
In Silico Analysis Highlights Potential Predictive Indicators Associated with Secondary Progressive Multiple Sclerosis

Abstract
:1. Introduction
2. Results
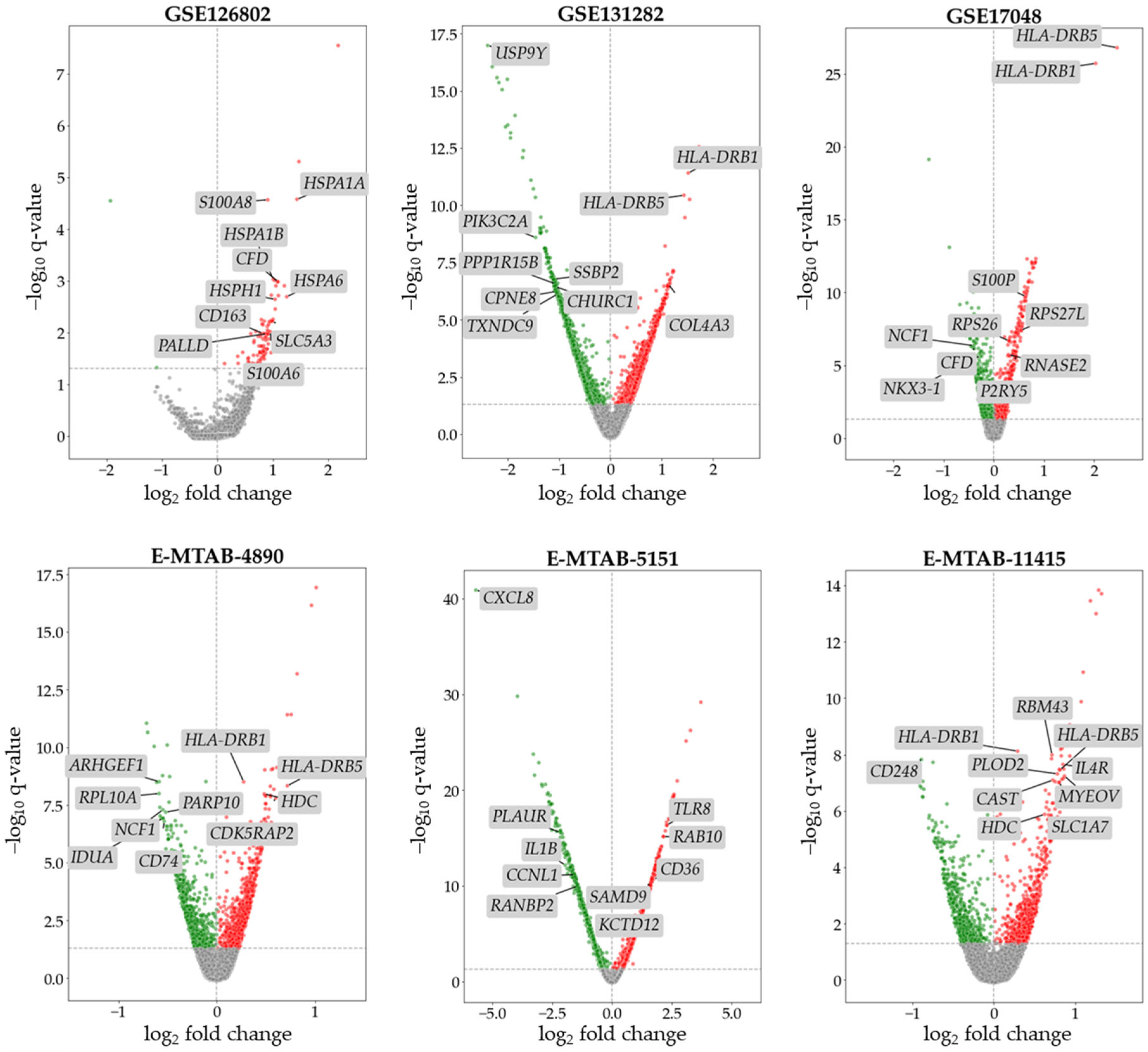
2.1. Differential Expression Analysis (DEA) of SPMS Data
2.2. DEG Filtering and Selection
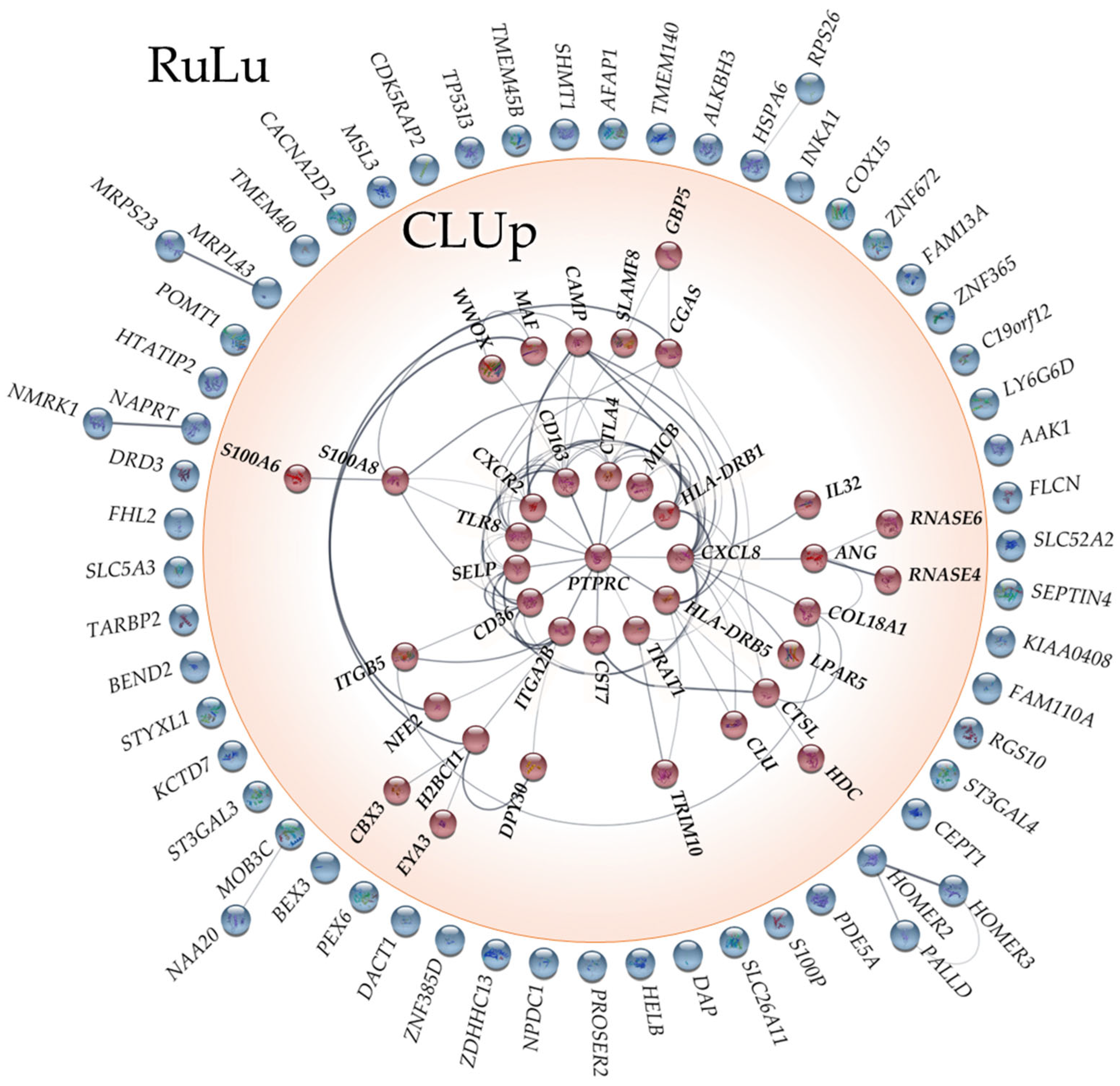
2.3. Network Analysis
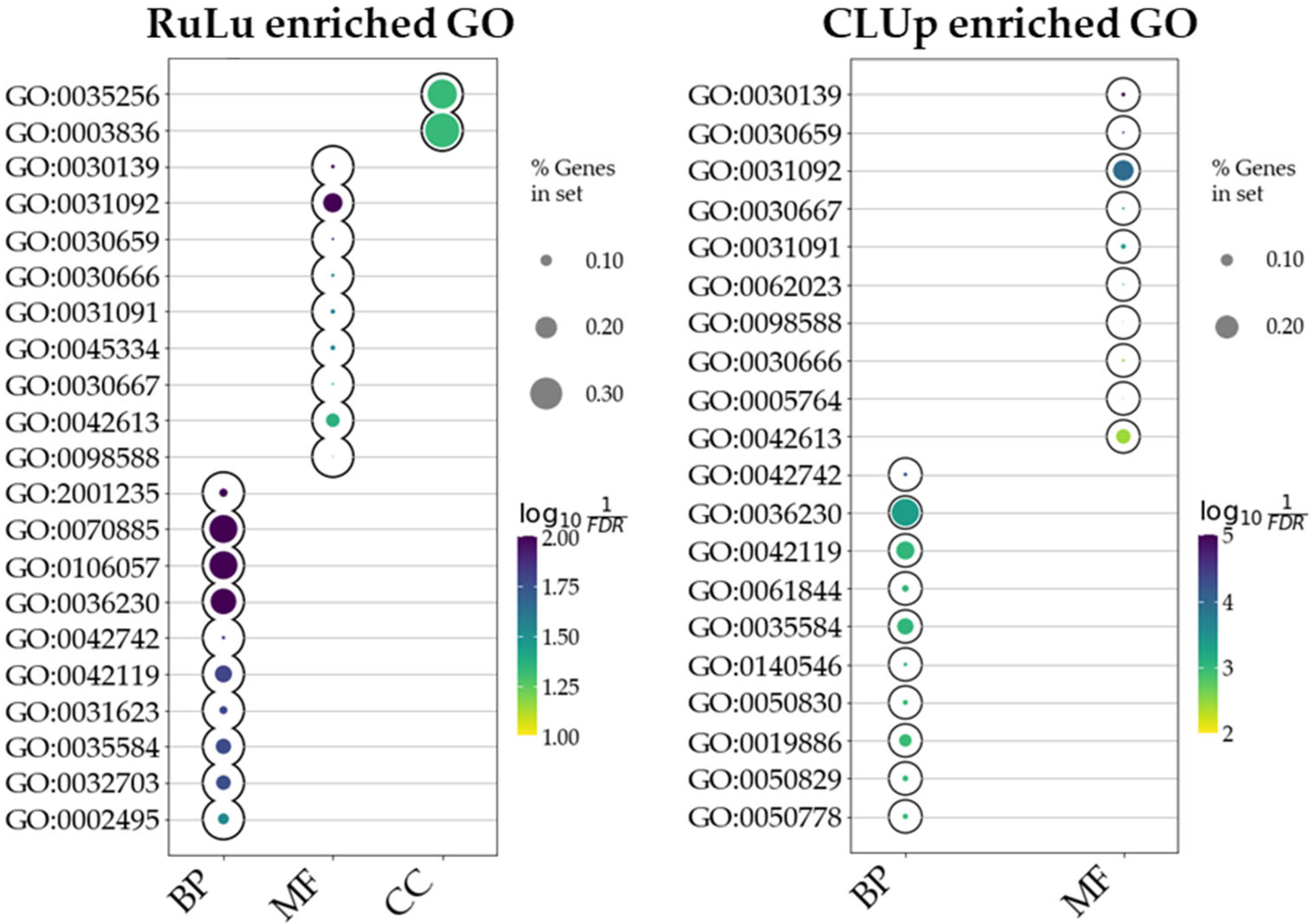
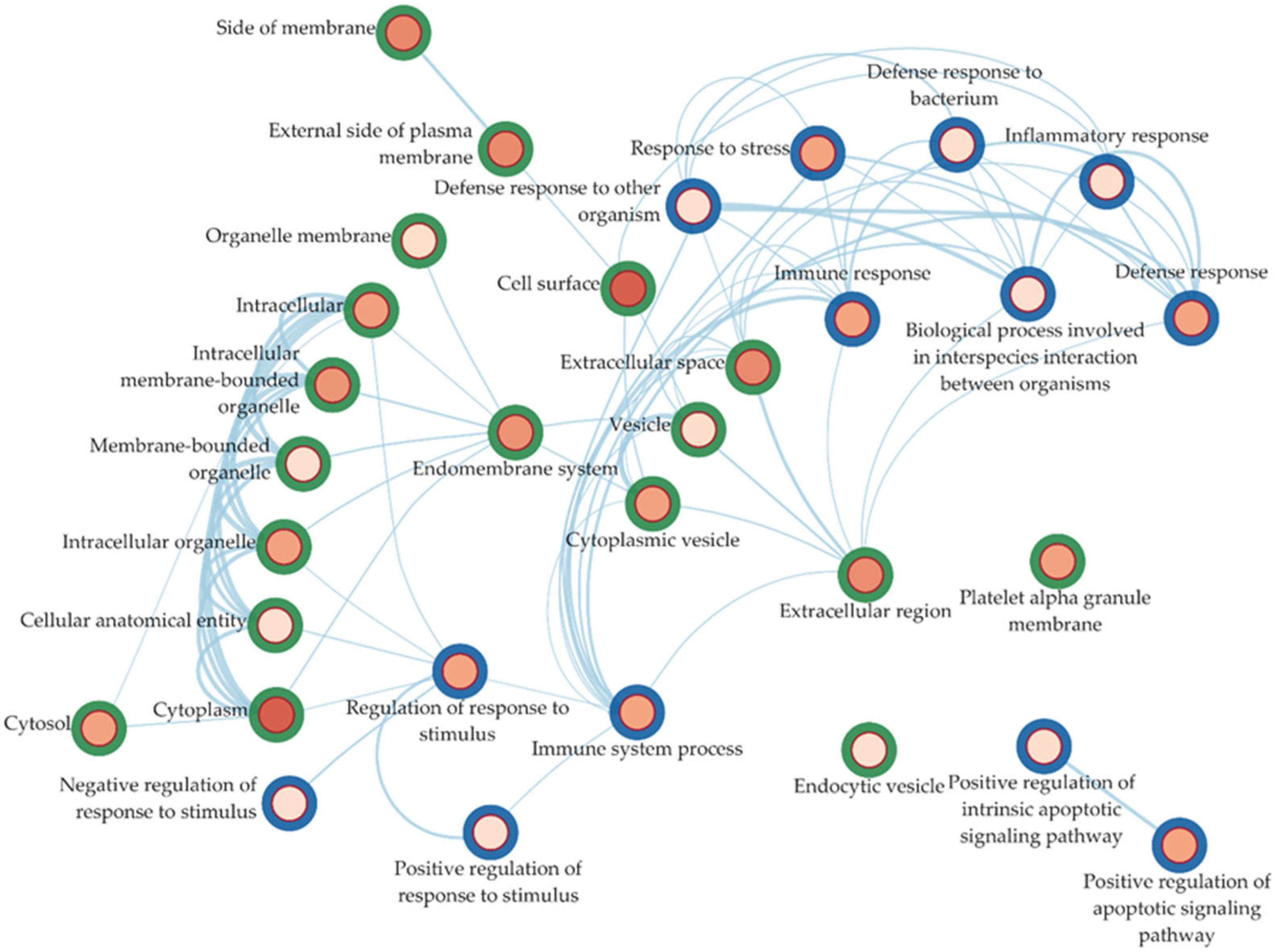
2.4. Ontology Enrichment
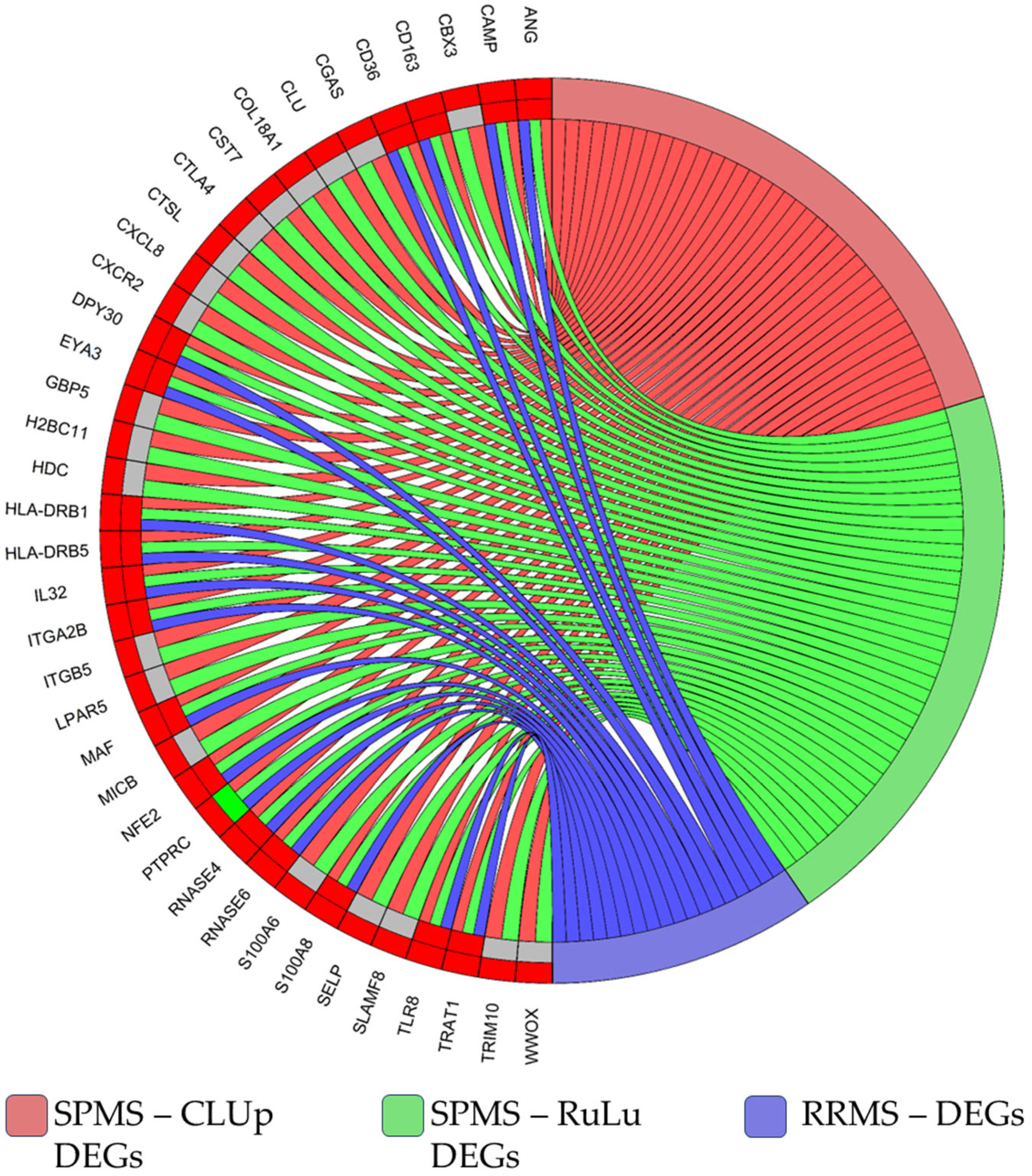
2.5. Comparison with RRMS Patients’ Data
3. Discussion
4. Materials and Methods
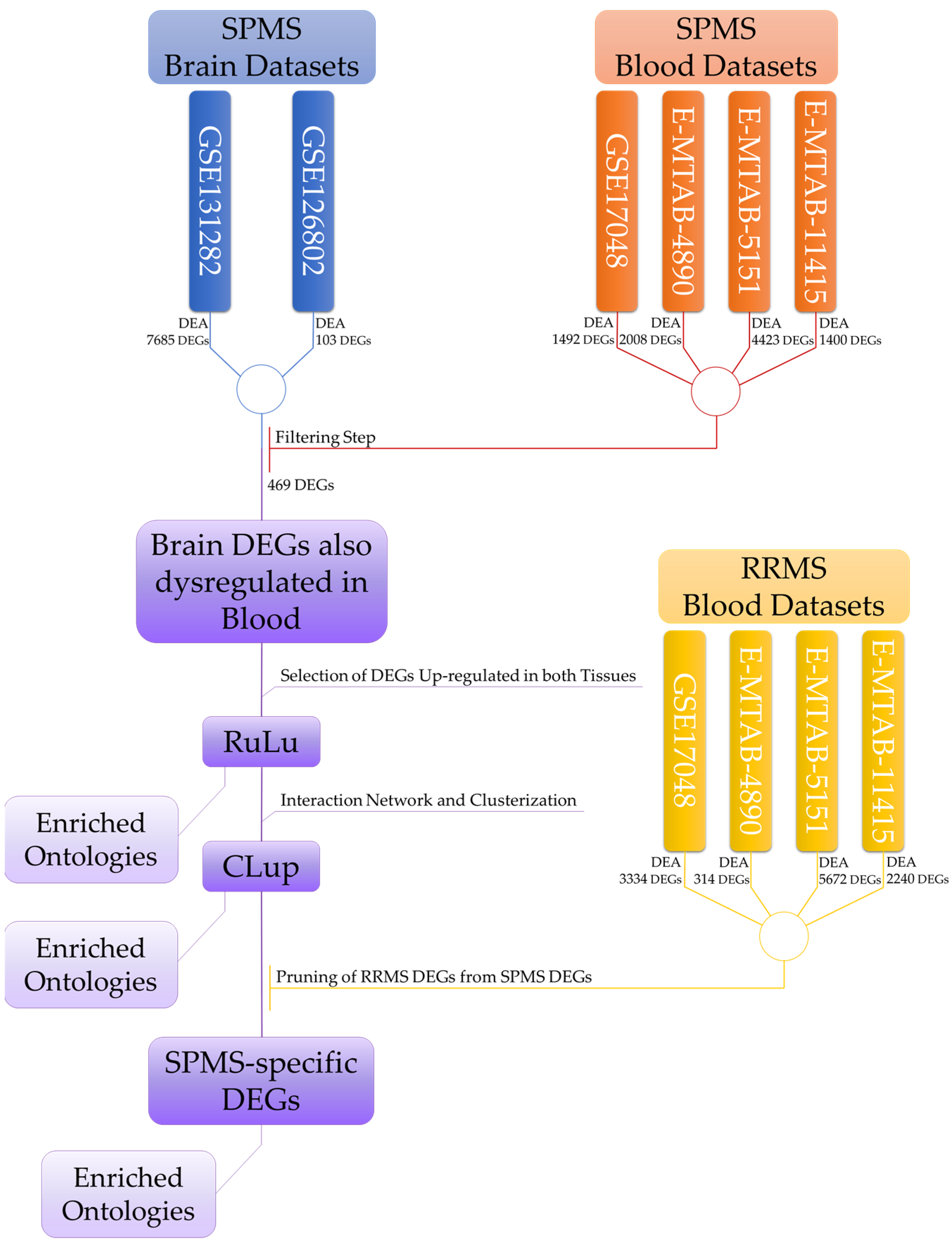
4.1. Flowchart of the Analysis
4.2. Dataset Selection
4.3. Dataset Description
4.4. Annotation of Probes and Genes
4.5. Bioinformatics Analysis
4.6. Differential Expression Analysis Specifics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Belbasis, L.; Bellou, V.; Evangelou, E.; Ioannidis, J.P.; Tzoulaki, I. Environmental risk factors and multiple sclerosis: An umbrella review of systematic reviews and meta-analyses. Lancet Neurol. 2015, 14, 263–273. [Google Scholar] [CrossRef]
- Sawcer, S.; Franklin, R.J.; Ban, M. Multiple sclerosis genetics. Lancet Neurol. 2014, 13, 700–709. [Google Scholar] [CrossRef]
- Trapp, B.D.; Peterson, J.; Ransohoff, R.M.; Rudick, R.; Mork, S.; Bo, L. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 1998, 338, 278–285. [Google Scholar] [CrossRef]
- Confavreux, C.; Vukusic, S. Natural history of multiple sclerosis: A unifying concept. Brain A J. Neurol. 2006, 129, 606–616. [Google Scholar] [CrossRef]
- Pugliatti, M.; Rosati, G.; Carton, H.; Riise, T.; Drulovic, J.; Vecsei, L.; Milanov, I. The epidemiology of multiple sclerosis in Europe. Eur. J. Neurol. 2006, 13, 700–722. [Google Scholar] [CrossRef]
- Grytten Torkildsen, N.; Lie, S.A.; Aarseth, J.H.; Nyland, H.; Myhr, K.M. Survival and cause of death in multiple sclerosis: Results from a 50-year follow-up in Western Norway. Mult. Scler. 2008, 14, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.; Trevick, S.; Younger, D.S. Epidemiology of Multiple Sclerosis. Neurol. Clin. 2016, 34, 919–939. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Reingold, S.C. Defining the clinical course of multiple sclerosis: Results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology 1996, 46, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Gaitan, M.I.; Ysrraelit, M.C.; Fiol, M.P. Progressive multiple sclerosis: From pathogenic mechanisms to treatment. Brain A J. Neurol. 2017, 140, 527–546. [Google Scholar] [CrossRef] [PubMed]
- Scalfari, A.; Neuhaus, A.; Degenhardt, A.; Rice, G.P.; Muraro, P.A.; Daumer, M.; Ebers, G.C. The natural history of multiple sclerosis: A geographically based study 10: Relapses and long-term disability. Brain A J. Neurol. 2010, 133, 1914–1929. [Google Scholar] [CrossRef]
- Katz Sand, I.; Krieger, S.; Farrell, C.; Miller, A.E. Diagnostic uncertainty during the transition to secondary progressive multiple sclerosis. Mult. Scler. 2014, 20, 1654–1657. [Google Scholar] [CrossRef]
