
Bone Marrow: The Central Immune System


Immune-Oncological Center Cologne (IOZK), D-50674 Cologne, Germany
Immuno 2023, 3(3), 289-329; https://doi.org/10.3390/immuno3030019
Submission received: 15 April 2023
/
Revised: 24 July 2023
/
Accepted: 28 July 2023
/
Published: 3 August 2023
(This article belongs to the Section Cancer Immunology and Immunotherapy)
Abstract
:Bone marrow is known as the site of hematopoiesis. What is not being described in textbooks of immunology is the fact that bone marrow is not only a generative, but also an antigen-responsive, immune organ. It is also a major storage site for antigen-specific memory B and T cells. That bone marrow is a priming site for T cell responses to blood borne antigens was discovered exactly 20 years ago. This review celebrates this important discovery. The review provides a number of examples of medical relevance of bone marrow as a central immune system, including cancer, microbial infections, autoimmune reactions, and bone marrow transplantation. Bone marrow mesenchymal stem cell-derived stromal cells provide distinct bone marrow niches for stem cells and immune cells. By transmitting anti-inflammatory dampening effects, facilitating wound healing and tissue regeneration mesenchymal stem cells contribute to homeostasis of bone and other tissues. Based on the evidence presented, the review proposes that bone marrow is a multifunctional and protective immune system. In an analogy to the central nervous system, it is suggested that bone marrow be designated as the central immune system.
1. Introduction
It is exclusively in vertebrates that the adaptive immunity system was developed about 500 million years ago [1]. The phylogenetic roots of adaptive immunity go back to the lamprey, a cartilaginous fish, in which nature apparently invented somatic diversification of antigen-specific lymphocyte receptors [2]. New findings have revealed that Permian tetrapods, 60 million years ago, developed a limb-bone growth plate and a centralized marrow cavity organization containing hematopoietic stem cells (HSCs), which program hematopoiesis, the production of blood cells [3]. The growth plate (epiphyseal plate) is a hyaline cartilage plate in the metaphysis at each end of a long bone. It contains mesenchymal stem cells (MSCs), chondrocytes, and matrix, and has a zonal arrangement from the epiphysis to the diaphysis, the zones of ossification. Bone tissue is maintained by a balance between activities of HSC-derived and MSC-derived cells. Osteoclasts (macrophage-like phagocytic cells) derived from HSCs break down and resorb bone, while osteogenic cells from MSCs (osteoblasts and osteocytes) form the bone matrix and maintain mineralized bone tissue.
In developing embryos, blood formation occurs in aggregates of blood cells in the yolk sac. As development progresses, blood formation occurs in the spleen, liver, and lymph nodes. When bone marrow (BM) develops after month four of embryogenesis, it eventually assumes the task of forming most of the blood cells for the entire organism [4]. In the process of osteogenesis, MSCs develop into osteoblasts and begin to synthesize osteoid, an organic component of mainly collagen which, together with inorganic minerals, makes up the extracellular matrix (ECM). In this matrix, the osteoblasts transform into osteocytes and build osteoid braces, which further develop into bone braces. Immediately after birth, hematopoiesis occurs in the BM. HSCs are located in niches mainly formed by MSCs and their descendants, and their function is associated with ECM molecules, hematopoietic cytokines, and chemokines [4].
Bones represent the inner skeleton of vertebrates. They protect various other organs of the body, produce red and white blood cells, store minerals, provide structure and support for the body, and enable mobility. The central cavity of bone, the medulla, is surrounded by a compact outer matrix which is covered by thin tissues, the periosteum at the outer side, and the endosteum at the inner side. These contain MSCs which can always build new osteoblasts if required, thus allowing flexibility in bone reconstruction and repair. The medulla is filled with a spongy structure, the substantia spongiosa. This is made up of supportive strands of connective tissue, called trabeculae, and of soft tissue, the BM. The trabeculae are aligned towards the mechanical load distribution that a bone like the femur experiences. Spongy bone is like a biofoam. It accounts for 20% of total bone mass, but has nearly ten times the surface area of compact bone. BM is vascularized by blood, not by lymphatic vessels [5], and is part of the lymphocyte recirculation network [6]. In support of this, depletion of conditional vascular cell adhesion molecule 1 (VCAM-1) in mice impaired lymphocyte migration to BM [6].
In mammals, B cells develop in the BM at the core of most bones into B cell receptor (BCR)-expressing cells. They further differentiate in the spleen into mature naïve B cells, then become activated and enter a germinal center in the lymph nodes. T cells develop in the BM up to the pre-T cell stage, and then further differentiate in the thymus to T cell receptor (TCR)-expressing mature naïve T cells. Mature B and T lymphocytes express highly diverse antigen-specific receptors. The BCR complex of mature B cells is composed of membrane immunoglobulin (Ig) molecules that bind antigen and associated Igα and Igβ proteins that deliver signals for B cell activation. The T cell TCR complex is a heterodimer consisting of two transmembrane polypeptide chains, TCRα and TCRβ, covalently linked to each other. The TCR-associated signaling molecules are CD3 and ζ [7]. Germline Ig and TCR genes are composed of multiple DNA segments that are spatially separate in all cells and are combined in developing lymphocytes. The total potential repertoire with junctional diversity is for Ig about 1011, and for TCRα,β, about 1016 [7].
The organs of the immune system are traditionally divided into generative or primary lymphoid organs (BM and thymus), where lymphocytes mature, and peripheral or secondary organs (lymph nodes, spleen, and parts of the mucosal immune system), where naïve lymphocytes are activated by antigens [7]. Extracellular fluid (lymph) is constantly drained from the peripheral tissues through lymphatic vessels into lymph nodes, and thereafter reaches the blood via the thoracic duct. This largest lymphatic vessel transports around 75% of the lymph from the entire body. Blood-transported self-antigens (SAs) carried to the BM and thymus are involved in negative selection of potentially self-reactive B or T cells.
The bone includes a series of blood vessels organized in a specific order to provide nutrients, regulatory factors, and oxygen to the cortex and medulla [8]. Blood flow also removes metabolic waste products such as carbon dioxide and acid. Nutrient arteries penetrate to the medulla and connect to the smaller periostal arterial supply to enable perfusion of the cortical bone. The arteries are longitudinally aligned along the diaphysis of long bones and infiltrate into BM via branching to small arterioles. The arterioles progress into endosteal regions in the BM and, at the same time, undergo thinning. The sinusoids lie close to the endosteal regions following arterioles and have a diameter higher than the arteries and arterioles. Sinusoids are surrounded by stromal cells bearing leptin receptors (LepR) and producing high levels of the receptor tyrosine kinase c-kit, the stromal cell derived factor 1 (SDF-1), and the chemokine CXCL-12. BM sinusoids form a capillary network with venous sinusoids, and the latter converge to a large sinus in the BM center [8]. Blood exits the medulla via multiple small veins that penetrate the cortex [9]. Billions of cells per day circulate between the BM and blood [10].
The oxygen concentration in BM is 1–6%. This is too low, but higher than the concentration required to initiate hypoxic responses. Oxygen concentration is at its maximum (6%) around sinusoids, where most of the immune reactions occur, while the lowest concentration (1%) is observed in endosteal regions [8].
BM tissue distributed inside the different long, short, and flat bones constitutes one of the largest organs in humans, accounting for 4–5% of the total body weight (TBW) [10]. In comparison, the entire network of secondary lymphoid organs makes up only 1–1.5% of TBW [9]. BM is the most prominent source of de novo cellular generation, reaching rates of 4–5 × 1011 cells per day in an adult human [10]. Remarkably, at any given moment, 90% of the total cells in the organism originate from or reside in the BM, with anuclear erythrocytes and platelets accounting for the majority [10].
Like the brain, with its network of neurons, the immune system has the capacity to learn and to develop memory. Unlike the brain, with its mostly immobile network of neurons, the immune system is based on a network of mostly mobile cells. These two learning systems are interconnected: autonomic and somatosensory nervous systems regulate the development of immune cells. They have an impact on hematopoiesis as well as on priming, migration, and cytokine production. In reverse, specific immune subsets contribute to homeostatic neural circuits such as those controlling metabolism, hypertension, and the inflammatory reflex [11]. While neuronal synapses transmit electrical impulses directly via gap junctions (about 3.5 nm distance) or indirectly via neurotransmitters (20–40 nm distance), immunological synapses (ISs) (about 13 nm distance) transmit biochemical signals. Three-dimensional micro-anatomical investigations of BM have revealed that Nestin-GFPhi neuron-glial antigen-2 (NG2+) elongated cells run adjacent to arteries and arterioles. Bundles of nonmyelinating Schwann cells ensheathed these adrenergic nerves [10]. A new type of innervation in lymphoid organs was recently suggested. Adrenergic nerve fiber terminals were visualized to end at microtubular and microfilament walls in the cytoplasm of a subset of dendritic cells (DCs) [11].
This review summarizes pioneering work and recent discoveries regarding the immune system of the BM. It presents BM as a hematopoietic organ and also as an antigen-responsive lymphatic organ. The review celebrates 20 years since the discovery of BM as an antigen-responsive organ. Therefore, the pioneering works receive particular detail and attention. New discoveries are included concerning survival niches for HSCs and memory lymphocytes in BM parenchyma, as well as remodeling of the BM compartment upon dietary restriction. One chapter deals with BM T regulatory cells (Tregs), as well as BM DCs, and their role in tissue regulation, microbial infection, and cancer. A final chapter is devoted to BM MSCs and BM stromal cells, and their importance in regenerative medicine.
2. Bone Marrow: A Hematopoietic and Antigen-Responsive Lymphatic Organ
2.1. BM: A Central Organ Protected by Bone
BM is unique in comparison to secondary lymphatic organs in many aspects. One is its central location, another its protection by solid bone. During millions of years of adaptation, this combination provided protection against environmental changes, mechanical insults, and UV light irradiation.
A man weighing 73 kg has around 3.7 kg of BM [10]. This is in the range of total peripheral blood. In fact, 12% of all lymphoid cells in human are found in the BM at any given time as compared to 2% in the peripheral blood, and 20–50% of mononuclear cells from BM and peripheral blood are T lymphocytes [12,13].
In adult humans, BM is located in the skull; the vertebrae; the ribs, sternum, and pelvis; and in bones of the extremities, such as the humerus, femur, and tibia. The vertebral body consists of a trabecular bone, which contains red marrow surrounded by a thin external layer of compact bone. The vertebral arch forms the vertical (spinal) canal, which contains the spinal cord [14]. Intercostal nerves and plexuses accompany the thorax, and peripheral neurons communicate with and regulate immunological processes [15].
2.2. BM: A Central Hematopoietic Organ
BM is composed of red (hematopoietic) and yellow (adipose) tissue and contains two types of stem cells: HSCs and MSCs. All the different types of blood cells are derived from HSCs, which reside in niches of the BM parenchyma and develop via committed precursors and late precursors into their mature forms. HSCs are maintained in a perivascular niche, in which LepR+ stromal cells and endothelial cells synthesize factors required for HSC maintenance, including stem cell factor (SCF), the ligand of c-kit. Restricted hematopoietic progenitors and erythropoiesis require SCF from LepR+ niche cells in the BM [16]. Other factors of importance for hematopoiesis are CXCL12, vascular endothelial growth factor receptor 2, E-selectin, jagged-1, and pleiotrophin [8,10].
Hematopoiesis is organized like a tree, leading to the development of the major lineages of blood cells. Myeloid progenitor cells lead to neutrophils, monocytes, dendritic cells, eosinophils, basophils, mast cells, megakaryocytes, and erythrocytes. Common lymphoid progenitor cells lead to pre-T cells, pre-B and B cells, NK cells, and innate lymphoid cells [7].
Novel techniques of optical imaging and bioimage-based analysis software tools allow for organ-wide three-dimensional (3D) reconstructions of a variety of tissues, including BM [10]. High-resolution 3D imaging of a reduced field of view showed the stromal scaffolds in BM: sinusoidal vessels and a network of perivascular bodies of CXCL12+ reticular cells forming a dense matrix through the emissions of abundant cytoplasmic projections [10]. Megakaryocytes are found in close proximity to the endothelial surfaces of sinusoidal vessel walls. They traverse these in the form of protrusions, from which proplatelets are continuously shed into the venous circulatory system [10]. Red blood cell development takes place in so-called erythroblastic islands where erythroid precursors proliferate, enucleate, and terminally differentiate [10]. Early lymphoid progenitors (Lin-, IL-7Rα+) are found in contact with IL-7 expressing CXCL12+ stromal reticular cells, sometimes proximal to mature, bone-lining osteoblasts [10].
2.3. BM: A Central Antigen Responsive Lymphatic Organ
That BM is a priming site for T-cell responses to blood-borne antigens was first described in 2003 [17].
The reasons for this late discovery are anatomical restrictions. The thick bone cortex that surrounds the BM impedes direct observation and experimental manipulation. This is a main reason for the relative paucity of data on BM physiology. One notable exception is the calvaria of the murine skull, where hematopoietically active BM is only covered by a thin lamella of bone that is sufficiently translucent to allow a detailed in situ analysis of the BM microcirculation by epifluorescence microscopy. This technique allowed for studies to be conducted on adhesion and homing of blood-borne cells in BM microvessels [18]. A second method consisted of visualization of T cells and DCs by immunohistology of frozen tissue sections, a procedure that required careful isolation of intact BM tissue from murine femurs [17,19,20].
For functional studies, DCs and T lymphocytes from murine [17,20] and human [19] BM were derived from isolated mononuclear cells, expanded in tissue culture, and specifically separated as described [17,19,20].
Figure 1 provides an outline of this chapter.
2.4. Active Control of Proliferating Tumor Cells by CD8+ Memory T Cells Leading to Tumor Dormancy in BM
A first hint to the potential importance of the BM in immunosurveillance came from experimental studies in mice [21]. The highly aggressive murine lymphoma ESb, when inoculated into the ear pinna of syngeneic mice, was found to be incapable of growing due to an induced strong immune response. When ESb cells were transfected with the bacterial lacZ gene coding for the enzyme β-galactosidase (Gal), it was possible to follow single tumor cells in tissues such as the lymph nodes, spleen, and BM of ESb-lacZ ear pinna-inoculated mice. These studies revealed that in these immunized mice, cancer-reactive CD8+ memory T cells (MTCs) actively controlled tumor dormancy in the BM and established long-term systemic immune resistance upon subcutaneous tumor cell challenge. Low numbers of tumor cells that homed to the BM expressed the proliferation-associated Ki67 antigen, but their proliferation was kept under control by the cancer-reactive CD8+ MTCs [21,22]. Upon CD8+ T cell depletion, the BM tumor cells expanded, and the mice died from metastases [22]. These early experiments demonstrated that BM is a privileged site where potentially lethal tumor cells can be controlled in a dormant state by CD8+ MTCs [22].
2.4.1. Antigen-Presentation Capacity of BM DCs to Naïve T Cells
Antigen presentation to T cells in BM is mediated via resident CD11c+ DCs. They are highly efficient in taking up blood-borne antigen and processing it via major histocompatibility complex (MHC) class I and class II pathways [17,19,20]. After intravenous injection of soluble ovalbumin (OVA) into naïve B6 mice, sorted BM derived CD11c+ DCs cross-presented OVA via MHC I to naïve (CD621high, CD44low, CD69neg) T cells from OT-I mice encoding a transgenic TCR specific for H2Kb-pOVA (257–264) [17].
Similarly, antigen processing and presentation capacity via MHC II by BM DCs was demonstrated, using antigen myoglobin and human C-reactive protein (hCRP) as models. The transfused responding T cells were either (i) T cell hybridoma specific for an MHC II-restricted myoglobin peptide or (ii) specific T cells from hCRP transgenic mice [20].
2.4.2. Primary T Cell Responses in Bone Marrow
Naïve CSFE-labeled, OVA-specific transgenic T cells from OT-I or OT II mice were injected intravenously into B6 mice. T cell activation was measured by high CD69 expression and loss of CFSE. The first responses occurred in the BM and concomitantly in the spleen. The responses in peripheral blood and lymph nodes lagged behind. CD69 upregulation was observed 4 h after OVA challenge, and the first cell division 26 h later [17].
Primary CD8+ T cell responses in BM were also measured in mice with normal naïve TCR repertoires. The mice were injected with syngeneic ESb-Gal tumor cells into the ear pinna, a site where the Gal-transfected live tumor cells could not grow [21]. T cells from the BM, spleen, and lymph nodes were tested for tetramer (Ld/Gal(876–884)-binding CD8+ T cells. Ten days after Gal immunization, a peak of activated specific T cells was observed in the BM, but not in the spleen or lymph nodes. The response then declined towards day 15 [17].
Primary CD4+ T cell responses were measured in mice inoculated with naïve CSFE-labeled OVA-specific T cells from OT-II mice: 24 h after T-cell reconstitution, the mice were challenged i.v. with OVA, and 5 h later, CD69 upregulation could be seen in cells from the BM and spleen, but not in cells from peripheral blood [20].
Further studies also revealed the generation of OVA-specific cytotoxic T cells (CTLs) in BM, as well as generation of systemic immunity and memory against B16-OVA tumor cells [17].
2.4.3. Two-Photon Dynamic Imaging Revealing Cross-Presentation of Blood-Borne Antigens to Naïve T Cells in BM
Ten years after these pioneering discoveries, some of the findings were confirmed and extended by in situ 2-photon microscopy. The studies revealed promiscuous cross-presentation of blood-borne antigens to naïve T cells in the BM [23]. Naïve CD8+ T cells crawled rapidly at a steady state, but arrested immediately upon sensing antigenic peptides. Antigen-specific T cells decelerated, clustered, upregulated CD69, and were observed dividing in situ to yield effector cells. Uniquely for BM, alongside DCs, other myeloid cells participated in cross-presentation. Potential antigen-presenting cells included phagocytic monocytes and macrophages, but not B cells [23].
2.4.4. Targeting Glycan Modified OVA to DC-SIGN
DCs have gained much interest in the field of anti-cancer vaccine development because of their central function in immune regulation. One of the receptors that facilitates DC-specific targeting of antigens is the DC-specific C-type lectin DC-SIGN. Targeting glycan-modified OVA to murine DC-SIGN transgenic DCs revealed a 10-fold enhanced antigen-presentation capacity for both CD4 and CD8 T cell responses in comparison to unmodified OVA [24].
2.4.5. Cluster Formation in BM Parenchyma of Antigen-Presenting DCs and Antigen-Specific T Cells
Immunohistology of frozen sections of BM from mouse femurs showed colocalization in the parenchyma of host CD3+ T cells with CD11c+ DCs. Without antigen-specific activation, the transferred CSFE labeled CD8+ OT-I T cells were scattered throughout the parenchyma. In contrast, 38 h after OVA injection, the BM parenchyma showed cell clusters containing approximately 10–30 vβ5+ transgenic T cells expressing CD69 [17].
Immunohistology of BM parenchyma also revealed a microenvironment for T cell homing and interaction with DCs. The expression of VCAM-1 on the sinus lining and stroma cells co-localized with CD3 T cells from OT-II mice. Co-localization was also seen of mucosal vascular addressin cell adhesion molecule 1 (MadCAM-1) with CD3 T cells in lymphoid-like follicles, and of CD69+ cells with CD80+ antigen-presenting cells [17].
Cognate interactions between CD4 T cells and antigen-presenting cells (APCs) were studied in situ after transfer of CFSE-labeled CD4 T cells specific for hCRP. Following antigenic challenge, T-APC clusters could be isolated from the BM and visualized, and 69% of such lymphostroma rosettes contained hCRP-specific proliferating CD4 T cell blasts [20].
In conclusion, it can be stated that BM is an antigen-responsive lymphatic organ. In 2003, T-cell priming in BM and its potential for long-lasting protective anti-tumor immunity was summarized and discussed [25]. The spontaneous induction and therapeutic potential of cancer-reactive MTCs from BM was further discussed in 2015 [26].
3. Comparison between BM and Blood
3.1. Comparison of DC Generation from Mononuclear Cells and Their Function
DCs from bone marrow (BM-DCs) and peripheral blood (BL-DCs) were generated in parallel from the same healthy normal donors by culturing the cells in serum-free medium containing granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-4. Then, their phenotypes and functions were compared. BM-DC generation occurred in 14 days, and involved proliferative expansion from CD34+ stem cells and differentiation, while BL-DC generation occurred in 7 days from CD14+ monocytes and involved only differentiation [27]. A 7- to 25-fold higher number of DCs was obtained from BM than from blood. The DCs had similar phenotypes, but differed in function. The capacity to stimulate autologous MTC responses to tetanus toxoid or tuberculin (purified protein derivative PPD) was higher with BM-DCs than with BL-DCs [27].
3.2. Enrichment of Memory T Cells in BM of Breast Cancer Patients
BM-derived cells from primary operated untreated breast cancer patients (n = 90) were compared with those from healthy donors (n = 10), and also with cells from respective blood samples [13]. The proportion of MTCs among the CD4 and CD8 T cells was much higher in the BM of cancer patients than in healthy donors, and the extent of MTC increase was related to the size of the primary tumor. The results suggest that BM is a valuable additional compartment to blood for immune diagnosis in pathological conditions, and possibly for follow-up treatment strategies [15].
With regard to the specificity of the BM MTC pool in breast cancer patients, further studies revealed the shaping of a polyvalent and highly individual T-cell repertoire. Peptide-HLA-A2-restricted reactivity was tested against 10 breast cancer-associated tumor antigens (TAs) and 3 normal breast tissue-associated SAs by short-term IFN-γ ELISpot analysis, and 67% of the patients recognized TAs with a mean frequency of 144 TA-reactive cells per 1 million T cells. The reactivity to SAs was lower, with a mean frequency of 85 per 1 million T cells. Healthy individuals also contained TA-reactive T cells, but this repertoire was more restricted [28]. A new metastasis-associated TA was recognized in breast cancer patients via BM MTCs, namely, heparanase (Hpa) [29]. The fact that such cells were detected in a high proportion of patients, but not in healthy people, suggests that Hpa is an attractive new TA and its HLA-A2-restricted peptides ought to be good candidates for peptide vaccination.
3.3. Generation of Tumor-Specific CTL from BM, but Not PBL, of Breast Cancer Patients
Mononuclear cells from the peripheral blood (PBL) or BM of individual primary operated, but untreated, breast cancer patients were co-incubated for seven days with autologous DCs pulsed with lysate from MCF-7 breast cancer cells or from unrelated U937 cancer cells. PBL failed to induce cytotoxic T lymphocyte (CTL) activity against MCF-7, whereas BM T cells derived from the same patients developed HLA-dependent CTL activity against MCF-7 cells [19].
