveisi, S. Significant Geometry and Charge Difference Between the E54− Bare Clusters of Group 14 Zintl anions and Their Coordinated Form in [E5[M(CO)3]2]4− (E = Si, Ge, Sn, Pb; M = Cr, Mo, W) complexes. New J. Chem. 2019, 43, 7797–7805. [Google Scholar] [CrossRef]
- Tate, D.P.; Knipple, W.R.; Augl, J.M. Nitrile Derivatives of Chromium Group Metal Carbonyls. Inorg. Chem. 1962, 1, 433–434. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. Section A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. App. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON SQUEEZE: A Tool for the Calculation of the Disordered Solvent Contribution to the Calculated Structure Factors. Acta Cryst. Section C 2015, 71, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Neese, F. The ORCA Program System. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software Update: The ORCA Program System, Version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [Green Version]
- Hertwig, R.H.; Koch, W. On the Parameterization of the Local Correlation Functional. What is Becke-3-LYP? Chem. Phys. Lett. 1997, 268, 345–351. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Grimme, S. Semiempirical Hybrid Density Functional with Perturbative Second-Order Correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef] [Green Version]
- Neese, F.; Schwabe, T.; Grimme, S. Analytic Derivatives for Perturbatively Corrected “Double Hybrid” Density Functionals: Theory, Implementation, and Applications. J. Chem. Phys. 2007, 126, 124115. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic Regular Two-Component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic Total Energy Using Regular Approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar] [CrossRef]
- van Lenthe, E.; Ehlers, A.; Baerends, E.J. Geometry Optimizations in the Zero Order Regular Approximation for Relativistic Effects. J. Chem. Phys. 1999, 110, 8943–8953. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Rolfes, J.D.; Neese, F.; Pantazis, D.A. All-Electron Scalar Relativistic Basis Sets for the Elements Rb–Xe. J. Comp. Chem. 2020, 41, 1842–1849. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Neese, F. All-Electron Scalar Relativistic Basis Sets for the 6p Elements. Theor. Chem. Acc. 2012, 131, 1292. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Klamt, A.; Schüürmann, G. COSMO: A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and its Gradient. J. Chem. Soc. Perkin Trans. 