- Miljkovic, D.; Spasojevic, I. Multiple sclerosis: Molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2013, 19, 2286–2334. [Google Scholar] [CrossRef]
- Chiricosta, L.; Blando, S.; D’Angiolini, S.; Gugliandolo, A.; Mazzon, E. A Comprehensive Exploration of the Transcriptomic Landscape in Multiple Sclerosis: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 1448. [Google Scholar] [CrossRef]
- Bielekova, B.; Martin, R. Development of biomarkers in multiple sclerosis. Brain A J. Neurol. 2004, 127, 1463–1478. [Google Scholar] [CrossRef]
- Anderson, N.L.; Polanski, M.; Pieper, R.; Gatlin, T.; Tirumalai, R.S.; Conrads, T.P.; Veenstra, T.D.; Adkins, J.N.; Pounds, J.G.; Fagan, R.; et al. The human plasma proteome: A nonredundant list developed by combination of four separate sources. Mol. Cell. Proteom. MCP 2004, 3, 311–326. [Google Scholar] [CrossRef]
- Melief, J.; Orre, M.; Bossers, K.; van Eden, C.G.; Schuurman, K.G.; Mason, M.R.J.; Verhaagen, J.; Hamann, J.; Huitinga, I. Transcriptome analysis of normal-appearing white matter reveals cortisol- and disease-associated gene expression profiles in multiple sclerosis. Acta Neuropathol. Commun. 2019, 7, 60. [Google Scholar] [CrossRef] [PubMed]
- Enz, L.S.; Zeis, T.; Schmid, D.; Geier, F.; van der Meer, F.; Steiner, G.; Certa, U.; Binder, T.M.C.; Stadelmann, C.; Martin, R.; et al. Increased HLA-DR expression and cortical demyelination in MS links with HLA-DR15. Neurol. Neuroimmunol. Neuroinflamm/ 2020, 7, e656. [Google Scholar] [CrossRef]
- Riveros, C.; Mellor, D.; Gandhi, K.S.; McKay, F.C.; Cox, M.B.; Berretta, R.; Vaezpour, S.Y.; Inostroza-Ponta, M.; Broadley, S.A.; Heard, R.N.; et al. A transcription factor map as revealed by a genome-wide gene expression analysis of whole-blood mRNA transcriptome in multiple sclerosis. PLoS ONE 2010, 5, e14176. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Di Dario, M.; Menon, R.; Valente, M.M.; Bassani, C.; Sarno, N.; Mazza, D.; Montini, F.; Moiola, L.; Comi, G.; et al. HNF4alpha, SP1 and c-myc are master regulators of CNS autoimmunity. J. Autoimmun. 2023, 138, 103053. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Di Dario, M.; Russo, A.; Menon, R.; Brini, E.; Romeo, M.; Sangalli, F.; Costa, G.D.; Rodegher, M.; Radaelli, M.; et al. Dysregulation of MS risk genes and pathways at distinct stages of disease. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e337. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Severa, M.; Rizzo, F.; Menon, R.; Brini, E.; Mechelli, R.; Martinelli, V.; Hertzog, P.; Salvetti, M.; Furlan, R.; et al. Transcriptional dysregulation of Interferome in experimental and human Multiple Sclerosis. Sci. Rep. 2017, 7, 8981. [Google Scholar] [CrossRef] [PubMed]
- Ha, N.; Pham, D.H.; Shahsafaei, A.; Naruse, C.; Asano, M.; Thai, T.H. HP-1gamma Controls High-Affinity Antibody Response to T-Dependent Antigens. Front. Immunol. 2014, 5, 271. [Google Scholar] [CrossRef] [PubMed]