3.4. Superior Therapeutic Efficiency In Vivo of Reactivated MTCs from BM in Comparison to Blood of Breast Cancer Patients
Immunotherapeutic effects were studied in a newly established NOD/SCID mouse model that allowed for outgrowth of subcutaneously implanted human tumor xenografts. A single intraperitoneal transfer of restimulated BM T cells caused regression of autologous tumor xenotransplants associated with infiltration by human T cells, as well as tumor-cell apoptosis and necrosis. PBL T cells showed much lower anti-tumor activity in vivo [19].
4. BM Storage Capacity for Memory B and Memory T Cells
4.1. Survival Niches for Memory B and Memory Plasma Cells in BM Parenchyma
B lymphocytes and plasma cells provide the humoral side of adaptive immune responses. They contribute to the effectiveness of prophylactic vaccination to prevent infections. Memory B cells and memory plasma cells survive in dedicated stromal niches of the BM [30]. The BM has been described as a sanctuary for memory cells such as plasma cells [31]. This has implications for adaptive immunity and vaccinology [32]. In autoimmunity and chronic inflammation, memory B cells and memory plasma cells can be important players and require special attention [30].
4.2. Memory T Cells
Priming of naïve T cells leads to association of the tyrosine kinase Lck with the CD8 coreceptor, thereby enhancing TCR signaling. The association beween Lck, which phosphorylates CD3 immunoreceptor tyrosine-based activation motifs (ITAMs), and CD8 is maintained in MTCs, which explains their enhanced sensitivity to antigen re-exposure. During a primary immune response, IL-7Rα (CD127) is downregulated on most CD8 T cells. Only a small subset of cells that are CD127hi contribute to the pool of MTCs (for review see, [25,26,32]).
The duration of TCR signaling required for activation is about 20–30 h for naïve T cells, but less than 1 h for effector T cells and MTCs. The life span of naïve T cells is 5–7 weeks, while that of effector T cells is only 2–3 days. In contrast, the life span of MTCs, in particular stem-like MTCs with self-renewal capacity (see Section 4.4), can last up to 50 years (for review, see [25,26,32]).
4.3. Survival Niches for Memory T Cells in BM Parenchyma
BM was reported to be (i) a nest for migratory MTCs [33], (ii) a major reservoir and site of recruitment for central memory CD8+ T cells [34], (iii) a preferred site for homeostatic proliferation of memory CD8+ T cells [35], and (iv) a reservoir for “enhanced effector memory” CD8+ T cells with potent recall function [36].
High frequencies of less differentiated and more proliferative CD8+ MTCs specific for mutated and overexpressed Wilms’ tumor suppressor gene 1 (WT1) were detected in the BM of tumor-bearing patients [37]. Similarly, high frequencies of functional tumor-reactive T cells were found in the BM of pancreatic cancer patients [38], and the BM of patients with multiple myeloma was enriched with functional CD8 MTCs specific for the TA mucin glycoprotein 1 (MUC1) [39]. These findings from cancer patients corroborate and extend the earlier reports from breast cancer patients described above [15,19,28,29].
Blood T cells enter vascular sinuses in BM and transmigrate via diapedesis through the endothelium into BM parenchyma. This involves traffic/adhesion molecular interactions, chemokines, and cytokines, and is true for circulating T cells and tumor cells. The chemokine axis CXCL12/CXCR4 plays an intriguing guiding role in the homing of central MTCs to secondary lymphatic organs [40]. The collagen II receptor CD49b on stromal cells was described to be required for migration of memory CD4 T-cell precursors into their survival niches in BM [41].
BM MTCs express key survival receptors, namely, IL-7Rα and IL-15Rβ. BM stromal niches provide the corresponding cytokines IL-7 [42] and IL-15 [43] for MTC survival. IL-15 is a pleiotropic cytokine which, along with IL-2, IL-4, IL-7, IL-9, and IL-21, shares the common cytokine receptor γ. It is trans-presented to immune cells and is vital for the development, survival, and expansion of CD8+ MTCs, as well as for natural killer cells. Il-15 binds to IL-15Rα, which is expressed on a number of cell types and is then trans-presented to responding cells that express IL-2Rβ and γ. This distinctive mechanism for IL-15 relates to its role in signaling in the context of cell–cell interactions and signaling synapses [44].
4.4. Tissue-Resident Memory T Cells in BM Parenchyma
Peripheral and systemic antigens were described to elicit an expandable pool of tissue resident memory CD8+ T cells in the BM [45]. These cells develop against various pathogens, independently of BM infection or local antigen recognition. They are polyfunctional cytokine producers, and are dependent on IL-15 and on transcription factors such as Blimp-1 and Hobit. This work extends the role of the BM in the maintenance of CD8+ T cell memory to include an expandable pool of functional, non-recirculating MTCs, which develop in response to a large variety of peripheral antigens [45]. Similarly, circulating, blood-borne SARS-CoV-2-reactive CD4+ MTCs have been described in unexposed people [46]. Such pre-existing cross-reactive MTCs derive from a BM pool, are polyfunctional, and may play an important role in shaping the systemic immune response to SARS-CoV-2 [47].
4.5. Stem-like Memory T Cells in BM Parenchyma
A unique population of BM MTCs is stem-like MTCs (TSCM). TSCM are characterized by C-type lectin CD69 and high IL-7Rα (CD127) expression [48]. Preferential homing to BM has been described for tumor-specific and functional CD8+ TSCM [48]. They express stem cell antigen-1 (Sca-1, CD122) and B-cell lymphoma protein-2 (Bcl-2), and they relocate, after their vigorous response to blood-borne antigens, to the BM by adhesion molecules VCAM-1, P-selectin glycoprotein 1, and P-selectin or E-selectin [49]. The natural CD4+ TSCMs in the BM colocalize with VCAM-1+, IL-15+, IL-7+, and CXCL-12+ stromal cells [50]. In contrast to spleen-resident CD4+ TSCMs, the BM resident TSCMs are able to assist in the maturation of antibodies [50].
TSCMs combine phenotypes of naïve and memory T cells. An analysis of 16 human subjects revealed that TSCMs show unique features in terms of their T-cell receptor repertoire [51]. They have increased diversity across all stretches of the TCRβ repertoire structure compared to those of naïve and other CD4+ MTCs. The top 1000 clonotypes in TSCMs were more public and grouped in more clusters, implying more epitope types [46].
In contrast to activated T cells, which, after adoptive T cell therapy, may rapidly become tolerant in the tumor microenvironment, due to anergy, senescence, and/or exhaustion [52], TSCMs are strongly resistant to tolerance. In vitro culture systems have been described for the generation of human TSCMs from naïve precursors [53] or from activated T cells [54].
TSCM phenotypes can be induced by suppressing genes related to T cell differentiation, such as T-bet, basic leucin zipper transcription factor (BATF), and eomesodermin, and by upregulating genes related to stemness, such as T cell factor 1 (TCF1) and lymphoid enhancer binding factor 1 (LEF1) [51]. More detailed information about the mechanisms of long-term protective anti-tumor immunity, the role of the BM, and epigenetic regulation of MTCs has been reviewed [32].
4.6. Enrichment of Virus-Specific MTCs in Human BM Parenchyma
Selective accumulation of virus-specific CD8+ T cells with unique homing phenotypes has been described within human BM [55]. CD8+ T cells specific for Epstein–Barr virus (EBV) lytic antigens were enriched threefold in BM compared to blood. BM T cells were found to exhibit a unique CCR5+CXCR6+CXCR3- homing phenotype which was not observed on T cells from secondary lymphoid organs or peripheral organs [55]. EBV is a human pathogenic herpesvirus (HHV4) that can lead to infectious mononucleosis.
BM samples of individuals persistently infected by hepatitis C virus (HCV) have been reported to be enriched with memory CD8+ T cells specific for current and historical HCV antigens [56]. Infections with HCV have a high rate of becoming chronic, with negative effects on the liver, such as liver cirrhosis.
Increased IL-15 production and accumulation of highly differentiated CD8+ effector/memory T cells was described in the BM of persons infected with the herpes virus cytomegalovirus (CMV) [57]. Infections with human CMV occur often during pregnancy and can be transmitted to the child. Human BM has been proposed as a source for the generation of CMV-specific CD4+ T cells with multifunctional capacity [58]. An open-label, first-in-human trial assessed the safety and therapeutic potential of adoptive cellular therapy (ACT) with CMV-specific T cells [59]. The procedure appeared to be a safe adjuvant immunotherapy for primary glioblastoma multiforme (GBM). If offered before recurrence, it is suggested to improve overall survival [59].
4.7. Cognate Re-Activation of TA-Specific BM MTCs Ex Vivo and In Situ
Primary operated breast cancer patients contain, in their BM, cancer-reactive MTCs for multiple TAs that can be re-activated ex vivo by DC-based APCs and exert therapeutic potential in human tumor xenotransplant models, as reported in 2001 [19].
Re-activation of MTCs from BM and recruitment to the site of vaccination has been described for the use of the Newcastle disease virus (NDV)-modified autologous tumor cell vaccine (ATV-NDV). This can explain the long-term survival benefit of colon cancer patients observed in a randomized controlled clinical study [32]. It appears to be the presence of a cognate antigen within the autologous vaccine and the danger signals provided by NDV infection that cause the re-activation of quiescent cancer-reactive MTCs from the BM of the vaccinated colon cancer patients. Only those BM MTCs primed spontaneously against colon cancer TAs in individual patients become re-activated by autologous TAs of the vaccine. The importance of costimulatory and danger signals provided by infection with NDV has been described [32].
4.8. Hypotheses for the Maintenance of Long-Term Memory in the BM
The organization of long-term immunological memory in the BM provides a number of challenges. The cells need to be able to self-renew, to persist for the long term, and to give rise to highly proliferative progeny while staying capable of quickly mounting a recall response upon reinfection [31].
A hypothesis on life-long T cell memory proposed the existence of two niches in the BM [60]. The maintenance of quiescent immune B and T cell memory in the BM has been described as follows [61].
- Quiescence: Following the successful resolution of an immune reaction, antibody-secreting memory plasma cells and memory B and T cells persist as quiescent cells (non-proliferative, non-migratory) in dedicated survival niches organized by BM stromal cells. The immune memory cells dock individually onto dedicated stromal cells, which control their maintenance. The number of available dedicated stromal cells defines the size of the memory compartment [61].
- Cognate re-activation of BM memory cells. Upon re-encounter with the antigen, which enters directly via the blood into the vascularized BM or is transported there by APCs, antigen-specific memory B and MTCs are re-activated. MTCs proliferate locally, form immune clusters, and provide local protection. Others exit the BM and contribute to secondary immune reactions in the periphery. BM clusters in the parenchyma can develop into large follicles. These include memory B and memory plasma cells in addition to CD4+ MTCs, suggesting T–B cell interaction [62]. Once a BCR binds its T cell-dependent antigen, the antigen is taken up into the B cell through receptor-mediated endocytosis. This is then degraded and presented to T cells as a p-MHC II complex at the cell membrane. Memory B cells in immune follicles might receive stimulatory signals from antigen-specific helper T cells upon T–B synapse formation. More than one antigenic determinant of a protein is required for such antigen-specific T-B cell interactions [63], one interacting with the BCR, the other with the TCR. Thus, activated memory B cells may directly differentiate into antibody-secreting cells in the BM, providing rapid enhancement of humoral immunity [61,62].
5. Bone Marrow Vaccination or Allogeneic BM Cell Injection: Novel Approaches to Enhance or Reduce Antigen-Specific Immunity
Since MTCs within the BM have distinct phenotypic and functional properties when compared to MTCs from other sites, a novel approach has been proposed to enhance antigen-specific immunity [64]. In a murine model, the bent knee joints of anesthetized mice were used for needle injection of the vaccine directly into the BM cavity of the tibia [64].
Intra-BM (IBM) vaccination was successfully exploited for the purpose of inducing enhanced protective antitumor immunity against human papillomavirus (HPV)-associated cancer [65]. IBM vaccination with the MHC class I HPV-16E7 epitope induced large numbers of activated, IFN-γ-producing, E7-specific T lymphocytes in the BM. In a prophylactic tumor challenge setting, direct IBM vaccination protected against tumor formation in 80% of the mice. In a therapeutic setting, IBM vaccination induced tumor regression in three of ten vaccinated mice and delayed tumor growth in the remaining animals. Adoptive transfer of BM cells from IBM-vaccinated mice to naïve animals conferred complete protection against tumor growth [65]. It was suggested that targeting BM T cells with vaccines may improve responses to cancer immunotherapy and offer important clinical advantages [65].
Intra-BM injection of allogeneic BM cells has been reported as a powerful strategy for the treatment of intractable autoimmune disease in MR1/lpr mice [66]. It consisted of fractionated host irradiation (5.5 Gy × 2) and IBM injection of BM mononuclear cells, including HSPs and MSC-derived stromal cells from allogeneic normal mice. All the recipients treated in this way survived for more than 1 year and remained free from autoimmune disease. Successful cooperation was achieved among T cells, B cells, and APCs. A critical role was played by donor-derived stromal cells [66]. All of this was possible without recourse to immunosuppressants [66].
6. Interactions in BM between Three Types of Stem Cells and Immune Cells
6.1. Hematopoietic Stem Cells (HSCs) in Cross-Talk with T Cells and DCs
Considering the fact that BM harbors niches for both stem cells and MTCs, perhaps even in close proximity, it is conceivable that cross-talk may exist between them. In fact, cross-talk between T cells and HSPs has been reported during ACT for malignant glioma [67]. GBM tumors are largely devoid of resident migratory DCs to function as APCs during immunotherapy. Transfer of HSCs with concomitant ACT led to the production of activated CD86+CD11c+MHC-II+ cells consistent with a DC phenotype which functioned within the brain tumor microenvironment (TME). During ACT, the HSC-derived cells in gliomas relied on T-cell-released IFN-γ to differentiate into DCs. These DCs activated T cells and promoted intracranial tumor rejection [67].
A fundamental property of HSCs is their ability to respond to environmental cues. In a recent study, it was reported that IL-1β expression in BM DCs can be induced by TLR1/2 agonists. Such DCs could regulate HSC function and induce their expansion [68]. The data suggest a model in which TLR1/2 stimulation of DCs induces secretion of IL-1β and other inflammatory cytokines into the perivascular niche, which in turn regulates multipotent HSCs [68].
These are examples of cross-talk via cytokines (IFN-γ, IL-1β) between HSCs from the BM and cells from the innate and adaptive immunity system.
6.2. Extramedullary HSCs in Meninges of Adult Mice Providing Immune Surveillance of the CNS
Brain meninges contain both innate and adaptive immune cells, which provide immunosurveillance of the central nervous system (CNS) [69]. HSCs lodge in the meninges after birth with local expression of pro-hematopoietic niche factors. With a tissue-specific expression profile, meningeal HSCs can provide the CNS with a constant supply of leukocytes more adapted to the local microenvironment [69]. In sublethally irradiated recipients, the meningeal HSCs showed long-term, efficient, multi-lineage reconstitution and self-renewal capacity in the meninges, blood, spleen, and BM [69]. To achieve a steady state, the meningeal HSCs were likely to cross-talk with T cells and DCs.
6.3. BM Neural Crest-Derived Stem Cells Affecting B Cell Lymphopoiesis
The depletion of neural-crest (NC)-derived cells in double-transgenic mice led to a reduction in plasma noradrenaline and to alterations in B cell lymphopoiesis [70]. NC-derived cells contribute to the development of BM stromal cells, Schwann cells, and sympathetic nerve fibers [70].
BM receives sensory and sympathetic innervation from the peripheral nervous system [71]. NC-derived Schwann cells reside in a neurovascular niche in the BM in association with nerve fibers [71]. The phenotype of human BM NC cells is NESTIN+/SOX9+/TWIST+/SLUG+/P75NTR+/BRN3A+/MSI1+/SNAIL1+ [72]. Such cells are able to differentiate into melanocytes, Schwann cells, and neurons [72]. BM MSCs can differentiate into neural progenitor-like cells in the presence of basic fibroblast growth factor (bFGF) and epidermal growth factor (EGF) [73].
6.4. Mesenchymal Stem Cells in Cross-Talk with T Cells
BM MSCs are multipotent cells with strong tissue repair and immunomodulatory properties [74]. Due to their ability to repress pathogenic immune responses, in particular T cell responses, they show therapeutic potential for the treatment of autoimmune diseases. The transfer of MSCs to CD4+ T cells from influenza hemagglutinin-specific TCR transgenic mice reduced their diabetogenic potential [74].
MSCs have the remarkable ability to export their own mitochondria to neighboring cells in response to injury and inflammation [74]. The effects of MSC-transferred mitochondria were additive to those of MSC-secreted prostaglandin E2. CD4+ T cell co-culture with MSCs and transfer of isolated MSC mitochondria prevented the upregulation of T-bet, the master transcription factor of Th1 polarization [74].
7. Effect of Dietary Restriction (DR) on the BM
7.1. Effect of DR on Monocytes from the BM
Major chronic diseases (metabolic syndrome, cardiovascular diseases, neurodegenerative diseases, immune system disorders, and cancer) are characterized by mitochondrial dysregulation of the cellular energy supply and metabolism [75]. Recently, it was shown that short-term fasting (dietary restriction, DR) reduces monocyte metabolic and inflammatory activity and reduces monocyte mobilization from the BM. The regulation of peripheral monocyte numbers was dependent on dietary glucose and protein levels. This was due to activation of the low-energy sensor 5′-AMP-activated protein kinase (AMPK) in hepatocytes, and to the suppression of systemic CC-chemokine ligand 2 (CCL2) production by peroxisome proliferator-activator receptor alpha (PPARα). Fasting improved chronic inflammatory diseases without compromising emergency mobilization during acute infectious inflammation and tissue repair [76].
7.2. Effect of DR on Mucosal Immune Responses: Migration of Naïve B Cells from PPs to BM
Another recent study revealed that DR drastically reduces the numbers of lymphocytes in Peyer’s patches (PPs), the inductive site of the gut immune response [77]. A large proportion of germinal center and IgA+ B cells was lost via apoptosis during fasting. Naïve B cells migrated from PPs to the BM. During refeeding, stromal cells sensed nutritional signals and upregulated C-X-C motif chemokine 13 (CXCL13) expression to recruit naïve B cells into PPs. Thus, nutritional signals are critical to maintaining gut immune homeostasis [77].
This study uncovered a novel link between nutritional signals, immune cell dynamics, and their functionality. The intestinal mucosa in adult humans possesses a total surface area of 200 m2. PPs conduct immunosurveillance on the mucosal surface to eliminate potentially hostile agents. B cells comprise a major population of PPs, where the B cell/T cell ratio is fivefold higher than in peripheral lymph nodes. Metabolic reprogramming to aerobic glycolysis is critical for the production of CXCL13. This shift from oxidative phosphorylation to aerobic glycolysis is well characterized in activated M1 macrophages and monocytes, as well as in Th1 and Th17 cells [78].
7.3. Effects of DR on Memory T Cells: BM as a Refuge for Immune Memory
MTCs collapsed in secondary lymphoid organs in the context of DR, but dramatically accumulated within the BM [79]. This response was coordinated by glucocorticoids (GCs) and was associated with energy conservation. T cell activities such as cytoskeletal rearrangements, transendothelial migration, differentiation, proliferation, effector function, and memory require extra energy provided by the mitochondria [76]. The response to DR included an increase in T cell homing factors, erythropoiesis, and adipogenesis. Adipocytes, as well as CXCR4-CXCL12 and sphingosine-1-phosphate (S1P) interacting with its receptor S1P1R, contributed to the enhanced T cell accumulation in BM during DR. MTC homing to BM during DR was associated with enhanced protection against infections and tumors [79].
DR-induced stress evoked GCs, which apparently coordinate the described adaptive response of promoting MTC survival in the privileged environment of the BM. Increased adipogenesis in BM also occurs in other settings, such as irradiation and chemotherapy, suggesting that this could represent a general response following stress [79].
Three recent papers have demonstrated that monocytes, naïve B cells, and memory CD8 T cells use BM as a refuge to tide off periods during negative energy balance and to maintain immune responsiveness.
The main features of chapters 2 to 7 are summarized in Table 1.
8. Blood-Borne Antigens, Circulating Cells, and Subcellular Particles
8.1. Self and Non-Self Antigens
Blood-borne antigens can be self (auto)-antigens (SAs) or non-self-antigens (NSAs). They can be systemic antigens from blood, or can be derived from peripheral tissues via lymphatics entering the blood. Naïve mature lymphocytes continuously migrate from the blood into secondary lymphoid organs through high endothelial venules or into the BM, and return to the blood directly or through lymphatics. This process maximizes the rare chance of cognate encounters with their respective antigens to initiate immune responses [7].
Blood-borne SAs are recognized by immature BCR-expressing B cells in the BM, and by immature TCR-expressing T cells in the thymus [7]. Blood-borne SAs include self-macromolecules [80]. Specialized transendothelial DCs in the thymus provide developing T cells with SAs to induce negative selection and to maintain central tolerance. The DCs are positioned in immediate proximity to thymic microvessels, where they extend cellular processes across the endothelial barrier into the bloodstream [80]. Negative selection also occurs in the BM through the binding of SAs with the BCR of developing B cells. The continuous supply via blood of SAs to developing B and T cells ensures the maintenance of self-tolerance and the prevention of autoimmune reactivities.
While contact with SAs affects T and B cells pre-maturely, contact with NSAs affects T and B cells only after their full maturation. Thus, antigen–immune cell interactions occur at two different stages of maturation. Mature T cells not reacting to SAs and ready to react to NSAs egress from the thymus in dependency on S1P receptor 1 (S1PR1) [81].
Blood-borne NSAs can be derived (i) from viruses, which represent a residual risk in potential organ donors (e.g., hepatitis virus B and C, human immunodeficiency virus (HIV)) [82], (ii) from bacteria or other microbes [83], or (iii) from tumor cells (TAs, neoantigens). A large proportion of mature B cells occupy an anatomically and functionally distinct perisinusoidal niche in the BM. Such cells circulate freely, as revealed by parabiosis studies [83]. Unlike their counterparts in the follicular niches of lymph nodes, these cells are capable of being activated in situ by blood-borne microbes in a T-independent manner to generate specific IgM antibodies. The BM represents a unique type of secondary lymphoid organ in which mature B cells are strategically positioned in the path of circulating microbes [83].
Immune complexes (ICs) in blood are efficiently removed mainly by liver reticuloendothelial systems consisting of sinusoidal endothelial cells and Kupffer cells expressing Fc-γ receptors. ICs can also be taken up by BM endothelial cells via Fc-γR IIb2 receptors [84]. This occurs in an erythropoitin (Epo)-dependent manner [84].
8.2. Circulating Tumor Cells, Tumor-Associated Antigens, and Immunogenic Cell Death
Blood from cancer patients can contain circulatory tumor cells [85], extracellular vesicles (EVs) derived thereof, apoptotic bodies [86], and tumor-associated proteins [87]. Minimally invasive methods such as liquid biopsy of the blood, urine, and cerebrospinal fluid can be used to sample circulatory tumor DNA (ctDNA), RNA, EVs, and tumor-associated proteins [88].