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Andzelm, J.; Kölmel, C.; Klamt, A. Incorporation of Solvent Effects into Density Functional Calculations of Molecular Energies and Geometries. J. Chem. Phys. 1995, 103, 9312–9320. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, Structures, and Electronic Properties of Molecules in Solution with the C-PCM Solvation Model. J. Comp. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comp. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Zhurko G., A. Chemcraft—Graphical Program for Visualization of Quantum Chemistry Computations. Ivanovo, Russia. 2005. Available online: https://chemcraftprog.com (accessed on 15 November 2021).
- Kesanli, B.; Fettinger, J.; Eichhorn, B. The closo-[Sn9M(CO)3]4− Zintl Ion Clusters where M = Cr, Mo, W: Two Structural Isomers and Their Dynamic Behavior. Chem. Eur. J. 2001, 7, 5277–5285. [Google Scholar] [CrossRef]
- Handy, L.B.; Ruff, J.K.; Dahl, L.F. Structural Characterization of the Dinuclear Metal Carbonyl Anions [M2(CO)10]2− (M= chromium, molybdenum) and [Cr2(CO)10H]−. Marked Stereochemical Effect of a Linearly Protonated Metal-Metal Bond. J. Am. Chem. Soc. 1970, 92, 7312–7326. [Google Scholar] [CrossRef]
- Green, J.C.; Green, M.L.H.; Parkin, G. The Occurrence and Representation of Three-Centre Two-Electron Bonds in Covalent Inorganic Compounds. Chem. Commun. 2012, 48, 11481–11503. [Google Scholar] [CrossRef]
- Labinger, J.A. Does Cyclopentadienyl Iron Dicarbonyl Dimer Have a Metal–Metal bond? Who’s Asking? Inorg. Chim. Acta 2015, 424, 14–19. [Google Scholar] [CrossRef]
- McGrady, J.E. Molecular Metal-Metal Bonds: Compounds, Synthesis, Properties; Wiley: New York, NY, USA, 1999. [Google Scholar]
- Spivak, M.; Arcisauskaite, V.; López, X.; McGrady, J.E.; de Graaf, C. A Multi-Configurational Approach to the Electronic Structure of Trichromium Extended Metal Atom Chains. Dalton Trans. 2017, 46, 6202–6211. [Google Scholar] [CrossRef] [Green Version]
- Lichtenberger, N.; Franzke, Y.J.; Massa, W.; Weigend, F.; Dehnen, S. The Identity of “Ternary” A/Tl/Pb or K/Tl/Bi Solid Mixtures and Binary Zintl Anions Isolated From Their Solutions. Chem. Eur. J. 2018, 24, 12022–12030. [Google Scholar] [CrossRef]
- McGrady, J.E. A Unified Approach to Electron Counting in Main-Group Clusters. J. Chem. Ed. 2004, 81, 733. [Google Scholar] [CrossRef]
- Goh, L.Y.; Wong, R.C.S.; Chu, C.K.; Hambley, T.W. Reaction of [(Cr(cp)(CO)3)2] (cp = η5-C5H5) with Elemental Phosphorus. Isolation of [Cr2(cp)2(P5)] as a Thermolysis Product and its X-ray Crystal Structure. J. Chem. Soc. Dalton Trans. 1990, 3, 977–982. [Google Scholar]