- Vinuesa, C.G.; Cyster, J.G. How T cells earn the follicular rite of passage. Immunity 2011, 35, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Shulman, Z.; Gitlin, A.D.; Targ, S.; Jankovic, M.; Pasqual, G.; Nussenzweig, M.C.; Victora, G.D. T follicular helper cell dynamics in germinal centers. Science 2013, 341, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Huang, Y.J.; He, X.; Zhao, M.; Wang, X.; Liu, Z.S.; Xue, W.; Cai, H.; Zhan, X.Y.; Huang, S.Y.; et al. Acetylation Blocks cGAS Activity and Inhibits Self-DNA-Induced Autoimmunity. Cell 2019, 176, 1447–1460.e1414. [Google Scholar] [CrossRef] [PubMed]
- Skopelja-Gardner, S.; An, J.; Elkon, K.B. Role of the cGAS-STING pathway in systemic and organ-specific diseases. Nat. Rev. Nephrol. 2022, 18, 558–572. [Google Scholar] [CrossRef]
- Duarte, N.; Shafi, A.M.; Penha-Goncalves, C.; Pais, T.F. Endothelial type I interferon response and brain diseases: Identifying STING as a therapeutic target. Front. Cell Dev. Biol. 2023, 11, 1249235. [Google Scholar] [CrossRef]
- Cuadrado, E.; Vanderver, A.; Brown, K.J.; Sandza, A.; Takanohashi, A.; Jansen, M.H.; Anink, J.; Herron, B.; Orcesi, S.; Olivieri, I.; et al. Aicardi-Goutieres syndrome harbours abundant systemic and brain-reactive autoantibodies. Ann. Rheum. Dis. 2015, 74, 1931–1939. [Google Scholar] [CrossRef]
- Razia, R.; Majeed, F.; Amin, R.; Mukhtar, S.; Mehmood, K.; Baig, D.N. The analysis of dynamic gene expression patterns in peripheral blood of multiple sclerosis patients indicates possible diagnostic and prognostic biomarkers. Mol. Immunol. 2022, 147, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; Zoubeidi, A. Clusterin as a therapeutic target. Expert Opin. Ther. Targets 2017, 21, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kim, J.K.; Edwards, C.A.; Xu, Z.; Taichman, R.; Wang, C.Y. Clusterin inhibits apoptosis by interacting with activated Bax. Nat. Cell Biol. 2005, 7, 909–915. [Google Scholar] [CrossRef]
- Leskov, K.S.; Klokov, D.Y.; Li, J.; Kinsella, T.J.; Boothman, D.A. Synthesis and functional analyses of nuclear clusterin, a cell death protein. J. Biol. Chem. 2003, 278, 11590–11600. [Google Scholar] [CrossRef] [PubMed]
- Han, B.H.; DeMattos, R.B.; Dugan, L.L.; Kim-Han, J.S.; Brendza, R.P.; Fryer, J.D.; Kierson, M.; Cirrito, J.; Quick, K.; Harmony, J.A.; et al. Clusterin contributes to caspase-3-independent brain injury following neonatal hypoxia-ischemia. Nat. Med. 2001, 7, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Girolamo, F.; Coppola, C.; Ribatti, D.; Trojano, M. Angiogenesis in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol. Commun. 2014, 2, 84. [Google Scholar] [CrossRef] [PubMed]