The information obtained from liquid biopsy can be useful: (i) comprehensive liquid biopsy analysis is a new tool for the early detection of minimal residual disease [89]; (ii) exosomal microRNA signature has predictive value for tumor immunity in cervical cancer patients treated with chemoradiotherapy [90]; and (iii) advances in proteogenomic analysis of HLA ligandomes demonstrating a subset of cryptic peptides derived from oncogenic noncoding RNA in human colorectal cancer cells [91].
Blood-borne antigens can be derived from immunogenic cell death (ICD). This is a specific form of regulated programmed cell death. It causes an adaptive immune response specific for endogenous (cellular) or exogenous (viral or bacterial) antigens expressed by the dying cells [92]. Oncolytic virus infections, some chemotherapeutics, radiotherapy, hypericin-based photodynamic therapy, and moderate electrohyperthermia (mEHT) can induce ICD [92,93]. ICD is based on the release of Damage-Associated Molecular Patterns (DAMPs), which are recognized by respective receptors of immune cells. Six DAMPs play an important role: (i) expression of calreticulin on the membrane as “eat-me” signals for phagocytosis by macrophages, neutrophils, and DCs; (ii) secretion of ATP as a “find-me” signal for macrophages and DC precursors; (iii) secretion of high-mobility group box 1 (HMGB1) proteins, which bind mainly to TLR4 (“approach-me” signal); (iv) secretion of immune stimulatory interferon type I (IFN-I); (v) release of cancer-cell-derived nucleic acids which can be taken up by DCs, macrophages, and neutrophils; and (vi) expression of annexin A1, which specifically engages DCs via formyl peptide receptor 1 [92,93].
8.3. Circulatory Antigen-Presenting DCs and Their Homing to BM
Blood-circulating cells of the immune system, such as lymphocytes and DCs, can transport information from the periphery into the BM. Such cells home to BM depending on constitutively expressed VCAM-1 and endothelial selectins in BM microvessels. A subset of DCs can travel as antigen-presenting cells (APCs) from the periphery into the blood, from which they migrate to the spleen and BM, but not to the lymph nodes [94]. Two-photon intravital microscopy in BM cavities revealed that such antigen-bearing DCs formed stable antigen-dependent contacts with BM-resident central MTCs, thereby triggering them to recall responses [94].
All DCs derive from HSCs in the BM. They are critical for adaptive immune responses and immune tolerance. DCs are strategically positioned as immune sentinels in tissues throughout the body, poised to respond to invading pathogens. The molecular traffic signals that govern DC migration throughout their life cycles have been reviewed [95]. The major trafficking molecular interactions concerning DC interactions with BM are PSGL1-CD62E, PSGL1-CD62P, VLA-4-VCAM-1, CCR2-CCL2, and CXCR4-CXCL12 [95].
All BM-immigrating and -emigrating cells must traverse the BM vasculature. The endothelial cells lining these vessels regulate the migration of cells by expressing the appropriate ligands on their surfaces. ICAM1, VCAM1, and BP1 (eukaryotic translation initiation factor 4-binding protein 1) positive perivascular stromal cells expressing CXCL12, IL-7, IL-15, and other chemokines are the cells determining which cells can enter the BM, and, subsequently, its survival niches [31].
8.4. Circulatory Naïve T and Memory T Lymphocyte Subsets
The percentages and absolute numbers of circulatory T cells, examined from 309 healthy volunteers, were tested by means of ten-color flow cytometry based on a single-platform technology [96]. CD3+ T cells represented 67.9% of all lymphocytes and 1140 cells/mL. The CD4:CD8 ratio was 1.37. Memory T cells represented the major fraction of CD4+ T cells (64.9%) and of CD8+ T cells (45.4%). Naïve T cells represented a minority fraction of CD4+ T cells (4.3%) and of CD8+ T cells (3.2%). Stem-like TSMCs, defined as CD45RO-, CCR7+, CD45RA+, CD62L+, C\D27+, and CD28+, represented 17.6% of CD4+ and 13.7% of CD8+ T cells. The remaining subsets were central memory, effector memory, and terminal effector T cells [96].
In a previous study, the authors demonstrated that absolute numbers of CD3+, CD3+CD4+, CD3+CD8+, B, and NK cells decreased in patients with non-small-cell lung cancer, but their percentages were normal. Therefore, absolute cell counts were considered to be of clinical value [97].
The high percentage of CD3+ T cells, MTCs, and in particular TSCMs is remarkable and demonstrates the importance of these T lymphocyte subsets for general health.
It can be concluded from chapter 8 that there is a continuous and highly dynamic exchange of information regarding blood-borne antigens to immune cells bearing antigen-specific receptors. In a steady state, this system provides protection against NSAs and maintains tolerance to SAs.
8.5. CNS-Derived Antigens, CNS Immunosurveillance, and Cells Traveling through Cerebrospinal Fluid into Venous Blood
The CNS is lined by meninges, known as the dura, arachnoid, and pia mater. Recently, a fourth meningeal layer has been described, the subarachnoid lymphatic-like membrane (SLYM) [98]. It encases blood vessels and immune cells. The close apposition of SLYM with the endothelial lining of the meningeal venous sinus permits direct exchange of small solutes between cerebrospinal fluid and venous blood [98].
A calvarial hematopoietic niche was recently discovered in a region of the skull, acting as a myeloid cell reservoir to the underlying meninges [99]. Vascular channels traverse the inner skull of the cortex, providing a passageway for cells and cerebrospinal fluid-derived antigens. The review highlights the anatomical routes and physiological properties of the vascular structures the myeloid cells use for trafficking, spanning from the skull to the brain parenchyma [99].
A tightly controlled microglia network throughout the CNS parenchyma facilitates efficient immunosurveillance [100]. Each cell is constantly surveilling its microenvironment, screening for pathogens, but also removing cell debris and metabolites, grooming neighboring cells, and facilitating cellular crosstalk [100]. This “tissue surveillance” by microglia is an essential process for CNS homeostasis and development [101]. Microglia continuously monitor and sculpt synapses, allowing for the remodeling of brain circuits [102]. Glia-mediated neuroplasticity is driven by neuronal activity, controlled by a plethora of feedback signaling mechanisms, and crucially involves ECM remodeling in the CNS [102].
9. Neuro-Immune and Neuro-Osteogenic Links, Pathologies, and Interventions
9.1. Neuro-Immune Links
Immune cells and immune-derived molecules, endocrine glands and hormones, the nervous system, and neuro-derived molecules form the combined tridirectional neuroimmune network, which plays a significant role in communication pathways and regulation at the level of the whole organism and at local levels [103]. The details of such neuronal–immune cell units have been studied in allergic inflammation of the nose [103].
9.2. Pathologies and Interventions
- (i)
- CNS lymphoma. In primary CNS lymphoma, attention has turned to the long-term outcomes of consolidation therapies, and recent studies have highlighted the excellent disease control afforded by high-dose chemotherapy and stem cell transplantation [105]. Also, in patients with primary CNS lymphoma, chemoimmunotherapy with methotrexate, cytarabine, thiotepa, and rituximab (MATRix regimen) achieved impressive increases in complete remission rates [106].
- (ii)
- Malignant glioma (GBM). The glioma immune landscape has been described as a double-edged sword for treatment [107]. There are the effects of tumor cells on the tumor microenvironment, the immunosuppressive effects of myeloid immune cells, and the lymphocyte responses against the glioma cells [107]. Clinical and translational advances in malignant glioma immunotherapy have been summarized recently [108]. The review includes vaccine-based therapies, adoptive cell therapies, technical innovation, and outlook [108]. Synergy between temozolomide chemotherapy and individualized multimodal immunotherapy has been reported to improve the overall survival of IDH1 wild-type MGMT promoter-unmethylated GBM [109]. The concept of randomized controlled immunotherapy clinical trials for GBM has been challenged [110].
- (iii)
- Neuro-degenerative and neuro-autoimmune diseases. The role of T cells in brain inflammation has been reviewed [111]. The immune system is deeply involved in autoimmune diseases of the CNS, such as multiple sclerosis (MS), n-methyl-d-aspartate (NMDA) receptor encephalitis, and narcolepsy [111]. Additionally, the immune system is involved in neuro-degenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) [111]. The review focuses on the role of T cells, including CD4+ T cells, CD8+ T cells, and regulatory T cells (Tregs) in cerebral infarction and neuro-degenerative diseases [111]. Myelin oligodendrocyte glycoprotein (MOG) is an important auto (self)-antigen (SA) in inflammatory demyelinating diseases of the CNS [112].
- (iv)
- Chimeric antigen-receptor (CAR) T cells. Chimeric antigen receptor (CAR) T-cell therapy is a new and emerging cell therapy which has achieved remarkable success in the treatment of hematological malignancies [113]. The side effects include prolonged cytopenia (PC). Cytokine analysis after CAR T-cell infusion showed that CXCL12 and stem cell factor were significantly decreased in the BM of patients with PC, suggesting reduced niche cell function [114]. Another study revealed apoptosis of HSCs contributing to BM suppression following CAR T-cell therapy [115]. New technologies involving CAR-cytokine-induced killer cells (CAR-CIK) [116] and synapse-tuned CARs [117] demonstrated selective homing to BM niches [86i] and enhanced anti-tumor immune cell activity [116,117].
9.3. Neuro-Osteogenic Network
Skeletal tissue is highly innervated. The hallmarks of peripheral nerve function in bone regeneration were reviewed [118]. The review summarizes the ways in which the peripheral nervous system (PNS) communicates with bone-lineage cells, the vasculature, and immune cells in the bone microenvironment [118]. It was concluded that the PNS regulates bone regeneration through neuropeptides or neurotransmitters and cells in the peripheral nerves [118]. Another recent review described that peripheral nerves interact with bone through innervated axons, multiple neurotrophins, and bone resident cells [119].
10. Bone Marrow–Blood Interaction
10.1. BM Capacity for Cognate T Cell–APC Interactions
Cognate T-APC interactions represent the core of adaptive T-cell-mediated immune responses. Upon APC contact, the mobile T cells with their antigen-specific TCR scan the APC cell surface for the presence of non-self pMHC complexes with maximal fit. Maximal fit includes the TCR contact residue of the peptide and the polymorphic MHC/TCR contact residues. Random migration precedes stable T-APC interactions. The in vivo scanning process allows for four non-self pMHCs (NSAs) to be distinguished per TCR cluster from the vast majority of the approximately 10,000 normal self-pMHCs (SAs) of an APC [120]. Antigen availability and dose were shown to determine the T-APC interaction kinetics, T cell triggering, and memory fate decision [121].
The spatial organization of the TCR, its coreceptor (CD8 or CD4), LFA-1, and CD28 plays unique and complementary roles in T cell signaling and T cell cytoskeletal reorganization [122,123]. The proximity of TCR and its coreceptor causes a switch of T cells from a low- to a high-antigen sensitivity mode [122]. In recent years, we have witnessed important advances in our understanding of the regulatory processes that control and modulate the TCR signaling response [124].
Of utmost importance is the recent finding of mitochondrial priming by CD28 [125]. Costimulatory signals during the initial phase of T cell activation prime mitochondria with latent metabolic capacity, which is essential for future T cell responses [126]. TCR signaling without CD28 can elicit primary effector T cells, but MTCs generated during this process are anergic, thus failing to respond to secondary antigen exposure. Early CD28-dependent mitochondrial engagement is needed for T cells to remodel cristae, to develop enhanced spare respiratory capacity, to facilitate fatty acid oxidation, and to rapidly produce cytokines upon restimulation [76,106]. Costimulation also plays a role in the control of MTC homeostasis [126].
Activation of T cells by APCs occurs through several molecular interactions: TCR-pMHC, CD4/8-MHC, LFA-1-ICAM-1, and CD28-CD80 [123]. These lead to the folding of an immunological synapse (IS) [111] with supramolecular activation complexes (SMAC) in the periphery (p-SMAC) and within the center (c-SMAC). The synapses allow for actin polymerization, cytoskeletal reorganization and sustained T cell stimulation, a prerequisite for immunological memory [126,127,128]. TCR-pMHC binding leads to the exclusion of CD45, phosphorylation of CD3 ITAMs in cytoplasmic tails, and docking of Zap70 and Lck. This further enhances the recruitment of CD8 and Lat [129]. The segregated TCR and the local assembly of multimolecular signaling complexes merge into microclusters and move towards the center of the IS [130]. Similarly to neuronal synapses, ISs also transmit signals. While the former use electrical impulses, the latter use biochemical signals such as enzyme-mediated protein phosphorylation or de-phosphorylation.
During IS assembly, lymphocyte polarization occurs due to synaptic F-actin. It controls cytoskeletal changes via polymerization, leading to centrosome polarization [131]. Cytoskeleton changes are highly coordinated to allow for molecular traffic in T cell migration, activation, and effector function [132].
T cell migration occurs during APC scanning or during endothelial passage. TCR-driven transendothelial migration (TEM) of human effector memory CD4 T cells requires ZAP-70-dependent activation of a pathway involving Rho GTPases (Vav, Rac) and myosin IIA [133]. Interestingly, T cell costimulation can occur not only via DCs, but also via human endothelial cells, which can, thus, modulate antigen-dependent recruitment of circulating T lymphocytes [134].
Mechanical forces and waves of actin polymerization initiate the centripetal movement of signaling microcluster complexes (Figure 1) toward the p-SMAC [132]. The F-actin-rich ring acts as a scaffold for microcluster assembly and stabilization [132].
The importance of T-cell costimulation became evident due to the results of a phase I clinical study. Strong T-cell costimulation via vaccination of 14 colorectal carcinoma patients with late-stage disease was capable to re-activate anergized TA-specific MTCs [135]. The vaccine contained a bispecific anti-CD28 fusion protein attached to an autologous NDV virus-modified tumor cell vaccine [135].
Adhesion cascades, cytoskeletal rearrangement, and IS shape depend on the encountered cellular target [116]. T cell microvilli are actin-rich membrane protrusions that puncture cell barriers, such as the glycocalix. They thereby actively place the considerably smaller TCRs in close proximity to APC-presented pMHCs. This process has been designated as mechanosurveillance [136]. It has also been described as tiptoeing, and plays a role in target cell killing by CD8+ CTLs [137].
T cell microvilli are highly fragile and easily separated as membrane particles, forming a new class of EVs. Released T cell microvilli-derived particles can act as vectors, transmitting T cell messages to cognate APCs [138]. During T-APC interaction, T cell microvilli might function in two directions, as information sensors and information senders [138]. This could have played a role in the bi-directional stimulation reported upon cognate interaction of BM-derived MTCs from breast cancer patients with DC-derived APCs [139]: IFNα induced in DCs by T cells has a reciprocal effect on T cells by inducing expression of IL-12R β, thus enabling the T cells to respond to IL-12 and to differentiate into Th1 cells [25].
Figure 2 illustrates bone marrow as a central immune system.
10.2. Effects of MSC-Derived Stromal Cells in the BM
BM MSC-derived stromal cells provide survival niches for memory cells of the adaptive immunity system. Some of the mechanisms behind this property have been elucidated:
- (i)
- Stromal cell–immune cell contact-dependent PI3K and APRIL induces NF-kB signaling and prevents mitochondrial and ER stress of memory plasma cells [140];
- (ii)
- Stromal cell CD80/CD86 expression provides CD28 stimulation in BM-resident plasma cells, leading to sustained antibody responses [141];
- (iii)
- Stromal cells providing superior bio-availability of IL-15 cause upregulation of glucocortioid-induced TNF receptor (GITR) on CD8 MTCs [142];
- (iv)
- Stromal-cell-derived Il-7 mediates homeostasis of naïve and memory CD8 T cells in vivo [143];
- (v)
- Stromal-cell-expressed VCAM-1 holds immune cells in the niche and maintains plasma cell longevity [144].
In addition to providing survival niches for HSC-derived immune cells, stromal cells also organize niches for MSC-derived cells such as chondrocytes, osteocytes, and adipocytes. Several transcription factors control the fate of specific MSC-derived stromal BM lineages [4]. Based on gene signatures, 17 subpopulations of such stromal cells were identified [4]. One branch originating from the MSC cluster diverges into adipocyte precursor and pre-adipocyte clusters; another branch from the MSC cluster diverges into osteoblast and chondroblast precursors, which are followed by pre-osteoblast and pre-chondrocyte clusters, finally diverging into pro-osteoblast and chondrocyte clusters [4].
MSC differentiation in the BM is of primary importance for the maintenance of hematopoiesis, and is controlled by a complex signaling network. Runx2 and Osterix are key transcription factors for osteogenic differentiation, while PPARγ and CEBPα stand for adipogenic differentiation [4]. Stress-related factors directly affect MSC differentiation. Aging, radiation, and chemotherapy can trigger a number of events that regulate the key transcription factors Runx2 and PPARγ, leading to a shift in the adipo-osteogenic balance [4].
10.3. Autonomous BM-Derived Adaptive Immune Response
To exclude a contribution by secondary lymphoid organs to the T-cell response observed in BM, T cell transfer and activation experiments were repeated in splenectomized mutant Map3k14aly/aly mice, which lack lymph nodes and PPs [17].
In these mutant mice, 58% of the CD8+ T cells from transferred OT-1/Rag1−/− animals were CD69+ 6 h after the OVA antigen challenge, a value similar to that obtained with OT-I T cells in normal mice. Furthermore, 40% of the transferred CSFE-labeled CD4+ transgenic OT-II T cells upregulated CD69 expression 5 h after OVA antigen challenge, a value similar to that of the BM in normal mice [17].
Thus, mice devoid of secondary lymphoid organs can activate CD8+ and CD4+ T cells in their BM upon cognate antigen stimulation [17].
To conclude:
- (i)
- Animals devoid of secondary lymphatic organs are viable, while animals devoid of BM cannot survive and, therefore, do not exist;
- (ii)
- BM is self-sufficient with regard to T cell-mediated immune responses against blood-borne antigens.
The main features of chapters 8 to 10 are summarized in Table 2.
11. Bone Marrow: T Regulatory Cells and Dendritic Cells Reacting to Cancer and Microbial Infection
Tolerance to self-antigens is a fundamental property of the adaptive immune system. Failure of self-tolerance leads to autoimmune diseases. Central tolerance is induced in the thymus and BM when immature lymphocytes encounter SAs (self pMHCs). Peripheral tolerance occurs when mature lymphocytes recognize SAs in peripheral tissues under particular conditions; in CD4+ T cells, anergy is induced by antigen recognition without adequate costimulation or by engagement of inhibitory receptors such as cytotoxic T lymphocyte antigen 4 (CTLA-4) and programmed cell death protein-1 (PD-1). Tregs inhibit immune responses by means of multiple mechanisms [7].
11.1. Epigenetic Regulation
The genome-wide DNA-methylation landscape has been described to define specialization of Tregs in tissues [145]. Tregs maintain self-tolerance and support organ homeostasis by differentiating into specialized-tissue Tregs [145]. Epigenetic modifications define the molecular characteristics of tissue Tregs. Many gene sites are associated with the Th2 subset of helper T cells, for instance the gene encoding cytokine IL-33 receptor ST2. Tissue Tregs integrate multiple waves of epigenetic reprogramming that define their tissue-restricted specialization [145].
11.2. BM and Treg Cells
Emerging evidence has revealed that Tregs also play a crucial role in maintaining the self-renewal and multi-lineage differentiation capacity of stem cells in different tissues [145]. Following allogeneic BM transplantation, Tregs co-localize with infused allogeneic HSCs in unique niches to maintain their quiescence and development [146]. The BM Tregs are enriched in BM and serve a dual function of immunosuppression and maintenance of HSCs [145]. BM Tregs orchestrate stem cell niches crucial for hematopoiesis [146]. The transcription factor BATF sustains homeostasis and functionality of BM Tregs to preserve the homeostatic regulation of hematopoiesis and the development of B cells [147]. In comparison to the spleen, BM contains a higher frequency of Tregs. By secreting high levels of IL-10, BM Tregs maintain the homeostasis and function of mesenchymal stromal cells and HSCs [147].
Treg reconstitution is essential for re-establishing tolerance and maintaining homeostasis following HSC transplantation [148]. Compared to peripheral Tregs (pTreg), BM-Tregs are more quiescent, express lower FoxP3, and are highly enriched for co-inhibitory markers and more profoundly depleted than splenic Tregs in chronic graft-versus-host disease (cGVHD). Analyses have identified upregulated expression of IL-9R, IL-33R, and IL-7R in BM-Tregs [148]. Plasmacytoid DC (pDC) depletion was found to hamper BM, but not splenic Treg homeostasis. The unique properties of BM Tregs were recommended for new therapies to reconstitute Tregs and re-establish tolerance following HSC transplantation [148].
11.3. GvL without GvH
A murine GvL/GvH tumor model was used to study the effect of adoptive MTC therapy by BM-derived cells in comparison to spleen- or peritoneal-cavity-derived immune cells. Tumor-resistant donor mice (strain B10.D2) were immunized against ESb-MP lymphoma cells from host mice (strain DBA/2), and immune cells were removed from the previously mentioned tissues. Fifteen million immune cells were transferred i.v. into DBA/2 recipient mice bearing 4-week-established large ESb-MP tumors and liver and kidney metastases derived thereof. 1 day before cell transfer, separate groups of the recipient mice (n = 10) were whole-body irradiated by either 5 Gy or 7.5 Gy to suppress host-versus-graft (HvG) reactivity [149]. The results demonstrated that BM-derived immune cells were superior to spleen-derived immune cells in terms of conferring a GvL response, and conferred significant protection, while immune cells from the peritoneal cavity exhibited a significant detrimental GvH effect [150].
Further studies revealed that both short-term and long-term BM-derived MTCs exhibited GvL without GvH reactivity. A marked difference was observed in the phenotype of CD4+ T cells: while the BM of naïve donor mice harbored over 70% of CD44hiCD62L-CD4+ T cells, less than 20% of equivalent cells (MTCs) were detected in the spleen. When donor mice were immunized with the lymphoma cells at day 0 and boosted at day 7, 14 days later, their BM CD8+ T cells expressed to >80% CD69 in comparison to 20% at day 0. There was also a clear-cut increase in CD25- and CD44hi-expressing cells [150].
GvH disease (GvHD) is caused by alloreactive donor T cells that trigger host tissue injury [149]. A human skin explant GVHD model was used to evaluate the effects of Tregs [149]. Donor-derived CD8 T cells were stimulated with HLA-unmatched recipient DCs (priming phase). Then, they were co-cultured with recipient skin to induce GvH tissue damage (effector phase). Donor T regs present during priming effectively suppressed CD8 T cell-mediated GVHD [149]. Another study reported that host-reactive CD8+ stem-like MTCs played an important role in GVHD [151].