- Tremel, W.; Hoffmann, R.; Kertesz, M. Inorganic Rings, Intact and Cleaved, Between Two Metal Fragments. J. Am. Chem. Soc. 1989, 111, 2030–2039. [Google Scholar] [CrossRef]
- Lepetit, C.; Fau, P.; Fajerwerg, K.; Kahn, M.L.; Silvi, B. Topological Analysis of the Metal-Metal Bond: A Tutorial Review. Coord. Chem. Rev. 2017, 345, 150–181. [Google Scholar] [CrossRef] [Green Version]
- Macchi, P.; Proserpio, D.M.; Sironi, A. Experimental Electron Density in a Transition Metal Dimer: Metal-Metal and Metal-Ligand Bonds. J. Am. Chem. Soc. 1998, 120, 13429–13435. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
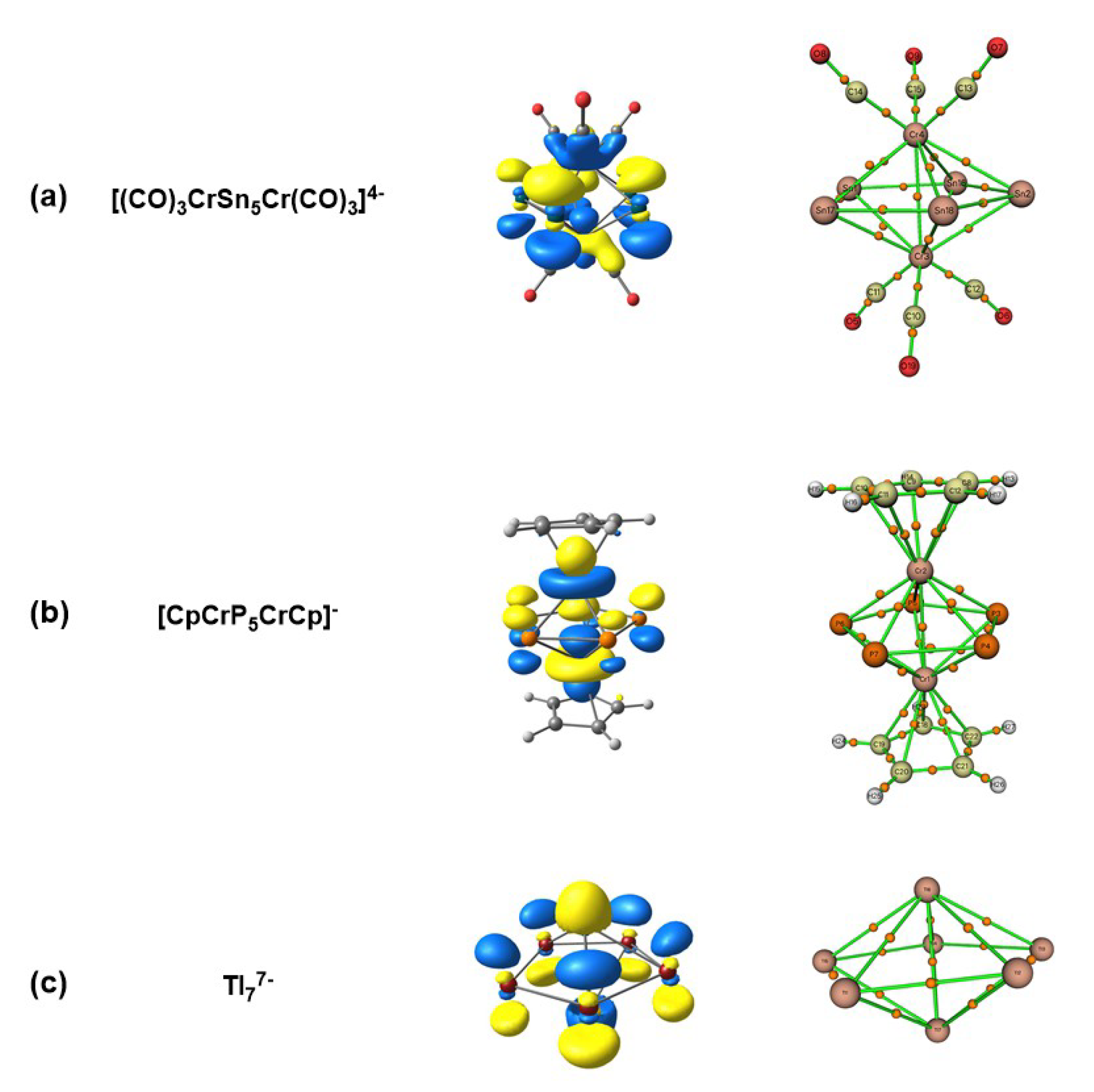{kind=link}
| M-M | M-Tt | Tt-Tt | ||
|---|---|---|---|---|
| [(CO)3CrSn5Cr(CO)3]4− | X-ray | 2.95 | 2.86 | 2.88 |
| PBE | 2.97 | 2.88 | 2.91 | |
| B3LYP | 3.07 | 2.91 | 2.91 | |
| M062X | 2.97 | 2.86 | 2.88 | |
| B2PLYP | 2.85 | 2.87 | 2.93 | |
| [(CO)3MoSn5Mo(CO)3]4− | X-ray | 3.14 | 2.95 | 2.93 |
| PBE | 3.19 | 2.99 | 2.98 | |
| B3LYP | 3.27 | 3.02 | 2.98 | |
| M062X | 3.20 | 2.97 | 2.95 | |
| B2PLYP | 3.15 | 2.98 | 2.97 | |
| [(CO)3MoPb5Mo(CO)3]4− | X-ray | 3.21 | 3.05 | 3.04 |
| PBE | 3.22 | 3.08 | 3.09 | |
| B3LYP | 3.31 | 3.11 | 3.10 | |
| B2PLYP | 3.17 | 3.06 | 3.09 |
| [(CO)3CrSn5Cr(CO)3]4− | [CpCrP5CrCp]− | Tl77− | |
|---|---|---|---|
| r/Å | 2.97 | 2.62 | 3.32 |
| BO | 0.39 | 0.72 | 0.31 |
| 0.40 | 0.93 | 0.52 | |
| /au | 0.028 | 0.055 | 0.020 |
| /au | 0.010 | 0.026 | 0.009 |
| /au | −0.015 | −0.045 | −0.010 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mondal, S.; Chen, W.-X.; Sun, Z.-M.; McGrady, J.E. Synthesis, Structure and Bonding in Pentagonal Bipyramidal Cluster Compounds Containing a cyclo-Sn5 Ring, [(CO)3MSn5M(CO)3]4− (M = Cr, Mo). Inorganics 2022, 10, 75. https://doi.org/10.3390/inorganics10060075
Mondal S, Chen W-X, Sun Z-M, McGrady JE. Synthesis, Structure and Bonding in Pentagonal Bipyramidal Cluster Compounds Containing a cyclo-Sn5 Ring, [(CO)3MSn5M(CO)3]4− (M = Cr, Mo). Inorganics. 2022; 10(6):75. https://doi.org/10.3390/inorganics10060075
Chicago/Turabian StyleMondal, Sourav, Wei-Xing Chen, Zhong-Ming Sun, and John E. McGrady. 2022. "Synthesis, Structure and Bonding in Pentagonal Bipyramidal Cluster Compounds Containing a cyclo-Sn5 Ring, [(CO)3MSn5M(CO)3]4− (M = Cr, Mo)" Inorganics 10, no. 6: 75. https://doi.org/10.3390/inorganics10060075