- Puxeddu, I.; Capecchi, R.; Pratesi, F.; Cianchetti, S.; Tavoni, A.; Migliorini, P. Mediators of angiogenesis and fibrosis in IgG4-related disease. Clin. Exp. Med. 2018, 18, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Bellocchi, C.; Assassi, S.; Lyons, M.; Marchini, M.; Mohan, C.; Santaniello, A.; Beretta, L. Proteomic aptamer analysis reveals serum markers that characterize preclinical systemic sclerosis (SSc) patients at risk for progression toward definite SSc. Arthritis Res. Ther. 2023, 25, 15. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, C.J.; Furlong, S.J.; Doucette, C.D.; Chen, P.L.; Hoskin, D.W.; Easton, A.S. Bevacizumab diminishes experimental autoimmune encephalomyelitis by inhibiting spinal cord angiogenesis and reducing peripheral T-cell responses. J. Neuropathol. Exp. Neurol. 2012, 71, 983–999. [Google Scholar] [CrossRef] [PubMed]
- Argaw, A.T.; Asp, L.; Zhang, J.; Navrazhina, K.; Pham, T.; Mariani, J.N.; Mahase, S.; Dutta, D.J.; Seto, J.; Kramer, E.G.; et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Investig. 2012, 122, 2454–2468. [Google Scholar] [CrossRef]
- Obata-Onai, A.; Hashimoto, S.; Onai, N.; Kurachi, M.; Nagai, S.; Shizuno, K.; Nagahata, T.; Matsushima, K. Comprehensive gene expression analysis of human NK cells and CD8(+) T lymphocytes. Int. Immunol. 2002, 14, 1085–1098. [Google Scholar] [CrossRef]
- Ma, J.; Tanaka, K.F.; Shimizu, T.; Bernard, C.C.; Kakita, A.; Takahashi, H.; Pfeiffer, S.E.; Ikenaka, K. Microglial cystatin F expression is a sensitive indicator for ongoing demyelination with concurrent remyelination. J. Neurosci. Res. 2011, 89, 639–649. [Google Scholar] [CrossRef]
- Nakagawa, T.; Roth, W.; Wong, P.; Nelson, A.; Farr, A.; Deussing, J.; Villadangos, J.A.; Ploegh, H.; Peters, C.; Rudensky, A.Y. Cathepsin L: Critical role in Ii degradation and CD4 T cell selection in the thymus. Science 1998, 280, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Basile, M.S.; Bramanti, P.; Mazzon, E. The Role of Cytotoxic T-Lymphocyte Antigen 4 in the Pathogenesis of Multiple Sclerosis. Genes 2022, 13, 1319. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, N.; Bartholomaus, I.; Pesic, M.; Mues, M. An autoimmunity odyssey: How autoreactive T cells infiltrate into the CNS. Immunol. Rev. 2012, 248, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, E.M.; Bar-Or, A.; Waliszewska, A.I.; Cai, G.; Anderson, D.E.; Krieger, J.I.; Hafler, D.A. CTLA-4 dysregulation in the activation of myelin basic protein reactive T cells may distinguish patients with multiple sclerosis from healthy controls. J. Autoimmun. 2003, 20, 71–81. [Google Scholar] [CrossRef]
- Takahashi, T.; Tagami, T.; Yamazaki, S.; Uede, T.; Shimizu, J.; Sakaguchi, N.; Mak, T.W.; Sakaguchi, S. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 2000, 192, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.; Patton, K.T.; Anderson, D.E.; Freeman, G.J.; Hafler, D.A. Expansion of autoreactive T cells in multiple sclerosis is independent of exogenous B7 costimulation. J. Immunol. 1998, 160, 1532–1538. [Google Scholar] [CrossRef]
- Mohammadzadeh, A.; Rad, I.A.; Ahmadi-Salmasi, B. CTLA-4, PD-1 and TIM-3 expression predominantly downregulated in MS patients. J. Neuroimmunol. 2018, 323, 105–108. [Google Scholar] [CrossRef]