From these studies, it appears that in the GvL/GvH model [150], the priming of donor mice against the host-derived tumor cells induced not only CD4 and CD8 cancer-reactive MTCs, but also CD8 Tregs against host-derived antigens, in their BM. The model demonstrated effective immune rejection of advanced metastasized cancer [152] and provided an example of the reversion of dysregulation in late-stage cancer. This included cachexia, tumor tissue pH, rejection of primary tumor from the skin, and eradication of liver metastases [152].
Transient GvH reactivity was observed when using immune spleen cells from donor mice in 5 Gy pre-irradiated late-stage tumor-bearing hosts in the above model [150]. The peak of GvH reactivity was revealed in the liver exactly 5 days after immune cell transfer by loss of glycogen. Thereafter, liver regeneration was initiated by recruitment of BM-derived MSCs differentiating into fat-drop-bearing hepatic stellate cells (HeSCs) [153,154]. The SDF-1/CXCR4 axis was likely involved in liver recruitment of MSCs, in analogy with a rat pancreatitis model in which this axis regulated MSC to organ migration [155]. The liver regeneration likely involved the Hippo pathway, which controls organ size and tissue homeostasis [156].
11.4. Treg Cells in BM of Ewing Sarcoma Patients
Ewing sarcoma (ES) is thought to arise from MSCs, and is the second-most common bone sarcoma in pediatric patients and young adults. BM cells of 45 primary or relapsed ES patients treated with standardized protocols were analyzed for immune cell subsets by 6-color-flow cytometry [157]. A high proportion of BM T cells with regulatory phenotype (CD4+CD25hiFoxP3+) was found to be associated with immune escape and metastatic disease [157].
Immune escape factors in ES are the absence of MHC class I molecules from the tumor tissue and an immunosuppressive TME including myeloid-derived suppressor cells, F2 fibrocytes, and M2-like macrophages. A promising therapeutic strategy was reported. It consisted of reducing suppressive retinoblastoma protein complex expression by CDK 4/6 inhibitors and oncolytic adenovirus [158]. The combination potentiated the anti-tumor effect in murine ES xenografts [158].
11.5. Tumor-Specific BM Treg Cells in Breast Cancer Patients
Spontaneous antitumor effector T-cell responses and immune suppressive Tregs critically influence the prognoses of patients with cancer. On the basis of an analysis of the function, antigen specificity, and distribution of TA-reactive T cells and Tregs in patients with breast cancer and transgenic tumor models, it was shown that tumor-specific Tregs were selectively activated in the BM and egressed into the peripheral blood [159]. The BM was constantly depleted of TA-specific Tregs and was instead a site of increased induction and activity of tumor-reactive effector/memory T cells [159]. The egress of Tregs from the BM resulted in the accumulation of Tregs in breast tumor tissue. This could be demonstrated to be due to activation-induced expression of peripheral homing receptors, such as CCR2, and expression of the CCR2 ligand CCL2 in breast cancer tissue [159].
It was suggested (i) that the BM simultaneously represented a source of tumor-infiltrating Tregs and a site for the induction of endogenous TA-specific effector T-cell responses; and (ii) that both antitumor immunity and local immune suppression in the peripheral TME are orchestrated in the BM [159].
11.6. Effect of Microbial Infection and Inflammation on BM
11.6.1. Sepsis
Sepsis is a highly prevalent cause of death in intensive care units. A high-survival fecal-induced mouse peritonitis sepsis model was used to study acute and sustained alterations to the BM [160]. Single-cell RNA sequencing classified 3402 single cells from control mice into 14 clusters. One day following polymicrobial infection, cell composition changes and sepsis-induced transcriptional alterations were observed in most BM immune cell types. The changes failed to completely resolve 1 month after sepsis [160].
Recently an immune-related gene (IRG) signature was reported to predict 28-day mortality in patients with sepsis [161]. The prognostic IRG signature comprised three gene members (LTB4R, HLA-DMB, and IL-4R) [161].
Sepsis leads to the immunoparalysis of DCs. A recent study demonstrated that mitochondrial DNA (mtDNA) promotes the immunoparalysis of DCs via a stimulator of interferon gene (STING) signaling [162]. mtDNA transfection into BM-derived DCs inhibited the expression of costimulatory molecules and the release of IL-12p70 while increasing the secretion of Il-10. The deletion of STING reversed mtDNA-mediated immunoprophylaxis of DCs and improved the prognosis of endotoxemia and sepsis [162].
11.6.2. Severe Malaria
Malaria is an acute febrile illness caused by Plasmodium parasites, which are spread to people through the bites of infected female Anopheles mosquitos. In 2021, an estimated 247 million cases of malaria worldwide were reported by the World Health Organization, with a high proportion in regions of Africa. The estimated number of malaria deaths in 2021 stood at 619,000.
A recent study revealed that CD169+ macrophages orchestrate pDC arrest and retention for optimal priming in the BM of malaria-infected mice [163]. pDCs are the most potent producers of IFN-I. It was demonstrated that TLR7 sensing in pDC and chemotaxis sensing via STING signaling were both required for pDC arrest in the BM. The results link pDC acquisition of IFN-I secreting capacity upon malaria infection and functional communication with CD169+ macrophages with changes in their motility and homing behavior [163]. It was proposed that targeting this pDC–macrophage interaction may help to modulate the IFN-I response to improve the outcomes of microbial infections and autoimmune diseases [163].
11.6.3. Colitis
Inflammatory bowel diseases (IBD) can be chronic or acute. Colitis ulcerosa and Morbus Crohn are two types of colitis based on autoimmunity.
Melatonin (MLT) has been reported to have therapeutic effects on IBD. In a mouse model of dextran sulfate sodium-induced colitis, RNA sequencing was employed to investigate the effects of MLT on BM-derived DCs [164]. MLT promoted the transformation of BM-DCs into tolerant phenotypes. Non-coding RNAs affected the PI3K-Akt pathway [164].
In conclusion, in all four reports from 2022 about microbial infection and inflammation in the periphery, DCs from the BM play an important role.
12. Tumor Dormancy in BM and Maintenance of Tumor-Specific MTCs
Immune compartmentalization and redistribution of T-cell subpopulations between the BM and peripheral tissues were achieved by vaccination with adenoviral vector-encoded TRP-2 as TA in a TET transgenic mouse model of spontaneous malignant melanoma [165]. Disseminated BM-dormant TRP-2+ tumor cells co-localized with memory CD8+ T cells. It was suggested that these dormant melanoma cells in the BM play an important role in the maintenance of MTCs in the BM [165].
Evidence had been provided before for spontaneous immune T cell reactivity in the BM from the well-defined ESb-Gal tumor model [21,22]. A novel model was established for the study of long-term protective anti-tumor immunity and immune T cell memory [166]. Long-term immune memory and tumor protection could be maintained over four successive transfers of Gal-primed T cells between tumor-inoculated recipient nude (nu/nu) mice. The Gal-specific CD8+ MTCs from the first transfer were able to be activated and recruited into the peritoneal cavity by ip tumor cell challenge. From there, they were harvested for a second adoptive transfer together with tumor cell challenge. About four weeks later, the Gal-specific MTCs had returned into a resting state and were detectable in the BM. This long-term experiment (>6 months) demonstrated the longevity and functionality of the MTCs [167]. A low level of persisting Gal antigen appeared to favor the selection of Gal-specific MTCs over irrelevant MTCs in BM niches of CD8+ MTCs [167].
The main features of chapters 11 and 12 are summarized in Table 3.
13. Clinical Studies Revealing the Importance of Stem-like MTCs
Ovarian cancer immunogenicity was reported to be governed by a narrow subset of progenitor tissue-resident MTCs [168]. Single-cell sequencing of CD3+CD8+CD103+CD69+ tissue-resident MTCs and immunohistochemistry of 122 high-grade serous ovarian cancer patients showed that only progenitor (TCF1low stem-like) T cells predicted ovarian cancer outcomes [168].
Peptide vaccine-treated and long-term surviving hepatocellular carcinoma patients were reported to harbor self-renewing tumor-specific CD8+ T cells [169]. Induced peptide-specific T cells could persist beyond 10 years following vaccination. It was concluded that effective anti-tumor immunity is governed by potentially proliferative MTCs, specific to TAs [169].
A pilot clinical study was performed to evaluate the feasibility and side effects of treatment of advanced metastasized breast cancer with BM-derived tumor-reactive MTCs [170]. The study protocol involved one transfusion of T cells, which were reactivated ex vivo with autologous DCs pulsed with lysate from MCF7 breast cancer cells, as a source of TAs. Twelve patients with proven pre-existing tumor-reactive MTCs were included. None of the advanced patients had Th1 cancer-reactive MTCs in their blood before the treatment. The treatment was feasible and well-tolerated. Six patients (immunological responders) showed de novo TA-specific IFN-γ secreting T cells in the blood after 7 days, according to an ELISPOT assay. These responder patients had received only 6.5 × 103 re-activated T cells from the memory restimulation culture. The observed blood-circulatory MTCs suggested an enormous in vivo expansion capacity of the transfused cells [170].
14. Bone Marrow Mesenchymal Stem Cells and Stromal Cells
The mesenchymal compartment of the BM is as diversified as its hematopoietic counterpart [9]. The International Society for Cell Therapy (ISCT) defined the MSCs as CD105+, CD73+, CD90+, and expressing the transcription factor Oct-4. They reside in a BM niche together with embryonal stem cells (ESC), multipotent adult stem cells (MASC), and mesodermal adult progenitor cells (MAPC) [172,173]. MSCs are adherent, proliferating, and colony-forming cells. The largest fraction of the BM mesenchymal component presents in the form of a dense weblike network of perivascular cells interconnected through large cytoplasmic projections that pervade entire BM tissues [10]. These fibroblast-like reticular cells are leptin-receptor- and CD140a-positive and secrete large amounts of regulatory factors, such as IL-7, stem cell factor, and CXCL12 [9].
MSCs and stromal cells play important roles in the BM. They can differentiate into vascular endothelial cells, adipocytes, chondrocytes, osteocytes, and niche-forming stromal cells. Lineage commitment, signaling pathways, and the cytoskeleton systems in MSCs have been described [174]. The differentiation of MSCs is regulated via a complex network of interrelated signaling pathways, including the small Rho GTPases RhoA/ROCK, Akt/Erk, the large tumor-suppressor kinase LATS1/2, and the transcriptional co-activator yes-associated protein (YAP)/TAZ of the Hippo pathway [174,175]. MSCs communicate via cytoplasmic protrusions among themselves as well as with immune cells. They can secrete anti-microbial peptides and can initiate neoangiogenesis [175].
Mesenchymal connective tissue is a non-fibrillar reticular tissue derived from the embryonic mesoderm that also contains parts from the embryonic ectoderm and entoderm. It is involved in the formation of bones and cartilage, blood vessels, lymphatic vessels, the kidneys and adrenal cortex, smooth musculature, and cardiac muscle. BM also contains ectoderm neuronal stem cells [176].
14.1. Effects on Tumors
In TMEs, different sources of MSCs show tumor-promoting and -inhibiting effects mediated by different signaling pathways [177]. Cancer-associated MSCs recruited from the BM mainly show tumor-promoting and immunosuppressive effects [177]. The transformed cancer-associated MSCs preserve the characteristics of stem cells, but the TME supports their tumor-promoting properties.
14.2. Effects in the Liver: Hepatic Stellate Cells, MTCs, and Liver Fibrosis
BM can exert a controlling effect on long-lasting CD8+ MTCs not only directly within its own niches but also indirectly, via other cells, in niches of the liver. The liver is endowed with the capability to effectuate long-lasting functional T cell memory [181]. The homeostasis of memory CD8+ T cells in niches from the BM is thought to be mediated by IL-15/Il-15R heterodimer (15HD)-expressing myeloid cells. Nonmyeloid HeSCs, which can be replenished from MSCs of the BM, also express 15HD. A mouse strain was genetically engineered to express IL-15R exclusively in HeSCs. Such mice developed and maintained long-lasting systemic antigen-specific CD8+ MTCs that were efficacious against tumor growth and Listeria infection. The MTCs appeared uniquely in the form of clusters in a specialized structure, i.e., sinusoidal niches of the liver [181].
Activated HeSCs, portal fibroblasts, and myofibroblasts of BM origin have been identified as major collagen-producing cells in the injured liver. Recent studies have revealed that liver fibrosis, a major health problem worldwide, is reversible [182]. Activated HeSCs can revert to quiescent HeSCs when causative agents are removed. Overexpression of a micro-RNA (mir-483-5p/3p) cooperated to inhibit mouse liver fibrosis [183]. Also, it was shown that oncolytic NDV can repress the activation of human HeSCs and reverse the development of hepatic fibrosis in mice [184].
14.3. Effects in Regenerative Medicine
MSCs and the exosomes and extracellular vesicles (EVs) derived thereof exert promising effects in regenerative medicine. They have anti-inflammatory, anti-apoptotic, and immunomodulatory properties. A few examples of recent publications demonstrate the potential of MSCs.
- (i)
- Heart failure: Human MSCs are being used to treat patients for heart failure such as cardiomyopathy. A systematic review and meta-analysis revealed that this is an effective treatment modality [185].
- (ii)
- Neurological diseases: Intracerebral hemorrhage (ICH) is a common acute nervous system disease with high mortality and causing severe disability. BM MSC transplantation alleviates brain injury after ICH in mice through the Hippo signaling pathway [186]. Polarized anti-inflammatory MSCs increase hippocampal neurogenesis and improve cognitive function in aged mice [187]. The transplantation of nasal olfactory mucosa MSCs showed benefits in the AD mouse model of Alzheimer’s disease (AD) [188]. An exosomal microRNA (miR-146a) secreted from BM-MSCs is taken up into astrocytes [189]. Intracerebra-ventricularly injected BM-MSCs improve cognitive impairment by increasing the expression of miR-146a in the hippocampus [190]. This is due to an effect of astrocytes, which are key cells for the formation of synapses. Thus, restoration of astrocyte function may lead to synaptogenesis and correction of cognitive impairment [191]. MSCs are likely able to cross the blood–brain barrier. Therefore, MSC-based therapy is regarded as an important means of ameliorating neurological impairment [187,188,189,190,191].
- (iii)
- Lung injury and fibrosis: MSC-derived EVs attenuate radiation-induced lung injury via miRNA-214-3p [192]. MSCs with downregulated Hippo signaling are better than normal MSCs in terms of attenuating lung injury in mice with acute respiratory distress syndrome (ARDS) [193]. The therapeutic efficacy of MSCs has been reported for post-COVID pulmonary fibrosis [194].
- (iv)
- Hepatobiliary diseases and sepsis: Adipose-derived MSCs (AD-MSCs) have the potential to modulate inflammation, ameliorate ischemia–reperfusion injury, and support liver and biliary tract regeneration [195]. The anti-inflammatory potential of these cells also has paramount importance in the treatment of sepsis [196].
- (v)
- Kidney injury and diseases: Murine MSCs protect against sepsis-associated acute kidney injury [196]. Rat MSCs ameliorate chronic kidney disease injury via regulating the Nrf2-keap1/p53 pathway [197]. A multi-therapeutic role of MSCs has been reported in diabetic nephropathy [198], and engineered BM stem cell-sheets were shown to alleviate renal damage in a rat model of chronic glomerulonephritis [199].
- (vi)
- Osteoarthritis: Rheumatic diseases such as osteoarthritis (OA) are a major social and economic burden. MSCs provide a cell source for cartilage regeneration due to numerous advantages, comprising relative ease to isolate and culture, chondrogenic capacity, and anti-inflammatory effects. Preclinical studies for articular cartilage repair have been reviewed [200]. Clinical-grade embryonic stem cell-derived mesenchymal stromal cells were shown to ameliorate the progression of OA in a rat model [201].
- (vii)
- Vascular regeneration: A mechanical conditioning technique applied to human MSCs enhances vascular regeneration [202].
- (viii)
- (ix)
- Autoimmunity: Transplantation of human umbilical cord MSCs ameliorated systemic lupus erythematosus (SLE) in MRL/lpr mice [205]. MSC-derived exosomes ameliorated lupus in the same mouse model by inducing M2 macrophage polarization and Treg cell expansion [206]. Melatonin treatment improved human MSC therapy in a mouse model of type II diabetes mellitus (T2DM) via the PI3K/AKT signaling pathway [207]. Transplanted MSCs ameliorated the clinical course of experimental autoimmune encephalomyelitis (EAE) in a mouse model of multiple sclerosis (MS) [191]. Human umbilical cord-derived MSCs ameliorated psoriasis-like dermatitis by suppressing IL-17 producing γ, δ T cells [208].
The therapeutic potential of MSCs has been known for about three decades, and also includes spinal cord injury [209] and cartilage repair. It is remarkable that many of the applications of MSCs in regenerative medicine have been published in recent years. The use of MSC-derived products is regulated as an Advanced Therapy Medicinal Product (ATMP), a Tissue Engineering Product (TEP) or a Somatic Cell Therapy Medicinal Product (sCTMP).
The main features of chapter 14 are summarized in Table 4.
Figure 3 summarizes the major points of this article.
15. Highlights and Perspectives
This review describes bone marrow as a central immune system in an analogy to the central nervous system. It is localized in the center of the body, protected by bone, and performs a multitude of functions to protect the body when it is in danger.
The review celebrates 20 years since the discovery of bone marrow as a priming site for T cell responses to blood-borne antigens [13], and reports findings regarding progress made on this subject since then. Nearly 50% of the cited references are from 2020 to 2023.
Among all antigen-responsive immune organs (BM, spleen, lymph nodes), BM is the largest. Comprising 4–5% of the total body weight, it exceeds other major vital organs such as the brain (2%), liver (2.6%), heart (0.5%), and spleen and lymph nodes (1–1.5%). In addition, BM is the most prominent source of de novo cellular generation within the body. Considering these facts, the newly described functions of this immune organ are remarkable:
- (i)
- Antigen presentation capacity;
- (ii)
- T-APC interaction capacity;
- (iii)
- Memory cell storage capacity;
- (iv)
- Capacity of maintaining self-tolerance and immune homeostasis;
- (v)
- Capacity for tissue repair;
- (vi)
- Capacity to adapt to periods of energy crisis;
- (vii)
- Ability to sense signals via adrenergic nerve fibers from the autonomous peripheral nervous system;
- (viii)
- Capacity of immunosurveillance of the central nervous system.
Box 1 summarizes the main characteristics of the BM as a central, multifunctional, and protective immune system
Box 1. Characteristics of the bone marrow.
| 1. Central: |
|
| 2. Multifunctional: |
|
| 3. Protective: |
|
The evolution of vertebrates apparently went along with co-evolution of the bones, of the CNS and the CIS.
Box 2 contains a description of the BM and its major features.
Box 2. Functions of the bone marrow.
| Feature | Description |
| Hematopoiesis | HSCs program the major lineages of blood cells Proliferation and maturation of committed precursor cells via colony-stimulating factors, growth factors, cytokines, and chemokines BM stromal cells providing signals for the development of lymphocyte progenitors from HSCs and for subsequent differentiation of pre-B/B cells and pre-T cells Egress to the thymus of BM-derived pre-T cells for further T cell differentiation with positive and negative selection Homeostatic hematopoiesis provides the organism with oxygen, energy, and immune protective capacity |
| Osteogenesis | MSCs program the major lineages of osteogenesis Osteogenesis, like hematopoiesis, is constantly maintained at a steady state HSC-derived osteoclasts interact in balance with MSC-derived osteoblasts and osteocytes |
| Antigen-specific T cell response to blood-borne antigens | Microenvironment facilitates T-APC interactions between BM and blood Priming of naïve T cells Responses resulting in the generation of CTL activity, protective anti-tumor immunity, and immunological memory Cognate antigen-specific re-activation of memory T cells |
| Storage and protection of immunological memory | Multiple niches created by stromal cells providing quiescence and survival signals Stem-like MTCs with special properties |
| Adaptation to energy crisis (transient dietary restriction) | Mobilization of monocytes from the BM Migration of B cells from PPs to BM Recruitment of MTCs from the periphery Increase in T cell homing factors, erythropoiesis, and adipogenesis Reorganization of the BM compartment |
| Central regulatory function within the immune system | Maintenance of homeostasis in concerted interaction of regulatory immune cells with the neuronal network Feedback from the periphery (lymph nodes) leads to central regulatory means in the BM |
| Autonomous life-protecting function | Secondary immune functions of BM independent from the lymph nodes and spleen No animal model without BM exists No bone cortex without BM MSCs Most prominent source of de novo cell generation in the body In communication with the autonomous peripheral nervous system, BM is autonomous in the maintenance of homeostasis |
| BM mesenchymal stem cells and stromal cells for tissue repair and niche formation | MSCs: great potential in regenerative medicine, e.g., heart failure, neurological diseases, lung injury and fibrosis, liver regeneration, kidney injury, vascular regeneration, and autoimmune diseases Stromal cells: Important role in niche formation, e.g., niches for HSCs, MSCs, memory B cells, memory plasma cells, memory CD4+ T cells, memory CD8+ T cells, stem-like memory T cells, and neuronal stem cells |
What are the perspectives and medical implications of these new findings?
- As a major immune organ, the BM is a treasure and should receive more attention. Many of its secrets have not yet been resolved and require intensive research efforts. We know more about the immune functions in the lymph nodes and spleen than about those in the BM. It would be fascinating, for instance, to find out how many T-APC immune synapses occur in the BM at any time point and to compare this with other immune organs. Also, the question of adjuvant requirement for vaccination responses in lymph nodes and BM requires further investigations. If the requirements are less for the BM, intra-BM vaccination might reduce the risk of adjuvant-associated side effects.
- Most of the cytostatic anti-cancer drugs that have been approved in the past have been proven to have detrimental effects on the BM (e.g., chlorambucil, melphalan, busulfan, thioguanin, cyclophosphamide, ifosfamide, imatinib, vinblastin, cisplatin, etoposid, chloramphenicol). New drug approval should minimize detrimental drug effects on the BM.
- Among the available immunotherapeutic anti-cancer drugs, the most important are immune checkpoint inhibitors, anti-cancer vaccines, oncolytic viruses, and adoptive T cell therapies. All of these have to be assessed with regard to their side effects on the BM. Cancer vaccines and oncolytic viruses have been found to exert profoundly lower side effects in cancer patients than other systemic therapies.
- Adoptive T cell therapies against cancer should make use of BM-enriched cancer-reactive MTCs, including stem-like MTCs, and their synergistic effects with dendritic cells as APCs and hematopoietic stem cells.
- To achieve improvements to the graft-versus-leukemia reactivity of allogeneic donor cells, vaccination of donors against host tumor cells is recommended. Donor-derived immune BM mononuclear cells from vaccinated donors are likely to contain cancer-reactive MTCs and primed T regulatory cells, preventing graft-versus-host disease.
- Given the fact that hematopoietic and mesenchymal stem cells from the BM interact with T cells and DCs, it is conceivable that in BM, stem cell-derived committed precursors and late precursors also interact with mature immune cells and build informative feedback-regulatory networks.
- Viral and microbial infections, inflammatory reactivities by the immune system, and auto-immune reactivities are of major medical concern. Experimental models of sepsis, malaria, and colitis have revealed effects on BM-derived DCs. New possibilities for central intervention should be explored.
- BM mesenchymal stem cells have great potential for regenerative medicine. This review presents many examples from research conducted within the last few years.
- The BM and the brain are the two centers (i) of learning by experience, (ii) of memory establishment, and (iii) of memory storage.
- The fascinating area of communication between networks of the immune system and those of the neuronal system is a promising future research area in which artificial intelligence (AI) could be helpful. Deep learning in AI attempts to simulate nature’s neuronal network learning process. Positive impacts can be expected, in particular in the fields of neuro-degenerative and neuro-autoimmune diseases.
Funding
This paper is funded by the IOZK foundation (www.iozk-stiftung.org (accessed on 17 November 2020)).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the studies of the author.
Data Availability Statement
Not applicable.
Acknowledgements
The excellent and fast help with the graphical designs by Andrea Riegel ([email protected]) is gratefully acknowledged. I thank in particular Kash Khazaie [21] and Markus Feuerer [17,19] for their scientific input in the years 1994 and 2001–2003, respectively.
Conflicts of Interest
The author declares no conflict of interest. The author is affiliated with the Immunological and Oncological Center Cologne (IOZK). He has no other affiliations or financial involvement with any organization or entirety with a financial interest in or financial conflict with the subject matter in the manuscript.
Abbreviations
ACT = adoptive T cell therapy; AD = Alzheimer’s disease; AD-MSC = adipose-derived MSC; AMPK = 5’-AMP-activated protein kinase; ARDS = acute respiratory distress syndrome; ATMP = Advanced Therapy Medicinal Product; ATV-NDV = autologous tumor cell vaccine modified by infection with Newcastle disease virus; BATF = basic leucin zipper transcription factor; BCL2 = B-cell lymphoma protein-2; BCR = B-cell receptor; BL-DC = blood-derived dendritic cells; BM = bone marrow; BM-DC = BM-derived dendritic cell; CCL2 = CC-chemokine ligand 2; CMV = cytomegalovirus; CNS = central nervous system; CTL = cytotoxic T lymphocyte; CTLA-4 = cytotoxic T lymphocyte antigen 4; ctDNA = circulatory tumor DNA; CXCL13 = C-X-C motif chemokine 13; DAMP = damage-associated molecular pattern; DC = dendritic cell; DR = dietary restriction; EAE = experimental autoimmune encephalomyelitis; EBV = Epstein–Barr virus; ECM = extracellular matrix; ES = Ewing sarcoma; ESC = embryonal stem cell; Gal = bacterial β-galactosidase protein; GBM = glioblastoma multiforme; GC = glucocorticoid; GITR = glucocortioid-induced TNF receptor; GM-CSF = granulocyte-macrophage colony-stimulating factor; GvH = graft-versus-host effect; GvHD = GvH disease; GvL = graft-versus-leukemia effect; HBV = hepatitis B virus; HCV = hepatitis C virus; hCRP = human C-reactive protein; HeSC = hepatic stellate cell; HIV = human immunodeficiency virus; HMGB1 = high-mobility group box 1; Hpa = heparanase; HPV = human papilloma virus; HvG = host-versus-graft reaction; IBM = intra-BM; IBD = inflammatory bowel disease; IC = immune complex; ICD = immunogenic cell death; ICH = intracerebral hemorrhage; IFN-I = type I interferons (a and β); Ig = immunoglobulin; IS = immunological synapse; ITAM = immunoreceptor tyrosin-based activation motifs; LEF1 = lymphoid enhancer binding factor; LepR = leptin receptor; MadCAM = mucosa-associated cell adhesion molecule; MASC = multipotent adult stem cell; MAPC = mesodermal adult progenitor cell; mEHT = moderate electrohyperthermia; MHC = major histocompatibility complex; MLT = melatonin; MSC = mesenchymal stem cell; MS = multiple sclerosis; MTC = memory T cell; mtDNA = mitochondrial DNA; NDV = Newcastle disease virus; NG2 = neuron-glial antigen-2; NSA = non-self-antigen; OA = osteoarthritis; OV = oncolytic virus; OVA = ovalbumin; PBL = peripheral blood lymphocyte; PD1 = programmed cell death protein-1; pDC = plasmacytoid dendritic cell; PP = Peyer’s patch lymph node; PPAR = peroxisome proliferator-activator receptor; SA = self-antigen; S1P = sphingosine-1-phosphate; S1PR = receptor of S1P; Sca-1 = stem cell antigen-1; SDF-1 = stromal-cell-derived factor 1; SLE = systemic lupus erythematosus; STING = stimulator of interferon genes; TA = tumor antigen; TBW = total body weight; TCF1 = T cell factor 1; TCR = antigen-specific T cell receptor; TEM = TCR-driven transendothelial migration; TLR = toll-like receptor; TME = tumor microenvironment; TEP = tissue engineering product; T2DM = type II diabetes mellitus; TSCM = stem-like memory T cell; Treg = T regulatory cell; VCAM-1 = vascular cell adhesion molecule 1; WT1 = Wilms tumor suppressor gene 1.
References
- Flajnik, M.F.; Kasahara, M. Origin and evolution of the adaptive immunity system: Genetic events and selective pressures. Nat. Rev. Genet. 2010, 11, 47–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pancer, Z.; Amemiya, C.T.; Ehrhardt, G.R.A.; Ceitlin, J.; Gartland, G.L.; Cooper, M.D. Somatic diversification of variable lymphocyte receptors in the agnathan sea lamprey. Nature 2004, 430, 174–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estefa, J.; Tafforeau, P.; Clement, A.M.; Klembara, J.; Niedzwiedzki, G.; Berruyer, C.; Sanchez, S. New light shed on the early evolution of limb-bone growth plate and bone marrow. eLife 2021, 10, e51581. [Google Scholar] [CrossRef] [PubMed]
- Belyavsky, A.; Petinati, N.; Drize, N. Hematopoiesis during ontogenesis, adult life, and aging. Int. J. Mol. Med. 2021, 22, 9231. [Google Scholar] [CrossRef]
- Osmond, D.G. Production and selection of B lymphocytes in bone marrow: Lymphostromal interactions and apoptosis in normal, mutant and transgenic mice. Adv. Exp. Med. Biol. 1994, 355, 15–20. [Google Scholar]
- Koni, P.A.; Joshi, S.K.; Temann, U.A.; Olson, D.; Burkly, I.; Flavell, R.A. Conditional vascular cell adhesion molecule 1 depletion in mice impaired lymphocyte migration to bone marrow. J. Exp. Med. 1994, 355, 15–20. [Google Scholar]
- Abbas, A.; Lichtman, A.; Pillai, S. (Eds.) Cellular and Molecular Immunology, 10th ed.; Elsevier: Philadelphia, PA, USA, 2022. [Google Scholar]
- Shahrabi, S.; Rezaeeyan, H.; Ahmadzadeh, A.; Shahjahani, M.; Saki, N. Bone marrow blood vessels: Normal and neoplastic niche. Oncol. Rev. 2016, 10, 72–77. [Google Scholar] [CrossRef] [Green Version]
- Marenzana, M.; Arnett, T.B. The key role of the blood supply to bone. Bone Res. 2013, 1, 203–215. [Google Scholar] [CrossRef] [Green Version]
- Nombela-Arrieta, C.; Manz, M.G. Quantification and three-dimensional microanatomical organization of the bone marrow. Blood Adv. 2017, 1, 407–416. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Li, R.; Zhang, Y.; Liu, Y.; Wang, M.; Huang, J.; Zhu, C.; Zhang, J.; Yuan, X.; Liu, Q. The discovery of a new type of innervation in lymphoid organs. Physiol. Rep. 2023, 11, e15604. [Google Scholar] [CrossRef]
- Pabst, R. The bone marrow is not only a primary lymphoid organ: The critical role for T lymphocyte migration and housing of long-term plasma cells. Eur. J. Immunol. 2018, 48, 1096–1100. [Google Scholar] [CrossRef] [Green Version]
- Feuerer, M.; Rocha, M.; Bai, L.; Umansky, V.; Solomayer, E.F.; Bastert, G.; Diel, I.J.; Schirrmacher, V. Einrichment of memory T cells and other profound immunological changes in the bone marrow from untreated breast cancer Patients. Int. J. Cancer 2001, 92, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Waxenbaum, J.A.; Reddy, V.; Futterman, B. Anatomy, back, thoracic vertebrae. In StatPearls (Internet); StatPearls Publishing: Treasure Islands, FL, USA, 2023; Bookshelf ID: NBK459153. [Google Scholar] [PubMed]
- Ordovas-Montanes, J.; Rakoff-Nahoum, S.; Huang, S.; Riol-Blanco, L.; Barreiro, O.; von Andrian, U.H. The regulation of immunological processes by peripheral neurons in homeostasis and disease. Trends Immunol. 2015, 36, 578–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comazzetto, S.; Murphy, M.M.; Berto, S.; Jeffery, E.; Zhao, Z.; Morrison, S.J. Restricted hematopoietic progenitors and erythropoiesis require SCF from Leptin receptor+ niche cells in the bone marrow. Cell Stem Cell 2019, 24, 477–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feuerer, M.; Beckhove, P.; Garbi, N.; Mahnke, Y.; Limmer, A.; Hommel, M.; Hämmerling, G.J.; Kyewski, B.; Hamann, A.; Umansky, V.; et al. Bone marrow as a priming site for T-cell responses to blood-borne antigen. Nat. Med. 2003, 9, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Mazo, I.B.; von Andrian, U.H. Adhesion and homing of blood-borne cells in bone marrow microvessels. J. Leukoc. Biol. 1999, 66, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Feuerer, M.; Beckhove, P.; Bai, L.; Solomayer, E.F.; Bastert, G.; Diel, I.J.; Pedain, C.; Oberniedermayr, M.; Schirrmacher, V.; Umansky, V. Therapy of human tumors in NOD/SCID mice with patient-derived reactivated memory T cells from bone marrow. Nat. Med. 2001, 7, 452–458. [Google Scholar] [CrossRef]
- Feuerer, M.; Beckhove, P.; Mahnke, Y.; Hommel, M.; Kyewsky, B.; Hamann, A.; Umansky, V.; Schirrmacher, V. Bone marrow microenvironment facilitating dendritic cell: CD4 T cell interactions and maintenance of CD4 memory. Int. J. Oncol. 2004, 25, 867–876. [Google Scholar]
- Khazaie, K.; Prifti, S.; Beckhove, P.; Griesbach, A.; Russel, S.; Collins, M.; Schirrmacher, V. Persistence of dormant tumor cells in the bone marrow of tumor cell-vaccinated mice correlates with long-term immunological protection. Proc. Natl. Acad. Sci. USA 1994, 91, 7430–7434. [Google Scholar] [CrossRef]
- Müller, M.; Gounari, F.; Prifti, S.; Hacker, H.J.; Schirrmacher, V.; Khazaie, K. EblacZ tumor dormancy in bone marrow and lymph nodes: Active control of proliferating tumor cells by CD8+ immune T cells. Cancer Res. 1998, 58, 5439–5446. [Google Scholar]
- Milo, I.; Sapoznikov, A.; Kalchenko, V.; Tal, O.; Krauthammer, R.; van Rooijen, N.; Dudziak, D.; Jung, S.; Shakkar, G. Dynamic imaging reveals promiscuous crosspresentation of blood-borne antigens to naïve CD8+ T cells in the bone marrow. Blood 2013, 122, 193–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Stephani, J.; Schaefer, M.; Kalay, H.; Garcia-Vallejo, J.J.; den Haan, J.; Saeland, E.; Sparwasser, T.; van Kooyk, Y. Targeting glycan modified OVA to murine DC-SIGN transgenic dendritic cells enhances MHC class I and II presentation. Mol. Immunol. 2009, 47, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V.; Feuerer, M.; Fournier, P.; Ahlert, T.; Umansky, V.; Beckhove, P. T-cell priming in bone marrow: The potential for long-lasting protective anti-tumor immunity. Trends Mol. Med. 2003, 9, 526–534. [Google Scholar] [CrossRef]
- Schirrmacher, V. Cancer-reactive memory T cells from bone marrow: Spontaneous induction and therapeutic potential (Review). Int. J. Oncol. 2015, 47, 2005–2016. [Google Scholar] [CrossRef] [Green Version]
- Bai, L.; Feuerer, M.; Beckhove, P.; Umansky, V.; Schirrmacher, V. Generation of dendritic cells from human bone marrow mononuclear cells: Advantages for clinical application in comparison to peripheral blood monocyte derived cells. Int. J. Oncol. 2002, 20, 247–253. [Google Scholar] [CrossRef]
- Sommerfeldt, N.; Schütz, F.; Sohn, C.; Förster, J.; Schirrmacher, V.; Beckhove, P. The shaping of a polyvalent and highly individual T-cell repertoire in the bone marrow of breast cancer patients. Cancer Res. 2006, 66, 8258–8265. [Google Scholar] [CrossRef] [Green Version]
- Sommerfeldt, N.; Beckhove, P.; Ge, Y.; Schütz, F.; Choi, C.; Bucur, M.; Domschke, C.; Sohn, C.; Schneeweis, A.; Rom, J.; et al. Heparanase: A new metastasis-associated antigen recognized in breast cancer patients by spontaneously induced memory T lymphocytes. Cancer Res. 2006, 66, 7716–7723. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Mei, H.; Dörner, T.; Hiepe, F.; Radbruch, A.; Fillatreau, S.; Hoyer, B.F. Memory B and memory plasma cells. Immunol. Rev. 2010, 237, 117–139. [Google Scholar] [CrossRef] [PubMed]
- Slamanig, S.A.; Nolte, M.A. The bone marrow as sanctuary for plasma cells and memory T-cells: Implications for adaptive immunity and vaccinology. Cells 2021, 10, 1508. [Google Scholar] [CrossRef]
- Schirrmacher, V. New insights into mechanisms of long-term protective anti-tumor immunity induced by cancer vaccines modified by virus infection. Biomedicines 2020, 8, 55. [Google Scholar] [CrossRef] [Green Version]
- Di Rosa, F.; Pabst, R. The bone marrow: A nest for migratory memory T cells. Trends Immunol. 2005, 26, 360–366. [Google Scholar] [PubMed]
- Mazo, I.B.; Honczarenko, M.; Leung, H.; Cavanagh, L.L.; Bonasio, R.; Weninger, W.; Engelke, K.; Xia, L.; McEver, R.P.; Koni, P.A.; et al. Bone marrow as a major reservoir and site of recruitment for central memory CD8+ T cells. Immunity 2005, 22, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Becker, T.C.; Coley, S.M.; Wherry, E.J.; Ahmed, R. Bone marrow is a preferred site for homeostatic proliferation of memory CD8 T cells. J. Immunol. 2005, 174, 1269–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Dong, H.; Lin, W.; Voss, S.; Hinkley, L.; Westergren, M.; Tian, G.; Berry, D.; Lewellen, D.; Vile, R.G.; et al. Human bone marrow: A reservoir for “enhanced effector memory” CD8+ T cells with potent recall function. J. Immunol. 2006, 177, 6730–6737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murao, A.; Oka, Y.; Tsuboi, A.; Elisseeva, O.A.; Tanaka-Harada, Y.; Fujiki, F.; Nakajima, H.; Nishida, S.; Hosen, N.; Shirakata, T.; et al. High frequencies of less differentiated and more proliferative WT1-specific CD8+ T cells in bone marrow in tumor-bearing patients: An important role of bone marrow as a secondary lymphoid organ. Cancer Sci. 2010, 101, 848–854. [Google Scholar] [CrossRef]
- Schmitz-Winnenthal, F.H.; Volk, C.; Z’graggen, K.; Galindo, L.; Nummer, D.; Ziouta, Y.; Bucur, M.; Weitz, J.; Schirrmacher, V.; Büchler, M.W.; et al. High frequencies of functional tumor-reactive T cells in bone marrow and blood of pancreatic cancer patients. Cancer Res. 2005, 65, 10079–10087. [Google Scholar] [CrossRef] [Green Version]
- Choi, C.; Witzens, M.; Bucur, M.; Feuerer, M.; Sommerfeld, N.; Trojan, A.; Ho, A.; Schirrmacher, V.; Goldschmidt, H.; Beckhove, P. Enrichment of functional CD8 memory T cells specific for MUC1 in bone marrow of patients with multiple myeloma. Blood 2005, 105, 2132–2134. [Google Scholar] [CrossRef]
- Sung, J.H.; Zhang, H.; Moseman, E.A.; Alvarez, D.; Iannacone, M.; Henrickson, S.E.; de la Torre, J.C.; Groom, J.R.; Luster, A.D.; von Andrian, U.H. Chemokine guidance of central memory T cells is critical for antiviral recall responses in lymph nodes. Cell 2012, 150, 1249–1263. [Google Scholar] [CrossRef] [Green Version]
- Hanazawa, A.; Hayashizaki, K.; Shinoda, K.; Yagita, H.; Okumura, K.; Lohning, M.; Hara, T.; Tani-ichi, S.; Ikuta, K.; Eckes, B.; et al. CD49b-dependent establishment of T helper cell memory. Immunol. Cell Biol. 2013, 91, 524–531. [Google Scholar] [CrossRef]
- Alp, S.Ö.; Durlanik, S.; Schulz, D.; McGrath, M.; Grün, J.R.; Bardua, M.; Ikuta, K.; Sgouroudis, E.; Riedel, R.; Zehentmeier, S.; et al. Memory CD8+ T cells colocalize with IL-7+ stromal cells in bone marrow and rest in terms of proliferation and transcription. Europ. J. Immunol. 2015, 45, 975–987. [Google Scholar]
- Herndler-Brandstetter, D.; Landgraf, K.; Jenewein, B.; Tzankow, A.; Brunauer, R.; Brunner, S.; Parson, W.; Kloss, F.; Gassner, R.; Lepperdinger, G.; et al. Human bone marrow hosts polyfunctional memory CD4+ and CD8+ T cells with close contact to IL-15 producing cells. J. Immunol. 2011, 186, 6965–6971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldmann, T.A.; Waldmann, R.; Lin, J.-X.; Leonard, W.J. The implications of IL-15 trans-presentation on the immune response. Adv. Immunol. 2022, 156, 103–132. [Google Scholar] [CrossRef] [PubMed]
- Pascutti, M.F.; Geerman, S.; Collins, N.; Brasser, G.; Nota, B.; Stark, R.; Behr, F.; Oja, A.; Slot, E.; Panagioti, E.; et al. Peripheral and systemic antigens elicit an expandable pool of resident memory CD8+ T cells in the bone marrow. Eur. J. Immunol. 2019, 49, 853–872. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wang, L.; Liu, Y.; Zhu, Y.; Liu, Y. Characteristics of T-cell receptor repertoire of stem cell-like memory CD4+ T cells. PeerJ. 2021, 9, e11987. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Reinke, S.; Shen, Y.; Schollmeyer, L.; Liu, Y.-C.; Wang, Z.; Hardt, S.; Hipfl, C.; Hoffmann, U.; Frischbutter, S.; et al. A ubiquitous bone marrow reservoir of preexisting SARS-CoV-2-reactive memory CD4+ T lymphocytes in unexposed individuals. Front. Immunol. 2022, 13, 1004656. [Google Scholar] [CrossRef]
- Kudernatsch, R.F.; Letsch, A.; Guerreiro, M.; Löbel, M.; Bauer, S.; Volk, H.D.; Scheibenbogen, C. Human bone marrow contains a subset of quiescent early memory CD8+ T cells characterized by high CD127 expression and efflux capacity. Eur. J. Immunol. 2014, 44, 3532–3542. [Google Scholar] [CrossRef]
- Wu, K.; Li, Y.; Zhang, S.; Zhou, N.; Liu, B.; Pan, T.; Zhang, X.; Luo, H.; Huang, Z.; Li, X.; et al. Preferential homing of tumor-specific and functional CD8+ stem cell-like memory T cells to the bone marrow. J. Immunother. 2019, 42, 197–207. [Google Scholar] [CrossRef]
- Wu, K.; Wang, F.; Guo, G.; Li, Y.; Qiu, L.-J.; Li, X. CD4+ TSCMs in the bone marrow assist in maturation of antibodies against Influenza in mice. Mediat. Inflamm. 2019, 2019, 3231696. [Google Scholar] [CrossRef]
- Chi, X.; Luo, S.; Ye, P.; Hwang, W.-L.; Cha, J.-H.; Yan, X.; Yang, W.-H. T-cell exhaustion and stemness in anti-tumor immunity: Characteristics, mechanisms, and implications. Front. Immunol. 2023, 20, 4771. [Google Scholar] [CrossRef]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Cieri, N.; Camissa, B.; Cocchiarella, F.; Forcato, M.; Oliveira, G.; Prosvasi, F.; Bondanza, A.; Bordignon, C.; Peccatori, J.; Ciceri, F.; et al. IL-7 and IL-15 instruct the generation of human memory stem cells from naïve precursors. Blood 2013, 121, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Imura, Y.; Ando, M.; Chikuma, S.; Yoshimura, A. In Vitro conversion of activated T cells into stem cell memory-like T cells. Methods Mol. Biol. 2019, 2048, 41–51. [Google Scholar]
- Palendira, U.; Chinn, R.; Raza, W.; Piper, K.; Pratt, G.; Machado, L.; Bell, A.; Khan, N.; Hislop, A.D.; Steyn, R.; et al. Selective accumulation of virus-specific CD8+ T cells with unique homing phenotype within human bone marrow. Blood 2008, 112, 3293–3302. [Google Scholar] [CrossRef] [Green Version]
- Pangrazzi, L.; Naismith, E.; Meryk, A.; Keller, M.; Jenewein, B.; Trieb, K.; Grubeck-Loebenstein, B. Increased IL-15 production and accumulation of highly differentiated CD8+ effector/memory T cells in the bone marrow of persons with cytomegalovirus. Front. Immunol. 2017, 8, 715. [Google Scholar] [CrossRef] [Green Version]
- Racanelli, V.; Frassanito, M.A.; Leone, P.; Brunetti, C.; Ruggien, S.; Dammacco, F. Bone marrow of persistently hepatitis C virus-infected individuals accumulates memory CD8+ T cells specific for current and historical viral antigens: A study in patients with benign hematological disorders. J. Immunol. 2007, 179, 5387–5398. [Google Scholar] [CrossRef]