- Liszewski, M.K.; Kolev, M.; Le Friec, G.; Leung, M.; Bertram, P.G.; Fara, A.F.; Subias, M.; Pickering, M.C.; Drouet, C.; Meri, S.; et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity 2013, 39, 1143–1157. [Google Scholar] [CrossRef]
- Shibamura-Fujiogi, M.; Yuki, K.; Hou, L. Cathepsin L regulates pathogenicCD4 T cells in experimental autoimmune encephalomyelitis. Int. Immunopharmacol. 2021, 93, 107425. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Guo, J.; Tomlinson, S.; Yuan, G.; He, S. The role of the complosome in health and disease. Front. Immunol. 2023, 14, 1146167. [Google Scholar] [CrossRef]
- Langerholc, T.; Zavasnik-Bergant, V.; Turk, B.; Turk, V.; Abrahamson, M.; Kos, J. Inhibitory properties of cystatin F and its localization in U937 promonocyte cells. FEBS J. 2005, 272, 1535–1545. [Google Scholar] [CrossRef]
- Cui, L.Y.; Chu, S.F.; Chen, N.H. The role of chemokines and chemokine receptors in multiple sclerosis. Int. Immunopharmacol. 2020, 83, 106314. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Liu, X.; Wang, Y.; Zhang, X.; Wang, H.; Du, X.; Du, Z.; Yang, L.; Hou, J. Discovery of grey matter lesion-related immune genes for diagnostic prediction in multiple sclerosis. PeerJ 2023, 11, e15299. [Google Scholar] [CrossRef] [PubMed]
- Sampson, A.P. The role of eosinophils and neutrophils in inflammation. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2000, 30 (Suppl. S1), 22–27. [Google Scholar] [CrossRef]
- Nguyen, D.; Stangel, M. Expression of the chemokine receptors CXCR1 and CXCR2 in rat oligodendroglial cells. Dev. Brain Res. 2001, 128, 77–81. [Google Scholar] [CrossRef]
- Sales, K.J.; Maldonado-Perez, D.; Grant, V.; Catalano, R.D.; Wilson, M.R.; Brown, P.; Williams, A.R.; Anderson, R.A.; Thompson, E.A.; Jabbour, H.N. Prostaglandin F(2alpha)-F-prostanoid receptor regulates CXCL8 expression in endometrial adenocarcinoma cells via the calcium-calcineurin-NFAT pathway. Biochim. Biophys. Acta 2009, 1793, 1917–1928. [Google Scholar] [CrossRef]
- Lund, B.T.; Ashikian, N.; Ta, H.Q.; Chakryan, Y.; Manoukian, K.; Groshen, S.; Gilmore, W.; Cheema, G.S.; Stohl, W.; Burnett, M.E.; et al. Increased CXCL8 (IL-8) expression in Multiple Sclerosis. J. Neuroimmunol. 2004, 155, 161–171. [Google Scholar] [CrossRef]
- Baranzini, S.E.; Elfstrom, C.; Chang, S.Y.; Butunoi, C.; Murray, R.; Higuchi, R.; Oksenberg, J.R. Transcriptional analysis of multiple sclerosis brain lesions reveals a complex pattern of cytokine expression. J. Immunol. 2000, 165, 6576–6582. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.; Siegel, R.J.; Fox, D.A.; Ahmed, S. Interferon-stimulated GTPases in autoimmune and inflammatory diseases: Promising role for the guanylate-binding protein (GBP) family. Rheumatology 2021, 60, 494–506. [Google Scholar] [CrossRef]
- Li, Y.; Lin, X.; Wang, W.; Wang, W.; Cheng, S.; Huang, Y.; Zou, Y.; Ke, J.; Zhu, L. The Proinflammatory Role of Guanylate-Binding Protein 5 in Inflammatory Bowel Diseases. Front. Microbiol. 2022, 13, 926915. [Google Scholar] [CrossRef]