- Na, I.K.; Letsch, A.; Guerreio, M.; Bauer, S.; Noack, I.; Geginat, J.; Reinke, P.; Loesch, M.; Kienapfel, H.; Thiel, E.; et al. Human bone marrow as a source to generate CMV-specific CD4+ T cells with multifunctional capacity. J. Immunother. 2009, 32, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Lineburg, K.E.; Martins, J.P.; Ambalathingal, G.R.; Neller, M.A.; Morrison, B.; Matthews, K.K.; Rehan, S.; Crooks, P.; Panikkar, A.; et al. Autologous CMV-specific T cells are a safe adjuvant immunotherapy for primary glioblastoma multiforme. J. Clin. Investig. 2020, 130, 6041–6053. [Google Scholar] [CrossRef] [PubMed]
- DiRosa, F. Two niches in the bone marrow: A hypothesis on life-long T cell memory. Trends Immunol. 2016, 17, 503–512. [Google Scholar] [CrossRef]
- Chang, H.-D.; Radbruch, A. Maintenance of quiescent immune memory in the bone marrow. Eur. J. Immunol. 2021, 51, 1592–1601. [Google Scholar] [CrossRef]
- Chang, H.-D.; Tokoyoda, K.; Radbruch, A. Immunological memories of the bone marrow. Immunol. Rev. 2018, 283, 86–98. [Google Scholar] [CrossRef] [Green Version]
- Rajewsky, K.; Schirrmacher, V.; Nase, S.; Jerne, N. The requirement for more than one antigenic determinant for immunogenicity. J. Exp. Med. 1969, 129, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Fresnay, S.; Zhang, X.; Strome, S.; Sewell, D.A. Bone marrow vaccination: A novel approach to enhance antigen specific antitumor immunity. Vaccine 2011, 29, 8599–8605. [Google Scholar] [CrossRef]
- Kushida, T.; Inaba, M.; Ichioka, N.; Esumi, T.; Ogawa, R.; Iida, H.; Ikehara, S. Intra-bone marrow injection of allogeneic bone marrow cells: A powerful new strategy for treatment of intractable autoimmune diseases in MRL/lpr mice. Blood 2001, 97, 3292–3299. [Google Scholar] [CrossRef] [Green Version]
- Kushida, T.; Inaba, M.; Ichioka, N.; Esumi, T.; Ogawa, R.; Iida, H.; Ikehara, S. Crucial role of donor-derived stromal cells in successful treatment for intractable autoimmune diseases in mrl/lpr mice by bmt via portal vein. Stem Cells 2001, 19, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Wildes, T.J.; Grippin, A.; Dyson, K.A.; Wummer, B.M.; Damiani, D.; Abraham, R.S.; Flores, C.T.; Mitchell, D.A. Cross-talk between T cells and hematopoietic stem cells during adopitive cellular therapy for malignant glioma. Clin. Cancer Res. 2019, 24, 3955–3966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Yao, J.-C.; Oetjen, K.A.; Krambs, J.R.; Xia, J.; Zhang, J.; Schmidt, A.P.; Helton, N.M.; Fulton, R.S.; Heath, S.E.; et al. Il-1β expression in bone marrow dendritic cells is induced by TLR2 agonists and regulates HSC function. Blood 2022, 140, 1607–1620. [Google Scholar] [CrossRef]
- Niu, C.; Yu, J.; Zou, T.; Lu, Y.; Deng, L.; Yun, H.; Si, C.-Y.; Wu, X.; Jiang, H.; Guo, T.; et al. Identification of hematopoietic stem cells residing in the meninges of adult mice at steady state. Cell Rep. 2022, 41, 111592. [Google Scholar] [CrossRef]
- Tsunokuma, N.; Yamane, T.; Matsumoto, C.; Tsuneto, M.; Isono, K.; Imanaka-Yoshida, K.; Yamazaki, H. Depletion of neural crest-derived cells leads to reduction in plasma noradrenaline and alters B lymphopoiesis. J. Immunol. 2017, 198, 156–169. [Google Scholar] [CrossRef] [Green Version]
- Ott, L.C.; Han, C.Y.; Mueller, J.L.; Rahman, A.A.; Hotta, R.; Goldstein, A.M.; Stavely, R. Bone marrow stem cells derived from nerves have neurogenic properties and potential uitility for regenerative therapy. Int. J. Mol. Sci. 2023, 24, 5211. [Google Scholar] [CrossRef]
- Coste, C.; Neirinckx, V.; Sharma, A.; Agirman, G.; Rogister, B.; Foguenne, J.; Lallemend, F.; Gothot, A.; Wislet, S. Human bone marrow harbors cells with neural crest-associated characteristics like human adipose and dermis tissues. PLoS ONE 2017, 12, e0177962. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.A.; Huat, T.J.; Mutery, A.A.; El-Serafi, A.T.; Kacem, H.H.; Abdullah, S.H.; Reza, M.F.; Abdullah, J.M.; Jaafar, H. Significant transcriptomic changes are associated with differentiation of bone marrow-derived mesenchymal stem cells into neural progenitor-like cells in the presence of bFGF and EGF. Cell Biosci. 2020, 10, 126. [Google Scholar] [CrossRef]
- Akhter, W.; Nakhle, J.; Vaillant, L.; Garcin, G.; Le Saout, C.; Simon, M.; Crozet, C.; Djouad, F.; Jorgensen, C.; Vignais, M.-L.; et al. Transfer of mesenchymal stem cell mitochondria to CD4+ T cells contributes to repress Th1 differentiation by downregulating T-bet expression. Stem Cell Res. Ther. 2023, 14, 12. [Google Scholar] [CrossRef]
- Schirrmacher, V. Mitochondria at work: New insights into regulation and dysregulation of cellular energy supply and metabolism. Biomedicines 2020, 8, 526. [Google Scholar] [CrossRef]
- Jordan, S.; Tung, N.; Casanova-Acebes, M.; Chang, C.; Cantoni, C.; Zhang, D.; Wirtz, T.H.; Naik, S.; Rose, S.A.; Brocker, C.N.; et al. Dietary intake regulates the circulating inflammatory monocyte pool. Cell 2019, 178, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Noguchi, R.; Takahashi, D.; Morikawa, T.; Koshida, K.; Komiyama, S.; Ishihara, N.; Yamada, T.; Kawamura, Y.I.; Muroi, K.; et al. Fasting-refeeding impacts immune cell dynamics and mucosal immune responses. Cell 2019, 178, 1072–1087. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 178, 771–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, N.; Han, S.-J.; Enamorado, M.; Link, V.M.; Huang, B.; Moseman, A.; Kishton, R.J.; Shannon, J.P.; Dixit, D.; Schwab, S.R.; et al. The bone marrow protects and optimizes immunological memory during dietary restriction. Cell 2019, 178, 1088–1101. [Google Scholar] [CrossRef] [PubMed]
- Vollmann, E.H.; Rattay, K.; Barreiro, O.; Thiriot, A.; Fuhlbrigge, R.A.; Vrbanac, V.; Kim, K.-W.; Jung, S.; Tager, A.M.; von Andrian, U.H. Specialized transendothelial dendritic cells mediate thymic T-cell selection agains blood-borne macromolecules. Nat. Commun. 2021, 12, 6230. [Google Scholar] [CrossRef]
- Matioubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef]
- Waller, K.M.; De La Mata, N.L.; Kelly, P.J.; Ramachandran, V.; Rawlinson, W.D.; Wyburn, K.R.; Webster, A.C. Residual risk of infection with blood-borne viruses in potential organ donors at increased risk of infection: Systematic review and meta-analysis. Med. J. Aust. 2019, 211, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Cariappa, A.; Mazo, I.B.; Chase, C.; Shi, H.N.; Liu, H.; Rose, H.; Leung, H.; Cherayil, B.J.; Russell, P.; von Andrian, U.; et al. Perisinusoidal B cells in the bone marrow participate in T-independent responses to blood-borne microbes. Immunity 2005, 23, 397–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Kometani, K.; Minato, N.; Hamazaki, Y. Bone marrow endothelial cells take up blood-borne immune complexes via Fcg receptor IIb2 in an erythropoitin-dependent manner. J. Immunol. 2020, 205, 2008–2015. [Google Scholar] [CrossRef]
- He, S.; Ding, L.; Yuan, H.; Zhan, G.; Yang, X.; Wu, Y. A review of sensors for classification and subtype discrimination of cancer: Insights into circulating tumor cells and tumor-derived extracellular vesicles. Anal. Chim. Acta 2023, 1244, 340703. [Google Scholar] [CrossRef] [PubMed]
- Rak, J.; Strzadala, L. Heterogeneity of extracellular vesicles and particles: Molecular voxels in the blood borne “hologram” of organ function, disfunction and cancer. Arch. Immunol. Ther. Exp. 2023, 71, 5. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, J.; Hao, H.; Lu, F.; Wang, J.; Ma, M.; Jia, B.; Zhou, M.; Wang, J.; Chi, Y.; et al. Secreted proteins MDK, WFDC2, and CXCL14 as candidate biomarkers for early diagnosis of lung adenocarcinoma. BMC Cancer 2023, 23, 110. [Google Scholar] [CrossRef]
- Tripathy, A.; John, V.; Wadden, J.; Kong, S.; Sharba, S.; Koschmann, C. Liquid biopsy in pedriatic brain tumors. Front. Genet. 2023, 13, 1114762. [Google Scholar] [CrossRef]
- Stergiopoulou, D.; Markou, A.; Strati, A.; Zavridou, M.; Tzanikou, E.; Mastoraki, S.; Kallergi, G.; Georgoulias, V.; Lianidou, E. Comprehensive liquid biopsy analysis as a tool for the early detection of minimal residual disease in breast cancer. Sci. Rep. 2023, 13, 1258. [Google Scholar] [CrossRef]
- Someya, M.; Hasegawa, T.; Tsuchiya, T.; Kitagawa, M.; Fukushima, Y.; Gocho, T.; Mafune, S.; Ikeuchi, Y.; Kozuka, Y.; Idogawa, M.; et al. Predictive value of an exosomal microRNA-based signature for tumor immunity in cervical cancer patients treated with chemoradiotherapy. Med. Mol. Morphol. 2023, 56, 38–45. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Tokita, S.; Hirama, T.; Kochin, V.; Nakatsugawa, M.; Shinkawa, T.; Hirohashi, Y.; Tsukahara, T.; Hata, F.; Takemasa, I.; et al. CD8+ T-cell immune surveillance against a tumor antigen encoded by the oncogenic long noncoding RNA PVT1. Cancer Immunol. Res. 2021, 9, 1342–1353. [Google Scholar] [CrossRef]
- Galluzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Vanderberk, L.; Koks, C.; Verschuere, T.; Boon, L.; Van Gool, S.W.; Agostinis, P. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci. Transl. Med. 2016, 8, 328ra27. [Google Scholar] [CrossRef]
- Cavanagh, L.L.; Bonasio, R.; Mazo, I.B.; Halin, C.; Cheng, G.; van der Welden, A.W.M.; Cariappa, A.; Chase, C.; Russell, P.; Starnbach, M.N.; et al. Activation of bone marrow-resident memory T cells by circulating, antigen-bearing dendritic cells. Nat. Immunol. 2005, 6, 1029–1037. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, D.; Vollmann, E.H.; von Andrian, U.H. Mechanisms and consequences of dendritic cell migration. Immunity 2008, 29, 325. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Liu, A.; Li, W.; Liu, Y.; Zhang, G.; Ye, S.; Zhao, Z.; Shi, J.; Jia, Y.; Liu, X.; et al. Reference range of naïve T and T memory lymphocyte subsets in peripheral blood of healthy adult. Clin. Trial. 2022, 207, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Li, W.; Li, Y.; Yu, J. The clinical value of the changes of peripheral lymphocyte subsets absolute counts in patients with non-small cell lung cancer. Transl. Oncol. 2020, 13, 849. [Google Scholar] [CrossRef] [PubMed]
- Mollgard, K.; Beinlich, F.R.M.; Kusk, P.; Miyakoshi, L.M.; Delle, C.; Pla, V.; Hauglund, N.L.; Esmail, T.; Rasmussen, M.K.; Gomolka, R.S.; et al. A mesothelium divides the subarachnoid space into functional compartments. Science 2023, 378, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Mills, W.A., 3rd; Coburn, M.A.; Eyo, U.B. The emergence of the calvarial hematopoietic niche in health and disease. Immunol. Rev. 2022, 311, 26–38. [Google Scholar] [CrossRef]
- Petry, P.; Oschwald, A.; Kierdorf, K. Microglia tissue surveillance: The never-resting gardener in the developing and adult CNS. Eur. J. Immunol. 2023, 53, e2250232. [Google Scholar] [CrossRef]
- Arutyunov, A.; Klein, R.S. Microglia at the scene of the crime: What their transcriptomics reveal about brain health. Curr. Opin. Neurol. 2023, 36, 207–213. [Google Scholar] [CrossRef]
- Dzyubenko, E.; Hermann, D.M. Role of glia and extracellular matrix in controlling neuroplasticity in the central nervous system. Semin. Immunopathol. 2023, 45, 377–387. [Google Scholar] [CrossRef]
- Klimov, V.; Cherevko, N.; Klimov, A.; Novikov, P. Neuronal-immune cell units in allergic inflammation in the nose. Int. J. Mol. Sci. 2022, 23, 6938. [Google Scholar] [CrossRef]
- Ji, H.; Lai, D.; Tou, J. Neuroimmune regulation in Hirschsprung’s disease associated enterocolitis. Front. Immunol. 2023, 14, 1127375. [Google Scholar] [CrossRef] [PubMed]
- Barden, M.M.; Omuro, A.M. Top advances of the year: Neuro-oncology. Cancer 2023, 129, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Ferren, A.J.M.; Cwynarski, K.; Pulczynski, E.; Ponzoni, M.; Deckert, M.; Politi, L.S.; Torri, V.; Fox, C.P.; Rosée, P.L.; Schorb, E.; et al. Chemoimmunotherapy with methotrexate, cytarabine, thiotepa, and rituximab (MATRix regimen) in patients with primary CNS lymphoma: Results of the first randomisation of the International extranodal lymphoma study group-32 (IELSG32) phase 2 trial. Lancet Hematol. 2016, 3, e217–e227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, S.; Schmidt, M.H.H.; Schuman, U. The glioma immune landscape: A double-edged sword for treatment regimens. Cancers 2023, 15, 2024. [Google Scholar] [CrossRef]
- Bunse, L.; Bunse, T.; Krämer, C.; Chih, Y.-C.; Platten, M. Clinical and translational advances in glioma immunotherapy. Neurotherapeutics 2022, 19, 1799–1817. [Google Scholar] [CrossRef]
- Van Gool, S.W.; Makalowski, J.; Bitar, M.; Van de Vliet, P.; Schirrmacher, V.; Stuecker, W. Synergy between TMZ and individualized multimodal immunotherapy to improve overall survival of IDH1 wild-type MGMT promoter-unmethylated GBM patients. Genes Immun. 2022, 23, 255–259. [Google Scholar] [CrossRef]
- Van Gool, S.W.; Makalowski, J.; Fiore, S.; Sprenger, T.; Prix, L.; Schirrmacher, V.; Stuecker, W. Randomized-controlled immunotherapy clinical trials for GBM challenged. Cancers 2020, 13, 32. [Google Scholar] [CrossRef]
- Yoshimura, A.; Ohyagi, M.; Ito, M. T cells in the brain inflammation. Adv. Immunol. 2023, 157, 29–58. [Google Scholar] [CrossRef]
- Eliseeva, D.D.; Zakharova, M.N. Myelin oligodendrocyte glycoprotein as an autoantigen in inflammatory demyelinating disease of the Central nervous system. Biochemistry 2023, 88, 551–563. [Google Scholar] [CrossRef]
- Hu, Y.; Feng, J.; Gu, T.; Wang, L.; Wang, Y.; Zhou, L.; Hong, R.; Yin, E.T.S.; Zhang, M.; Lu, P.; et al. CAR T-cell therapies in China: Rapid evolution and a bright future. Lancet Hematol. 2022, 9, e930–e941. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, W.; Asada, N.; Naoi, Y.; Abe, M.; Fujiwara, H.; Ennishi, D.; Nishimori, H.; Fujii, K.; Fujii, N.; Matsuoka, K.-I.; et al. Bone marrow microenvironment disruption and sustained inflammation with prolonged haematologic toxicity after CAR T-cell therapy. Br. J. Haematol. 2023, 202, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Read, J.A.; Rouce, R.H.; Mo, F.; Mamonkin, M.; King, K.Y. Apoptosis of hematopoietic stem cells contributes to bone marrow suppression following chimeric antigen receptor T cell therapy. Transplant. Cell Ther. 2023, 29, e1–e165. [Google Scholar] [CrossRef] [PubMed]
- Biondi, M.; Tettamanti, S.; Galimberti, S.; Cerina, B.; Tomasoni, C.; Piazza, R.; Donsante, S.; Bido, S.; Perriello, V.M.; Broccoli, V.; et al. Selective homing of CAR-CIK cells to the bone marrow niche enhances control of the Acute myeloid leukemia burden. Blood 2023, 141, 2587–2598. [Google Scholar] [CrossRef]
- Chockley, P.J.; Ibanez-Vega, J.; Krenciute, G.; Talbot, L.J.; Gottschalk, S. Synapse-tuned CARs enhance immune cell anti-tumor activity. Nat. Biotechnol. 2023, 2013, 1–12. [Google Scholar] [CrossRef]
- Tao, R.; Mi, B.; Hu, Y.; Lin, S.; Xiong, Y.; Lu, X.; Panayi, A.C.; Li, G.; Liu, G. Hallmarks of peripheral nerve function in bone regeneration. Bone Res. 2023, 11, 6. [Google Scholar] [CrossRef]
- Liu, S.; Liu, S.; Li, S.; Liang, B.; Han, X.; Liang, Y.; Wie, X. Nerves within bone and their application in tissue engineering of bone regeneration. Front. Neurol. 2023, 13, 1085560. [Google Scholar] [CrossRef]
- Manz, B.N.; Jackson, B.L.; Petit, R.S.; Dustion, M.L.; Groves, J. T-cell triggering thresholds are modulated by the number of antigen within individual T-cell clusters. Proc. Natl. Acad. Sci. USA 2011, 108, 9089–9094. [Google Scholar] [CrossRef]
- Henrickson, S.E.; Perro, M.; Loughhead, S.M.; Senman, B.; Stutte, S.; Quigley, M.; Alexe, G.; Iannacone, M.; Flynn, M.P.; Omid, S.; et al. Antigen availability determines CD8+ T cell-dendritic cell interaction kinetics and memory fate decisions. Immunity 2013, 39, 496–507. [Google Scholar] [CrossRef] [Green Version]
- Borger, J.; Zamoyska, R.; Gakamsky, D.M. Proximity of TCR and its CD8 coreceptor controls sensitivity of T cells. Immunol. Lett. 2014, 157, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Sedwick, C.E.; Morgan, M.M.; Jusino, L.; Cannon, J.L.; Miller, J.; Burkhardt, J.K. TCR, LFA-1, and CD28 play unique and complementary roles in signaling T cell cytoskeletal reorganization. J. Immunol. 1999, 162, 1367–1375. [Google Scholar] [CrossRef]
- Gaud, G.; Lesourne, R.; Love, P.E. Regulatory mechanisms in T cell receptor signalling. Nat. Rev. Immunol. 2018, 18, 485–497. [Google Scholar] [CrossRef]
- Geltink, R.I.K.; O’Sullivan, D.; Corrado, M.; Bremser, A.; Buck, M.D.; Buescher, J.M.; Firat, E.; Zhu, X.; Niedermann, G.; Caputa, G.; et al. Mitochondrial priming by CD28. Cell 2017, 171, 385–397.e11. [Google Scholar] [CrossRef] [Green Version]
- Panda, A.K.; Kim, Y.; Shevach, E.M. Control of memory phenotype T lymphocyte homeostasis: The role of costimulation. J. Immunol. 2022, 208, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Martin-Cófreces, N.B.; Valpuesta, J.M.; Sánchez-Madrid, F. Folding for the immune synapse: CCT chaperonin and the cytoskeleton. Front. Cell Dev. Biol. 2021, 9, 658460. [Google Scholar] [CrossRef] [PubMed]
- Lanzavecchia, A.; Sallusto, F. From synapses to immunological memory: The role of sustained T cell stimulation. Curr. Opin. Immunol. 2000, 12, 92–98. [Google Scholar] [CrossRef]
- Waldman, M.M.; Rahkola, J.T.; Sigler, A.L.; Chung, J.W.; Willett, B.A.S.; Kedl, R.M.; Friedman, R.S.; Jacobelli, J. Ena/VASP protein-mediated actin polymerization contributes to naïve CD8+ T cell activation and expansion by promoting T cell-APC interactions in vivo. Front. Immunol. 2022, 13, 856977. [Google Scholar] [CrossRef]
- Kaitao, L.; William, R.; Zhou, Y.; Cheng, Z. Single-molecule investigations on T-cell activation. Curr. Opin. Biomed. Eng. 2019, 12, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Cassioli, C.; Baldari, C.T. Lymphocyte polarization during immune synapse assembly: Centrosomal actin joins the game. Front. Immunol. 2022, 13, 830835. [Google Scholar] [CrossRef] [PubMed]
- Mastrogiovanni, M.; Juzanz, M.; Alcover, A.; Di Bartolo, V. Coordinating cytoskeleton and molecular traffic in T cell migration, activation, and effector functions. Front. Cell Dev. Biol. 2020, 8, 591348. [Google Scholar] [CrossRef] [PubMed]
- Manes, T.D.; Pober, J.S. T cell receptor-driven transendothelial migration of human effector memory CD4 T cells involves Vav, Rac, and Myosin IIA. J. Immunol. 2013, 190, 3079–3088. [Google Scholar] [CrossRef] [PubMed]
- Manes, T.D.; Wang, V.; Pober, J.S. Costimulators expressed on human endothelial cells modulate antigen-dependent recruitment of circulating T lymphocytes. Front. Immunol. 2022, 13, 1016361. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V.; Schlude, C.; Weitz, J.; Beckhove, P. Strong T-cell costimulation can reactivate tumor antigen-specific T cells in late-stage metastasized colorectal carcinoma patients: Results from a phase I clinical study. Int. J. Oncol. 2015, 46, 71–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Göhring, J.; Schrangl, L.; Schütz, G.J.; Huppa, J.B. Mechanosurveillance: Tiptoeing T cells. Front. Immunol. 2022, 13, 886326. [Google Scholar] [CrossRef]
- Basu, R.; Whitlock, B.M.; Husson, J.; Le Floc’h, A.; Jin, W.; Oyler-Yaniv, A.; Dotiwala, F.; Giannone, G.; Hivroz, C.; Biais, N.; et al. Cytotoxic T cells use mechanical force to potentiate target cell killing. Cell 2016, 165, 100–110. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-R.; Jun, C.-D. T cell microvilli: Sensors or senders? Front. Immunol. 2019, 10, 1753. [Google Scholar] [CrossRef]
- Bai, L.; Beckhove, P.; Feuerer, M.; Umansky, V.; Choi, C.; Solomayer, F.S.; Diel, I.J.; Schirrmacher, V. Cognate interactions between memory T cells and tumor antigen presenting dendritic cells from bone marrow of breast cancer patients: Bidirectional cell stimulation, survival and antitumor activity in vivo. Int. J. Cancer 2003, 103, 73–83. [Google Scholar] [CrossRef]