- Shenoy, A.R.; Wellington, D.A.; Kumar, P.; Kassa, H.; Booth, C.J.; Cresswell, P.; MacMicking, J.D. GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science 2012, 336, 481–485. [Google Scholar] [CrossRef]
- Byun, D.J.; Lee, J.; Ko, K.; Hyun, Y.M. NLRP3 exacerbates EAE severity through ROS-dependent NET formation in the mouse brain. Cell Commun. Signal. CCS 2024, 22, 96. [Google Scholar] [CrossRef]
- Naegele, M.; Tillack, K.; Reinhardt, S.; Schippling, S.; Martin, R.; Sospedra, M. Neutrophils in multiple sclerosis are characterized by a primed phenotype. J. Neuroimmunol. 2012, 242, 60–71. [Google Scholar] [CrossRef]
- Karlsson, M.; Zhang, C.; Mear, L.; Zhong, W.; Digre, A.; Katona, B.; Sjostedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A single-cell type transcriptomics map of human tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef]
- Jia, J.; Han, Z.; Wang, X.; Zheng, X.; Wang, S.; Cui, Y. H2B gene family: A prognostic biomarker and correlates with immune infiltration in glioma. Front. Oncol. 2022, 12, 966817. [Google Scholar] [CrossRef]
- Sandoval-Talamantes, A.K.; Gomez-Gonzalez, B.A.; Uriarte-Mayorga, D.F.; Martinez-Guzman, M.A.; Wheber-Hidalgo, K.A.; Alvarado-Navarro, A. Neurotransmitters, neuropeptides and their receptors interact with immune response in healthy and psoriatic skin. Neuropeptides 2020, 79, 102004. [Google Scholar] [CrossRef]
- Branco, A.; Yoshikawa, F.S.Y.; Pietrobon, A.J.; Sato, M.N. Role of Histamine in Modulating the Immune Response and Inflammation. Mediat. Inflamm. 2018, 2018, 9524075. [Google Scholar] [CrossRef]
- Rafiee Zadeh, A.; Falahatian, M.; Alsahebfosoul, F. Serum levels of histamine and diamine oxidase in multiple sclerosis. Am. J. Clin. Exp. Immunol. 2018, 7, 100–105. [Google Scholar] [PubMed]
- Saligrama, N.; Case, L.K.; del Rio, R.; Noubade, R.; Teuscher, C. Systemic lack of canonical histamine receptor signaling results in increased resistance to autoimmune encephalomyelitis. J. Immunol. 2013, 191, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yang, K.; Zheng, J.; Sun, X.; Zhao, W. Establishment of a novel prognostic signature based on an identified expression profile of integrin superfamily to predict overall survival of patients with colorectal adenocarcinoma. Gene 2022, 808, 145990. [Google Scholar] [CrossRef] [PubMed]
- Mathew, D.; Torres, R.M. Lysophosphatidic Acid Is an Inflammatory Lipid Exploited by Cancers for Immune Evasion via Mechanisms Similar and Distinct from CTLA-4 and PD-1. Front. Immunol. 2020, 11, 531910. [Google Scholar] [CrossRef] [PubMed]
- Konen, J.M.; Rodriguez, B.L.; Wu, H.; Fradette, J.J.; Gibson, L.; Diao, L.; Wang, J.; Schmidt, S.; Wistuba, I.I.; Zhang, J.; et al. Autotaxin suppresses cytotoxic T cells via LPAR5 to promote anti-PD-1 resistance in non-small cell lung cancer. J. Clin. Investig. 2023, 133, e163128. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.; Brunkhorst, R.; de Bruin, N.; Mayer, C.A.; Haussler, A.; Ferreiros, N.; Schiffmann, S.; Parnham, M.J.; Tunaru, S.; Chun, J.; et al. Dysregulation of lysophosphatidic acids in multiple sclerosis and autoimmune encephalomyelitis. Acta Neuropathol. Commun. 2017, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Pillars Article: Activation of NK Cells and T Cells by NKG2D, a Receptor for Stress-Inducible MICA. Science 1999, 285, 727–729, Erratum in J. Immunol. 2018, 200, 2231–2233. [Google Scholar] [CrossRef] [PubMed]