- Cornelis, R.; Hahne, S.; Taddeo, A.; Petkau, G.; Malko, D.; Durek, P.; Thiem, M.; Heiberger, L.; Peter, L.; Mohr, R.; et al. Stromal cell-contact dependent PI3K and APRIL induced NF-kB signaling prevent mitochondrial and ER stress induced death of memory plasma cells. Cell Rep. 2020, 32, 107982. [Google Scholar] [CrossRef]
- Rozanski, C.H.; Arens, R.; Carlson, L.M.; Nair, J.; Boise, L.H.; Chanan-Khan, A.A.; Schoenberger, S.P.; Lee, K.P. Sustained antibody responses depend on CD28 function in bone marrow-resident plasma cells. J. Exp. Med. 2011, 208, 1435–1446. [Google Scholar] [CrossRef]
- Snell, L.M.; Lin, G.H.Y.; Watts, T.H. IL-15-dependent upregulation of GITR on CD8 memory phenotype T cells in the bone marrow relative to spleen and lymph node suggests the bone marrow as a site of superior bioavailability of IL-15. J. Immunol. 2012, 188, 5915–5923. [Google Scholar] [CrossRef] [Green Version]
- Schluns, K.S.; Kieper, W.C.; Jameson, S.C.; Lefrancois, L. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat. Immunol. 2000, 1, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Wols, H.A.M.; Underhill, G.H.; Kansas, G.S.; Witte, P.L. The role of bone marrow-derived stromal cells in the maintenance of plasma cell longevity. J. Immunol. 2002, 169, 4213–4221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delacher, M.; Imbusch, C.D.; Weichenhan, D.; Breiling, A.; Wagenblatt, A.H.; Träger, U.; Hofer, A.-C.; Kägebein, D.; Wang, Q.; Frauhammer, F.; et al. Genome-wide DNA-methylation landscape defines specialization of regulatory T cells in tissues. Nat. Immunol. 2017, 18, 1160–1172. [Google Scholar] [CrossRef] [PubMed]
- Ashman, J.; Mutsonziwa, N.; Romano, M.M.; Kordasti, S.; Lombardi, G.; Shangaris, P. Regulatory T cell niche in the bone marrow, a new player in hematopoietic stem cell transplantation. Blood Rev. 2022, 31, 101030. [Google Scholar] [CrossRef]
- Tikka, C.; Beasley, L.; Xu, C.; Yang, J.; Cooper, S.; Lechner, J.; Gutch, S.; Kaplan, M.H.; Capitano, M.; Yang, K. BATF sustains homeostasis and functionality of bone marrow Treg cells to preserve homeostatic regulation of hematopoiesis and development of B cells. Front. Immunol. 2023, 14, 1026368. [Google Scholar] [CrossRef]
- Nicholls, J.; Cao, B.; Le Texier, L.; Xiong, L.Y.; Hunter, C.R.; Llanes, G.; Aguliar, E.G.; Schroder, W.A.; Phipps, S.; Lynch, J.P.; et al. Bone marrow regulatory T cells are a unique population, supported by niche-specific cytokines and plasmacytoid dendritic cells, and required for chronic graft-versus-host disease control. Front. Cell Dev. Biol. 2021, 9, 737880. [Google Scholar] [CrossRef]
- Wang, X.; Hanuffa, M.A.; Holtick, U.; Collin, M.P.; Jackson, G.; Hilkens, C.M.U.; Holler, E.; Edinger, M.; Hoffmann, P.; Dickinson, A.M. Regulatory T-cell suppression of CD8+ T-cell-mediated graft-versus-host reaction requires their presence during priming. Transplantation 2009, 88, 188–197. [Google Scholar] [CrossRef]
- Schirrmacher, V.; Beckhove, P.; Choi, C.; Griesbach, A.; Mahnke, Y. Tumor-immune memory T cells from the bone marrow exert GvL without GvH reactivity in advanced metastasized cancer. Int. J. Oncol. 2005, 27, 1141–1149. [Google Scholar] [CrossRef]
- Zhang, Y.; Joe, G.; Hexner, E.; Zhu, J.; Emerson, S.G. Host-reactive CD8+ memory stem cells in graft-versus-host disease. Nat. Med. 2005, 11, 1299–1305. [Google Scholar] [CrossRef]
- Schirrmacher, V.; Beckhove, P.; Krüger, A.; Rocha, M.; Umansky, V.; Fichtner, K.-P.; Hull, W.E.; Zangemeister-Wittke, U.; Griesbach, A.; Jurianz, K.; et al. Effective immune rejection of advanced metastasized cancer. Int. J. Oncol. 1995, 6, 505–521. [Google Scholar] [CrossRef]
- Lee, K.; Hacker, H.; Umansky, V.; Schirrmacher, V.; Rocha, M. Changes in liver glycogen and lipid metabolism during transient graft-versus-host (GvH) and graft-versus-leukenia (GvL) reactivity. Int. J. Oncol. 1996, 9, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. T cell-mediated immunotherapy of metastases: State of the art in 2005. Expert Opin. Biol. Ther. 2005, 5, 1051–1068. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Meng, H.B.; Hua, J.; Song, Z.S.; He, Z.G.; Zhou, B.; Qian, M.P. The SDF-1/CXCR4 axis regulates migration of transplanted bone marrow mesenchymal stem cells towards the pancreas in rats with acute pancreatitis. Mol. Med. Rep. 2014, 9, 1575–1582. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [Green Version]
- Brinkrolf, P.; Landmeier, S.; Altvater, B.; Chen, C.; Pscherer, S.; Rosemann, A.; Ranft, A.; Dirksen, U.; Juergens, H.; Rossig, C. A high proportion of bone marrow T cells with regulatory phenotype (CD4+CD25hiFoxP3+) in Ewing sarcoma patients is associated with metastatic disease. Int. J. Cancer 2009, 125, 879–886. [Google Scholar] [CrossRef]
- Koch, J.; Schober, S.J.; Hindupur, S.V.; Schöning, C.; Klein, F.G.; Mantwill, K.; Ehrenfeld, M.; Schillinger, U.; Hohnecker, T.; Qi, P.; et al. Targeting the Retinoblastoma/E2F repressive complex by CDK4/6 inhibitors amplifies oncolytic potency of an oncolytic adenovirus. Nat. Commun. 2022, 13, 4689. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Böhm, H.-H.; Rathinasamy, A.; Xydia, M.; Hu, X.; Pincha, M.; Umansky, L.; Breyer, C.; Hillier, M.; Bonertz, A.; et al. Tumor-specific regulatory T cells from the bone marrow orchestrate antitumor immunity in breast cancer. Cancer Immunol. Res. 2019, 7, 1998–2012. [Google Scholar] [CrossRef] [Green Version]
- Cho, D.S.; Schmitt, R.E.; Dasgupta, A.; Ducharme, A.M.; Doles, J.D. Acute and sustained alterations to the bone marrow immune microenvironment following polymicrobial infection. Shock 2022, 58, 45–55. [Google Scholar] [CrossRef]
- Peng, Y.; Wu, Q.; Liu, H.; Zhang, J.; Han, Q.; Yin, F.; Wang, L.; Chen, Q.; Zhang, F.; Feng, C.; et al. An immune-related gene signature predicts the 28-day mortality in patients with sepsis. Front. Immunol. 2023, 14, 1152117. [Google Scholar] [CrossRef]
- Tu, Q.; Li, Y.; Zhu, J.; Guo, L.; Liu, C.; Liu, L.; Yuan, Y.; Zou, Y.; Chen, F.; Yao, L.; et al. Mitochondrial DNA mediates immunoparalysis of dendritic cells in sepsis via STING signalling. Cell Prolif. 2022, 55, e13328. [Google Scholar] [CrossRef]
- Moore-Fried, J.; Paul, M.; Jing, Z.; Fooksman, D.; Lauvau, G. CD169+ macrophages orchestrate plasmacytoid dendritic cell arrest and retention for optimal priming in the bone marrow of malaria-infected mice. eLife 2022, 11, e78873. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Xu, Z.; Zhang, Z.; Mo, Y.; Deng, Y.; Li, L.; Fei, S.; Wu, J.; Wang, K.; Zhang, Q.; et al. RNA-Seq approach to investigate the effects of melatonin on bone marrow-derived dendritic cells from dextran sodium-induced colitis mice. Toxicology 2022, 481, 153354. [Google Scholar] [CrossRef] [PubMed]
- Flores-Guzmaán, F.; Utikal, J.; Umansky, V. Dormant tumor cells interact with memory CD8+ T cells in RET transgenic mouse melanoma model. Cancer Lett. 2020, 474, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Mahnke, Y.D.; Schirrmacher, V. A novel tumor model system for the study of long-term protective immunity and immune T cell memory. Cell. Immunol. 2005, 221, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Mahnke, Y.D.; Schwendemann, J.; Beckhove, P.; Schirrmacher, V. Maintenance of lon-term tumour-specific T-cell memory by residual dormant tumour cells. Immunology 2005, 115, 325–336. [Google Scholar] [CrossRef]
- Anadon, C.M.; Yu, X.; Hänggi, K.; Biswas, S.; Chaurio, R.A.; Martin, A.; Payne, K.K.; Mandal, G.; Innamarato, P.; Harro, C.M.; et al. Ovarian cancer immunogenicity is governed by a narrow subset of progenitor tissue-resident memory T cells. Cancer Cell 2022, 40, 545–557.e13. [Google Scholar] [CrossRef]
- Mizukoshi, E.; Nakagawa, H.; Tamai, T.; Kitahara, M.; Fushimi, K.; Nio, K.; Terashima, T.; Lida, N.; Arai, K.; Vamashita, T.; et al. Peptide vaccine-treated, long-term surviving cancer patients harbor self-renewing tumor-specific CD8+ T cells. Nat. Commun. 2022, 13, 3123. [Google Scholar] [CrossRef]
- Schuetz, F.; Ehlert, K.; Ge, Y.; Schneeweiss, A.; Rom, J.; Inzkinweli, N.; Sohn, C.; Schirrmacher, V.; Beckhove, P. Treatment of advanced metastasized breast cancer with bone marrow-derived tumour-reactive memory T cells: A pilot clinical study. Cancer Immunol. Immunother. 2009, 58, 887–900. [Google Scholar] [CrossRef]
- Domschke, C.; Ge, Y.; Bernhardt, I.; Schott, S.; Keim, S.; Juenger, S.; Bucur, M.; Mayer, L.; Blumenstein, M.; Rom, J.; et al. Long-term survival after adoptive bone marrow T cell therapy of advanced metastasized breast cancer: Follow-up analysis of a clinical pilot trial. Cancer Immunol. Immunother. 2013, 62, 1053–1060. [Google Scholar] [CrossRef]
- Poliwoda, S.; Noor, N.; Downs, E.; Schaaf, A.; Cantwell, A.; Ganti, L.; Kaye, A.D.; Mosel, L.I.; Carroll, C.B.; Viswanath, O.; et al. Stem cells: A comprehensive review of origins and emerging clinical roles in medical practice. Orthop. Rev. 2022, 14, 37498. [Google Scholar] [CrossRef]
- Senkal, S.; Hayal, T.B.; Sagrac, D.; Sisli, H.B.; Asutay, A.B.; Kirath, B.; Sümer, E.; Rizvanov, A.A.; Sahin, F.; Dogan, A. Human ESC-derived neuromesodermal progenitors (NMPs) successfully differentiate into mesenchymal stem cells (MSCs). Stem Cell Rev. Rep. 2022, 18, 278–293. [Google Scholar] [CrossRef] [PubMed]
- Saidova, A.A.; Vorobjev, I.A. Lineage commitment, signaling pathways, and the cytoskeleton systems in mesenchymal stem cells. Tissue Eng. Part B Rev. 2020, 26, 13–25. [Google Scholar] [CrossRef]
- Qi, S.; Zhong, Z.; Zhu, Y.; Wang, Y.; Ma, M.; Wang, Y.; Liu, X.; Jin, R.; Jiao, Z.; Zhu, R.; et al. Two Hippo signaling modules orchestrate liver size and tumorigenesis. EMBO J. 2023, 42, e112126. [Google Scholar] [CrossRef]
- Yan, Y.-C.; Li, Y.-H.; Xiao, B.-G.; Wang, J.; Xi, J.-Y.; Yu, W.-B. Cellular and molecular mechanisms underly the combined treatment of fasudil and bone marrow derived-neuronal stem cells in a Parkinson’s disease mouse model. Mol. Neurobiol. 2023, 60, 1826–1835. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.-W.; Ding, D.-C. Role of cancer-associated mesenchymal stem cells in the tumor microenvironment: A review. Tzu Chi Med. J. 2022, 35, 24–30. [Google Scholar] [CrossRef]
- Keshavarz, M.; Ebrahimzadeh, M.S.; Mirt, S.M.; Dianat-Moghadam, H.; Ghorbanhosseini, S.S.; Mohebbi, S.R.; Keyvani, H.; Ghaemi, A. Oncolytic Newcastle disease virus delivered by mesenchymal stem cell-engineered system enhances the therapeutic effects altering tumor microenvironment. Virol. J. 2020, 17, 64. [Google Scholar] [CrossRef]
- Schirrmacher, V.; van Gool, S.; Stuecker, W. Counteracting immunosuppression in the tumor microenvironment by oncolytic Newcastle disease virus and cellular immunotherapy. Int. J. Mol. Sci. 2022, 23, 13050. [Google Scholar] [CrossRef]
- Schirrmacher, V. Cancer vaccines and oncolytic viruses exert profoundly lower side effects in cancer patients than other systemic therapies: A comparative analysis. Biomedicines 2020, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-T.; Su, Y.-C.; Or, Y.-E.; Cheng, C.-F.; Kung, T.K. CD8+ T cell memory is sustained in mice by hepatic stellate cells. Hepatology 2022, 77, 1486–1498. [Google Scholar] [CrossRef]
- Sun, M.; Kisseleva, T. Reversibility of liver fibrosis. Clin. Res. Hepatol. Gastroenterol. 2015, 39 (Suppl. S1), 60–63. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Ma, N.; Zhao, R.; Wu, G.; Zhang, Y.; Qiao, Y.; Han, D.; Xu, Y.; Xiang, Y.; Yan, B.; et al. Overexpression of mir-483-5p/3p cooperates to inhibit mouse liver fibrosis by suppressing the TGF-β stimulated HSCs in transgenic mice. J. Cell. Mol. Med. 2014, 18, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-L.; Wu, J.; Wei, D.; Zhang, D.-W.; Feng, H.; Chen, Z.-N.; Bian, H. Newcastle disease virus represses the activation of human hepatic stellate cells and reverses the development of hepatic fibrosis in mice. Liver Int. 2009, 29, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Kalou, Y.; Al-Khani, A.M.; Haider, K.H. Bone marrow mesenchymal stem cells for heart failure treatment: A systematic review and Meta-analysis. Heart Lung Circ. 2023, 32, 870–880. [Google Scholar] [CrossRef]
- Chen, X.; Xu, C.-X.; Liang, H.; Xi, Z.; Pan, J.; Yang, Y.; Sun, Q.; Yang, G.; Sun, Y.; Bian, L. Bone marrow mesenchymal stem cells transplantation alleviates brain injury after intracerebral hemorrhage in mice through the Hippo signaling pathway. Aging 2020, 12, 6306–6323. [Google Scholar] [CrossRef] [PubMed]
- Tfilin, M.; Gobshtis, N.; Fozailoff, D.; Fraifeld, V.E.; Turgeman, G. Polarized anti-inflammatory mesenchymal stem cells increase hippocampal neurogenesis and improve cognitive function in aged mice. Int. J. Mol. Sci. 2023, 24, 4490. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.-G.; Chen, M.-L.; Duan, R.; Wang, X.; Pang, Z.-L.; Ge, L.-T.; Lu, M.; Xie, H.; Liu, Z.-Z. Transplantation of nasal olfactory mucosa mesenchymal stem cells benefits Alzheimer’s disease. Mol. Neurobiol. 2022, 59, 7323–7336. [Google Scholar] [CrossRef]
- Qu, M.; Xing, F.; Xing, N. Mesenchymal stem cells for the treatment of cognitive impairment caused by neurological diseases. Biotechnol. Lett. 2022, 44, 903–916. [Google Scholar] [CrossRef]
- Nakano, M.; Kubota, K.; Kobayashi, E.; Chikenji, T.S.; Saito, Y.; Konan, N.; Fujimiya, M. Bone marrow-derived mesenchymal stem cells improve cognitive impairment in an Alzheimer’s disease model by increasing the expression of microRNA-146a in hippocampus. Sci. Rep. 2020, 10, 10772. [Google Scholar] [CrossRef]
- Vigo, T.; Voulgari-Kokota, A.; Errede, M.; Girolamo, F.; Ortolan, J.; Mariani, M.C.; Ferrara, G.; Virgintino, D.; Buffo, A.; de Rosbo, N.K.; et al. Mesenchymal stem cells instruct a beneficial phenotype in reactive astrocytes. Glia 2021, 69, 1204–1215. [Google Scholar] [CrossRef]
- Lei, X.; He, N.; Zhu, L.; Zhou, M.; Zhang, K.; Wang, C.; Huang, H.; Chen, S.; Li, Q.; Han, Z.; et al. Mesenchymal stem cell-derived extracellular vesicles attenuate radiation-induced lung injury via miRNA-214-3p. Antiox. Redox Signal. 2021, 35, 849–862. [Google Scholar] [CrossRef]
- Li, L.; Dong, L.; Zhang, J.; Gao, F.; Hui, J.; Yan, J. Mesenchymal stem cells with downregulated Hippo signalling attenuate lung injury in mice with lipopolysaccharide-induced acute respiratory distress syndrome. Int. J. Mol. Sci. 2019, 43, 1241–1252. [Google Scholar] [CrossRef] [Green Version]
- Vishnupriya, M.; Naveenkumar, M.; Manjima, K.; Sooryasree, N.V.; Saranya, T.; Ramya, S.; Winster, S.H.; Paulpandi, M.; Arul, B.N. Post-COVID pulmonary fibrosis: Therapeutic efficacy using with mesenchymal stem cells—How the lung heals. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 2748–2751. [Google Scholar] [CrossRef] [PubMed]
- Satilmis, B.; Cicek, G.S.; Cicek, E.; Akbulut, S.; Sahin, T.T.; Yilmaz, S. Adipose-derived stem cells in the treatment of hepatobiliary diseases and sepsis. World J. Clin. Cases 2022, 10, 4348–4356. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Luo, F.; Che, L.; Zhang, H.; Zhao, L.; Zhang, W.; Man, X.; Bu, Q.; Luan, H.; Zhou, B.; et al. Mesenchymal stem cells protect against sepsis-associated acute kidney injury by inducing Gal-9/Tim-3 to remodel immune homeostasis. Ren. Fail. 2023, 45, 2187229. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Fang, Q.; Shi, C.; Zhang, Y.; Xia, C.; Zhang, Y.; Wang, J.; Chen, F. Bone marrow mesenchymal stem cells inhibit ferroptosis via regulating the Nrf2-keap1/p53 pathway to ameliorate chronic kidney disease injury in the rats. J. Recept. Signal Transduct. Res. 2023, 43, 1–10. [Google Scholar] [CrossRef]
- Wang, Y.; Shan, S.-K.; Guo, B.; Li, F.; Zheng, M.-H.; Lei, L.-M.; Xu, Q.-S.; Ullah, M.H.E.; Xu, F.; Lin, X.; et al. The multi-therapeutic role of MSCs in diabetic nephropathy. Front. Endocrinol. 2021, 12, 671566. [Google Scholar] [CrossRef]
- Wang, B.; Kim, K.; Tian, M.; Kameishi, S.; Zhuang, L.; Okano, T.; Huang, Y. Engineered bone marrow stem cell-sheets alleviate renal damage in a rat chronic glomerulonephritis model. Int. J. Mol. Sci. 2023, 24, 3711. [Google Scholar] [CrossRef]
- Fernandez-Pernas, P.; Barrachina, L.; Marquina, M.; Rodellar, C.; Arufe, M.C.; Costa, C. Mesenchymal stromal cells for articular cartilage repair: Preclinical studies. Eur. Cell Mater. 2020, 40, 88–114. [Google Scholar] [CrossRef]
- Xing, D.; Wang, K.; Wu, J.; Zhao, Y.; Liu, W.; Li, J.J.; Gao, T.; Yan, D.; Wang, L.; Hao, J.; et al. Clinical-grade human embryonic stem-cell-derived mesenchymal stromal cells ameliorate the progression of osteoarthritis in a rat model. Molecules 2021, 26, 604. [Google Scholar] [CrossRef]
- Massidda, M.W.; Im, B.; Lee, J.; Baker, A.B. Mechanical conditioning of human mesenchymal stem cells for enhancing vascular regeneration. STAR Protoc. 2023, 4, 102103. [Google Scholar] [CrossRef]
- Hackel, A.; Vollmer, S.; Bruderek, K.; Lang, S.; Brandau, S. Immunological priming of mesenchymal stromal/stem cells and their extracellular vesicles augment their therapeutic benefits in experimental graft-versus-host disease via engagement of PD-1 ligands. Front. Immunol. 2023, 14, 1078551. [Google Scholar] [CrossRef] [PubMed]
- Sang, H.; Zhao, R.; Lai, G.; Deng, Z.; Zhuang, W.; Wu, M.; Wu, J. Bone marrow mesenchymal stem-cell-derived exosomes attenuate the maturation of dendritic cells and reduce the rejection of allogeneic transplantation. Adv. Clin. Exp. Med. 2023, 32, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Chun, W.; Tian, J.; Zhang, Y. Transplantation of mesenchymal stem cells ameliorates systemic lupus erythematosus and upregulates B10 cells through TGF-β1. Stem Cell Res. Ther. 2021, 12, 512. [Google Scholar] [CrossRef]
- Sun, W.; Yan, S.; Yang, C.; Yang, J.; Wang, H.; Li, C.; Zhang, L.; Zhao, L.; Zhang, J.; Cheng, M.; et al. Mesenchymal stem cells-derived exosomes ameliorate lupus by inducing M2 macrophage polarization and regulatory T cell expansion in MRL/lpr mice. Immunol. Investig. 2022, 51, 1785–1803. [Google Scholar] [CrossRef]
- Aierken, A.; Li, B.; Liu, P.; Cheng, X.; Kou, Z.; Tan, N.; Zhang, M.; Yu, S.; Shen, Q.; Du, X.; et al. Melatonin treatment improves human umbilical cord mesenchymal stem cell therapy in a mouse model of type II diabetes mellitus via the PI3K/AKT signaling pathway. Stem Cell Res. Ther. 2022, 13, 164. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, Y.; Zhou, X.; Zhao, Z.; Yu, Q.; Chen, Z.; Wang, Y.; Xu, P.; Yu, Z.; Guo, C.; et al. Human umbilical cord-derived mesenchymal stem cells ameliorate psoriasis-like dermatitis by suppressing IL-17-producing gd T cells. Cell Tissue Res. 2022, 388, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, Y.; Takagi, T.; Takeda, Y.; Rajbhandari, S.; Yoshida, Y.; Nakagomi, T.; Yoshimura, S. Identification of novel multipotent stem cells in mouse spinal cord following traumatic injury. Stem Cell Dev. 2022, 31, 555–568. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(A) provides a sketch of the human vertical column and pelvis. The iliac crest of the pelvis is often used to extract samples of bone marrow for clinical studies. (B) illustrates steps of T cell activation in BM parenchyma, exemplified from a sample of iliac crest. Mature circulating naive T cells can home to BM sinuses after they have passed through aortic arteries and arterioles. There, they transmigrate sinus endothelium and enter the parenchyma. This contains resident DCs with their capacity of antigen uptake, process sing, and presentation. Cognate interactions between antigen-specific T cells and antigen-presenting DCs (APCS) in BM parenchyma involve APC scanning, T-APC cluster formation, T cell activation, T cell proliferation, and T cell re-circulation to blood. This occurs in cases of recognition of non-self-antigens (NSA). ln cases of recognition of self-antigens (SA), the T cell response leads to tolerance.
Figure 1.
(A) provides a sketch of the human vertical column and pelvis. The iliac crest of the pelvis is often used to extract samples of bone marrow for clinical studies. (B) illustrates steps of T cell activation in BM parenchyma, exemplified from a sample of iliac crest. Mature circulating naive T cells can home to BM sinuses after they have passed through aortic arteries and arterioles. There, they transmigrate sinus endothelium and enter the parenchyma. This contains resident DCs with their capacity of antigen uptake, process sing, and presentation. Cognate interactions between antigen-specific T cells and antigen-presenting DCs (APCS) in BM parenchyma involve APC scanning, T-APC cluster formation, T cell activation, T cell proliferation, and T cell re-circulation to blood. This occurs in cases of recognition of non-self-antigens (NSA). ln cases of recognition of self-antigens (SA), the T cell response leads to tolerance.

Figure 2.
Cartoon demonstrating bone marrow as a central site for cognate interaction between antigen-specific T cells (T) and antigen-presenting cells (APCs). (A) shows a femoral bone. (B) shows the spongy architecture of bone marrow with MSC-derived stromal cells providing survival niches for HSC-derived cells, including memory B and T cells (Image: https://ashpublications.org/ (accessed on 8 July 2023)). (C) shows cognate T-APC interaction upon immunological synapse formation, with centrosome polarization and signaling events via signaling complexes (Image: https://www.frontiersin.org/ (accessed on 8 July 2023)).
Figure 2.
Cartoon demonstrating bone marrow as a central site for cognate interaction between antigen-specific T cells (T) and antigen-presenting cells (APCs). (A) shows a femoral bone. (B) shows the spongy architecture of bone marrow with MSC-derived stromal cells providing survival niches for HSC-derived cells, including memory B and T cells (Image: https://ashpublications.org/ (accessed on 8 July 2023)). (C) shows cognate T-APC interaction upon immunological synapse formation, with centrosome polarization and signaling events via signaling complexes (Image: https://www.frontiersin.org/ (accessed on 8 July 2023)).

Figure 3.
The cartoon shows bone marrow at the center of a variety of immune activities. In addition it pinpoints at various types of clinical application.
Figure 3.
The cartoon shows bone marrow at the center of a variety of immune activities. In addition it pinpoints at various types of clinical application.


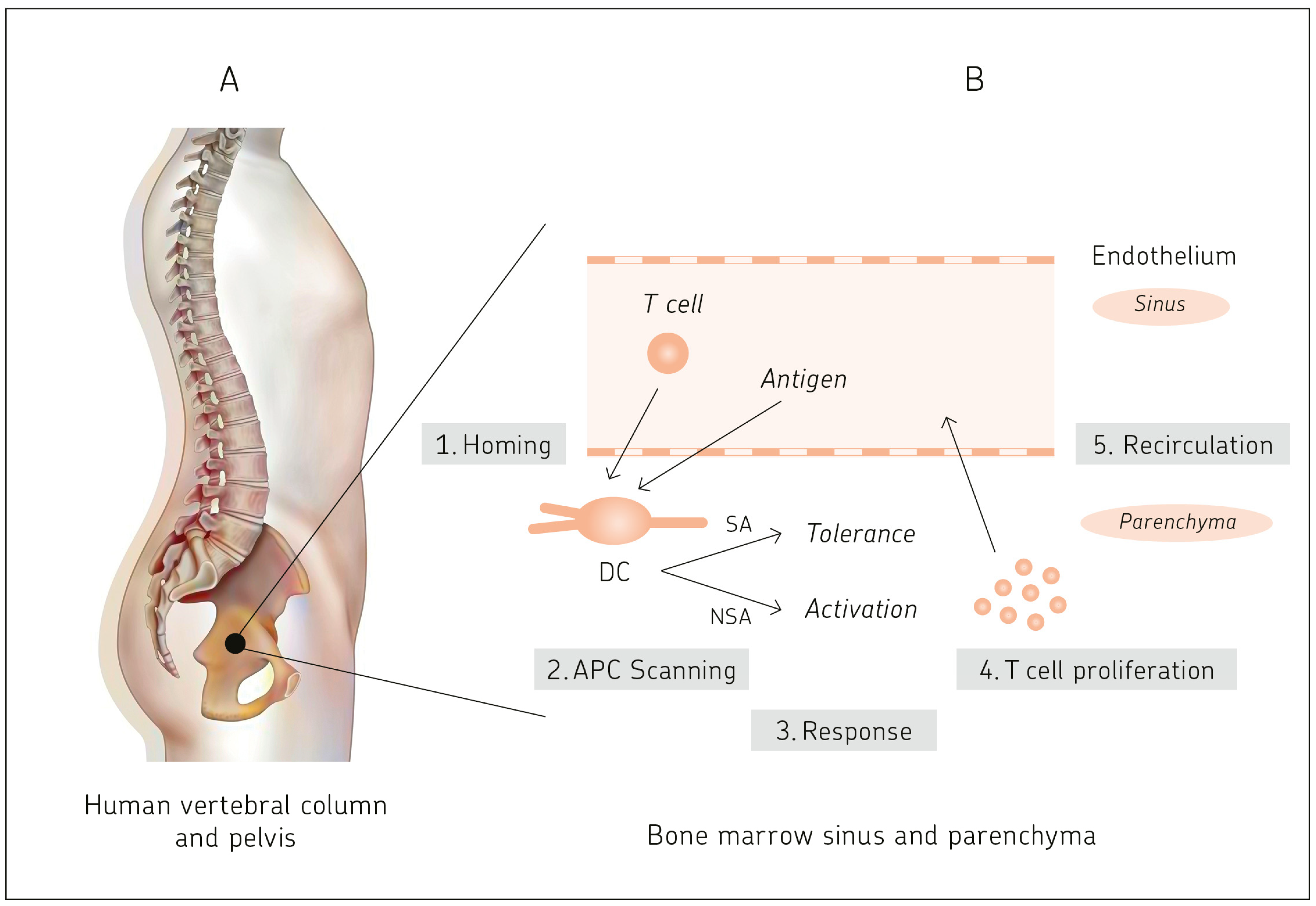{kind=link}
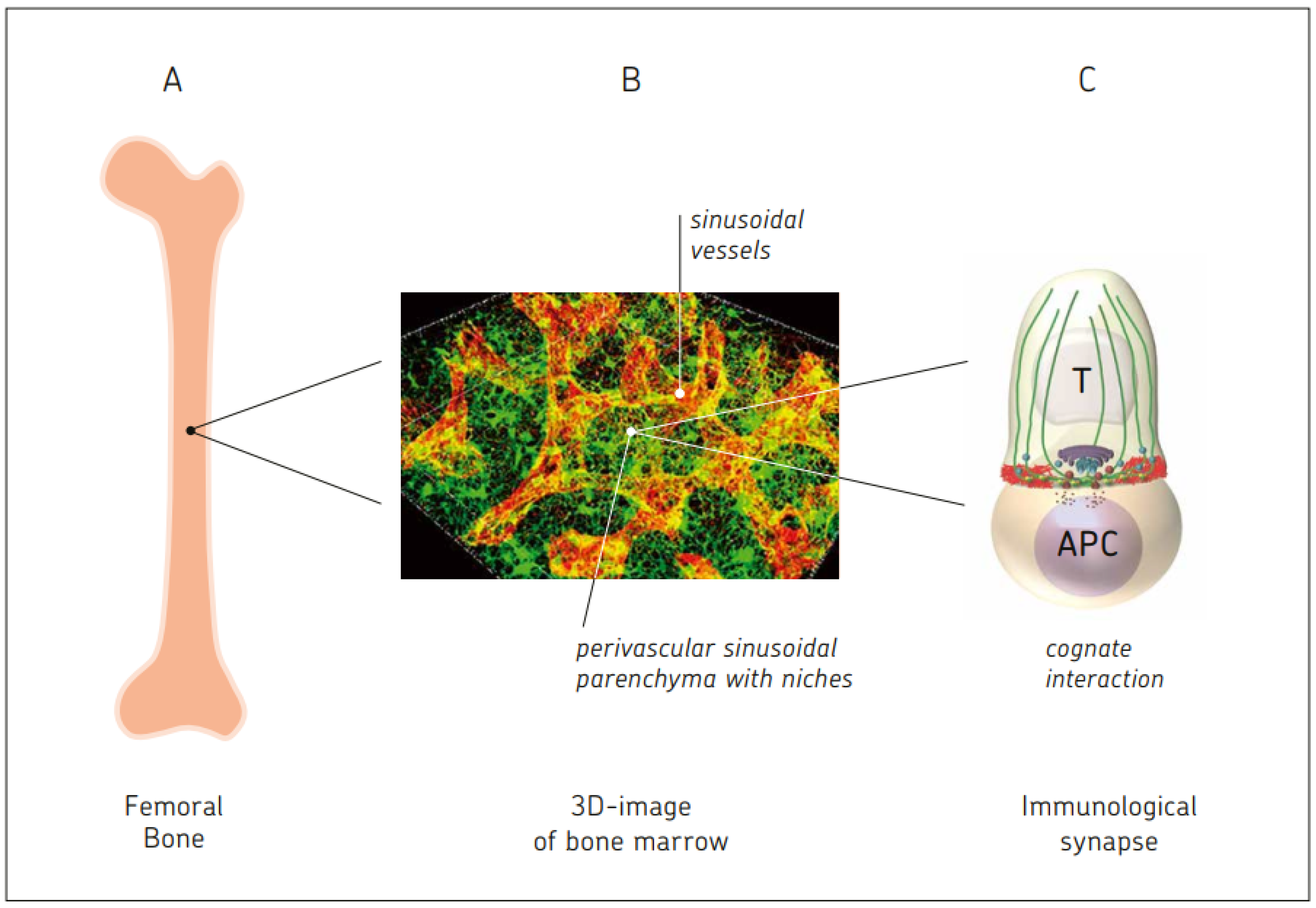{kind=link}
{kind=link}
Table 1.
Bone marrow: a unique lymphatic organ.
| Feature | Description | Ref. | Year |
|---|---|---|---|
| Generative lymphatic organ | -Organ of hematopoiesis and osteogenesis: from stem cells via committed precursors to late precursors and mature forms | [7] [9] | 2022 2013 |
| -Maintaining a constantly steady state of cells from the bone cortex and marrow | [8] | 2016 | |
| -Most prominent source of de novo cellular generation in the body: 4–5 × 1011/day | [10] | 2017 | |
| Antigen responsive lymphatic organ | -Bone marrow as a priming site for T-cell responses to blood-borne antigens | [17] | 2003 |
| -Two photon dynamic imaging in situ revealing T cell priming | [23] | 2013 | |
| Functional comparison to blood | -12% of all human lymphocytes found in the BM at any time compared to 2% in the peripheral blood | [9] | 2013 |
| -BM: 4–5% TBW; lymph nodes and spleen: 1–1.5% TBW | [10] | 2017 | |
| -Tumor dormancy in bone marrow | [21] | 1994 | |
| -Active control in BM by CD8+ memory T cells | [22] | 1998 | |
| -Enrichment of MTCs in BM of breast cancer patients | [13] | 2001 | |
| -Generation of CTLs from BM, but not PBL, of breast cancer patients | [17] | 2003 | |
| [13] | 2001 | ||
| -Superior therapeutic efficiency of BM MTCs from breast cancer patients | [19] | 2001 | |
| Survival niches for memory lymphocytes organized by bone marrow stromal cells in the parenchyma | -IgG+ memory B cells, IgG+ memory plasma cells, | [61] | 2021 |
| -IgA+ memory plasma cells | [62] | 2018 | |
| -Memory T: unique homing phenotype: CCR5 + CXCR6 + CXCR3- | [55] | 2008 | |
| -Memory CD4+ T cells, CD69+ | [20] | 2004 | |
| -Memory CD8+ T cells, CD69+ | [17] | 2003 | |
| -Stem cell-like memory T cells, CD69+ CD127+ | [47] | 2022 | |
| -Cognate re-activation of TA-specific BM-MTCs: ex vivo | [19] | 2001 | |
| in situ by vaccination with ATV-NDV | [32] | 2020 | |
| -Enrichment in BM of virus-specific MTCs: EBV | [55] | 2008 | |
| CMV | [58] | 2009 | |
| HCV | [47] | 2022 | |
| SARS-Cov-2 | [46] | 2021 | |
| Hematopoietic stem cell cross-talk with T cells and DCs | -Cross-talk with T cells: adoptive T cell therapy and transfer of HSC leading to potent intratumoral DCs in malignant glioma | [67] | 2019 |
| -Cross-talk with BM DCs: Il-1β regulating HSC function | [68] | 2022 | |
| Effect of dietary restriction | -Monocyte mobilization from BM | [76] | 2019 |
| -Migration of B cells from PPs to BM | [77] | 2019 | |
| -Memory T cells collapse in secondary lymphatic organs; in contrast, memory T cell accumulates in BM | [79] | 2019 | |
| -Adipocytes and CXCR4-CXCL12 interactions contribute to enhanced T cell accumulation and remodeling of the BM compartment | [79] | 2019 |
References are examples and not exclusive. BM = bone marrow; CMV = cytomegalovirus; CTL = cytotoxic T lymphocyte; DC = dendritic cell; EBV = Epstein–Barr virus; HCV = hepatitis C virus; MTC = memory T cell; PP = Peyer’s patch lymph node; TBW = total body weight.
Table 2.
Bone marrow–blood interaction.
| Feature | Description | Ref | Year |
|---|---|---|---|
| Transport of blood-borne antigens | -Peripheral and systemic antigens | [45] | 2019 |
| -Immune complexes | [84] | 2020 | |
| -Macromolecules (for central tolerance) | [80] | 2021 | |
| -Tumor-secreted proteins | [87] | 2023 | |
| Homing of blood circulating cells | -Blood-borne viruses, e.g., HCV, HBV, and HIV | [82] | 2019 |
| -Circulatory tumor cells and derived extracellular vesicles, apoptotic bodies, and tumor-associated proteins | [85] | 2023 | |
| -Circulating antigen-presenting cells | [94] | 2005 | |
| Egress of blood circulating cells | -Circulatory naïve and memory T cells | [96] | 2022 |
| Antigen presentation capacity | -BM DCs, prepulsed in vivo with a model tumor antigen, primed naïve T cells in vivo and induced long-lasting systemic tumor antigen-specific resistance. | [25] | 2003 |
| T–APC interaction capacity | -APC scanning | [120] | 2011 |
| -Immunological synapse formation | [128] | 2000 | |
| [127] | 2021 | ||
| -T cell costimulation and signaling | [126] | 2022 | |
| -Mitochondrial priming by CD28 | [125] | 2017 | |
| -Cytoskeletal reorganization | [123] | 1999 | |
| -Response polarization | [131] | 2022 | |
| -Bidirectional cell stimulation | [139] | 2003 | |
| -Follicular immune cluster formation in parenchyma | [17] | 2003 | |
| -T cell activation and proliferation | [20] | 2004 | |
| -Memory B cell activation and antibody production | [31] | 2021 | |
| Storage capacity of memory cells in BM survival niches | -Stromal-cell-expressed VCAM-1 holding immune cells in the niche | [144] | 2002 |
| -Stromal-cell-secreted IL-7 and IL-15 mediating survival of MTCs | [6] | 1994 | |
| -Stromal-cell-expressed CD80/CD86 binding CD28 on T cells and plasma cells | [143] | 2000 | |
| -Stromal cell contact and secreted APRIL/BAFF preventing mitochondrial and ER stress of plasma cells | [144] [141] | 2002 2011 | |
| Autonomic adaptive immune response | -Independence from secondary lymphoid organs | [17] | 2003 |
| -Map3k14aly/aly splenectomized mouse response similar to that of BM in normal mice | |||
| Neuro-immune links | -Allergic inflammation | [103] | 2022 |
| -Neuro-oncology | [105] | 2023 | |
| -Neuro-osteogenic network | [118] | 2023 |
References are examples and not exclusive. APC = antigen-presenting cell; BM = bone marrow; DC = dendritic cell; HBV = hepatitis B virus; HCV = hepatitis C virus; HIV = human immunodeficiency virus; VCAM-1 = vascular cell adhesion molecule 1.
Table 3.
Bone marrow: T regulatory cells and dendritic cells maintaining self-tolerance and reacting to microbial infection and cancer.
Table 3.
Bone marrow: T regulatory cells and dendritic cells maintaining self-tolerance and reacting to microbial infection and cancer.
| Feature | Description | Ref. | Year |
|---|---|---|---|
| Tissue-regulatory cells | -Maintenance of self-tolerance and support of organ homeostasis | [145] | 2017 |
| -Epigenetic regulation | |||
| BM niche for Treg cells | -Immunosuppression and maintenance of HSCs | [146] | 2022 |
| -Future role in HSC transplantation | |||
| BM Tregs being unique | -Supported by niche-specific cytokines and pDCs | [148] | 2021 |
| -Future role in control of GvH disease | |||
| -Role of transcription factor BATF | [147] | 2023 | |
| GvL without GvH | -Tumor-immune MTCs from allogeneic MHC-identical bone marrow exerting GvL without GvH | [150] | 2005 |
| -Suppression of GvH requiring Treg priming | [151] | 2005 | |
| Tumor-specific BM Treg cells | -Orchestration of antitumor immunity in breast cancer | [159] | 2019 |
| -Tregs, selectively activated in BM, egressing into the blood | [159] | 2019 | |
| Osseous Ewing sarcoma (ES) | -High proportion of BM Treg cells | [157] | 2009 |
| -Association with metastatic disease | |||
| Effect of microbial infection and inflammation | -Widespread transcriptional changes in BM immune cells in peritonitis sepsis model | [160] | 2022 |
| -Mitochondrial DNA mediating immunoparalysis in BM DCs via STING signaling in sepsis model | [162] | 2022 | |
| -Optimal priming of BM pDCs by CD169+ macrophages in mouse model of severe malaria | [163] | 2022 | |
| -Melatonin affecting BM DCs in DSS-induced colitis mice | [164] | 2022 | |
| Tumor dormancy | -Colocalization in BM of melanoma cells and CD8+ MTCs | [165] | 2020 |
| -Maintenance of CD8+ MTCs in BM niches by residual ESb-Gal lymphoma cells | [167] | 2005 |
References are examples and not exclusive. GvH = graft versus host reaction; DSS = dextran sodium sulfate; HSC = hematopoietic stem cell; MHC = major histocompatibility complex; MTC = memory T cell; pDC = plasmacytoid dendritic cell; STING = stimulator of interferon genes; Treg = T regulatory cell.
Table 4.
Bone marrow: mesenchymal stem and stromal cells in regenerative medicine.
| Feature | Description | Ref. | Year |
|---|---|---|---|
| Heart failure | -Meta-analysis revealing BM-MSC treatment as an effective intervention | [185] | 2023 |
| Neurological diseases | -MSC treatment for cognitive impairment | [189] | 2022 |
| -Instruction of a beneficial phenotype in reactive astrocytes | [191] | 2021 | |
| -Cognitive impairement in an Alzheimer’s disease model | [178] | 2020 | |
| -Hippocampal neurogenesis in aged mice by polarized MSCs | [190] | 2020 | |
| Lung injury and fibrosis | -MSC-derived EVs attenuate radiation-induced lung injury | [192] | 2021 |
| -Therapeutic efficiency in post-COVID pulmonary fibrosis | [194] | 2021 | |
| Liver regeneration | -Adipose-derived stem cells in hepatobiliary diseases | [195] | 2022 |
| -Reversion of activated to quiescent HeSCs by NDV | [184] | 2009 | |
| -Inhibition of liver fibrosis by mirRNA | [183] | 2014 | |
| -Sustainment of CD8+ T cell memory by IL-15+ HeSCs | [181] | 2022 | |
| Kidney injury | -Sepsis model, remodeling immune homeostasis | [196] | 2023 |
| -Inhibition of ferroptosis via Nrf2-keap1/p53 pathway | [197] | 2023 | |
| -Multi-therapeutic role in diabetic nephropathy | [198] | 2021 | |
| Osteoarthritis | -Amelioration of progression in a rat model | [201] | 2021 |
| -Articular cartilage repair in preclinical studies | [200] | 2020 | |
| Vascular regeneration | -A new conditioning technique to enhance vascular regenerative properties of hMSCs with increases in the expression of endothelial and pericyte markers | [202] | 2023 |
| Autoimmune diseases | -Amelioration of lupus erythematosus | [205] | 2021 |
| -Amelioration of psoriasis-like dermatitis | [208] | 2022 | |
| -Amelioration of type II diabetes mellitus | [207] | 2022 | |
| -Amelioration of experimental autoimmune encephalomyelitis | [191] | 2021 |
References are examples and not exclusive. HeSC = hepatic stellate cell; hMSC = human mesenchymal stem cell; MSC = mesenchymal stem cell; NDV = Newcastle disease virus.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Schirrmacher, V. Bone Marrow: The Central Immune System. Immuno 2023, 3, 289-329. https://doi.org/10.3390/immuno3030019
AMA Style
Schirrmacher V. Bone Marrow: The Central Immune System. Immuno. 2023; 3(3):289-329. https://doi.org/10.3390/immuno3030019
Chicago/Turabian StyleSchirrmacher, Volker. 2023. "Bone Marrow: The Central Immune System" Immuno 3, no. 3: 289-329. https://doi.org/10.3390/immuno3030019