- Mo, X.B.; Lei, S.F.; Qian, Q.Y.; Guo, Y.F.; Zhang, Y.H.; Zhang, H. Integrative analysis revealed potential causal genetic and epigenetic factors for multiple sclerosis. J. Neurol. 2019, 266, 2699–2709. [Google Scholar] [CrossRef] [PubMed]
- Beecham, A.H.; Amezcua, L.; Chinea, A.; Manrique, C.P.; Gomez, L.; Martinez, A.; Beecham, G.W.; Patsopoulos, N.A.; Chitnis, T.; Weiner, H.L.; et al. Ancestral risk modification for multiple sclerosis susceptibility detected across the Major Histocompatibility Complex in a multi-ethnic population. PLoS ONE 2022, 17, e0279132. [Google Scholar] [CrossRef] [PubMed]
- Abediankenari, S.; Yousefzadeh, Y.; Majidi, M.; Ghasemi, M.; Nasehi, M.M.; Ghaffari, J.; Habibi Saravi, R.; Abedini, M.; Elyasi, M. MICB gene expression on peripheral blood mononuclear cells and susceptibility to multiple sclerosis in north of Iran. Iran. J. Allergy Asthma Immunol. 2011, 10, 261–265. [Google Scholar]
- Vyshkina, T.; Leist, T.P.; Shugart, Y.Y.; Kalman, B. CD45 (PTPRC) as a candidate gene in multiple sclerosis. Mult. Scler. 2004, 10, 614–617. [Google Scholar] [CrossRef]
- Vidmar, L.; Maver, A.; Drulovic, J.; Sepcic, J.; Novakovic, I.; Ristic, S.; Sega, S.; Peterlin, B. Multiple Sclerosis patients carry an increased burden of exceedingly rare genetic variants in the inflammasome regulatory genes. Sci. Rep. 2019, 9, 9171. [Google Scholar] [CrossRef]
- Hecker, M.; Ruge, A.; Putscher, E.; Boxberger, N.; Rommer, P.S.; Fitzner, B.; Zettl, U.K. Aberrant expression of alternative splicing variants in multiple sclerosis—A systematic review. Autoimmun. Rev. 2019, 18, 721–732. [Google Scholar] [CrossRef]
- Bartkowska, K.; Swiatek, I.; Aniszewska, A.; Jurewicz, E.; Turlejski, K.; Filipek, A.; Djavadian, R.L. Stress-Dependent Changes in the CacyBP/SIP Interacting Protein S100A6 in the Mouse Brain. PLoS ONE 2017, 12, e0169760. [Google Scholar] [CrossRef]
- Karimi, N.; Motovali-Bashi, M.; Ghaderi-Zefrehei, M. Gene network reveals LASP1, TUBA1C, and S100A6 are likely playing regulatory roles in multiple sclerosis. Front. Neurol. 2023, 14, 1090631. [Google Scholar] [CrossRef]
- Tsai, M.H.; Lin, C.H.; Tsai, K.W.; Lin, M.H.; Ho, C.J.; Lu, Y.T.; Weng, K.P.; Lin, Y.; Lin, P.H.; Li, S.C. S100A6 Promotes B Lymphocyte Penetration Through the Blood-Brain Barrier in Autoimmune Encephalitis. Front. Genet. 2019, 10, 1188. [Google Scholar] [CrossRef] [PubMed]
- McEver, R.P. Selectins: Initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc. Res. 2015, 107, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Rammohan, K.; Lin, S.; Robinson, N.; Li, O.; Liu, X.; Bai, X.F.; Yin, L.; Scarberry, B.; Du, P.; et al. CD24 is a genetic modifier for risk and progression of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2003, 100, 15041–15046. [Google Scholar] [CrossRef]
- Patel, K.D.; Cuvelier, S.L.; Wiehler, S. Selectins: Critical mediators of leukocyte recruitment. Semin. Immunol. 2002, 14, 73–81. [Google Scholar] [CrossRef]
- Romero, X.; Sintes, J.; Engel, P. Role of SLAM family receptors and specific adapter SAP in innate-like lymphocytes. Crit. Rev. Immunol. 2014, 34, 263–299. [Google Scholar] [CrossRef] [PubMed]
- de Lange, K.M.; Moutsianas, L.; Lee, J.C.; Lamb, C.A.; Luo, Y.; Kennedy, N.A.; Jostins, L.; Rice, D.L.; Gutierrez-Achury, J.; Ji, S.G.; et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 2017, 49, 256–261. [Google Scholar] [CrossRef]
- Zou, C.Y.; Guan, G.F.; Zhu, C.; Liu, T.Q.; Guo, Q.; Cheng, W.; Wu, A.H. Costimulatory checkpoint SLAMF8 is an independent prognosis factor in glioma. CNS Neurosci. Ther. 2019, 25, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Rong, X.; Yu, C.; Jia, P.; Yang, J.; Zhou, G. Knockout of SLAMF8 attenuates collagen-induced rheumatoid arthritis in mice through inhibiting TLR4/NF-kappaB signaling pathway. Int. Immunopharmacol. 2022, 107, 108644. [Google Scholar] [CrossRef]
- Zhang, Q.; Cheng, L.; Qin, Y.; Kong, L.; Shi, X.; Hu, J.; Li, L.; Ding, Z.; Wang, T.; Shen, J.; et al. SLAMF8 expression predicts the efficacy of anti-PD1 immunotherapy in gastrointestinal cancers. Clin. Transl. Immunol. 2021, 10, e1347. [Google Scholar] [CrossRef]
- Guo, M.; Cao, W.; Chen, S.; Tian, R.; Wang, L.; Liu, Q.; Zhang, L.; Wang, Z.; Zhao, M.; Lu, Q.; et al. TRIM10 binds to IFN-alpha/beta receptor 1 to negatively regulate type I IFN signal transduction. Eur. J. Immunol. 2021, 51, 1762–1773. [Google Scholar] [CrossRef]
- Jia, X.; Zhao, C.; Zhao, W. Emerging Roles of MHC Class I Region-Encoded E3 Ubiquitin Ligases in Innate Immunity. Front. Immunol. 2021, 12, 687102. [Google Scholar] [CrossRef]
- Meyer, M.; Gaudieri, S.; Rhodes, D.A.; Trowsdale, J. Cluster of TRIM genes in the human MHC class I region sharing the B30.2 domain. Tissue Antigens 2003, 61, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Simon-Sanchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- Lanata, C.M.; Nititham, J.; Taylor, K.E.; Chung, S.A.; Torgerson, D.G.; Seldin, M.F.; Pons-Estel, B.A.; Tusie-Luna, T.; Tsao, B.P.; Morand, E.F.; et al. Genetic contributions to lupus nephritis in a multi-ethnic cohort of systemic lupus erythematous patients. PLoS ONE 2018, 13, e0199003. [Google Scholar] [CrossRef]
- Bednarek, A.K.; Keck-Waggoner, C.L.; Daniel, R.L.; Laflin, K.J.; Bergsagel, P.L.; Kiguchi, K.; Brenner, A.J.; Aldaz, C.M. WWOX, the FRA16D gene, behaves as a suppressor of tumor growth. Cancer Res. 2001, 61, 8068–8073. [Google Scholar]
- Aldaz, C.M.; Hussain, T. WWOX Loss of Function in Neurodevelopmental and Neurodegenerative Disorders. Int. J. Mol. Sci. 2020, 21, 8922. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yang, Y.; Zhou, C.; Mei, X.; Liu, J.; Luo, K.; Zhou, J.; Qin, C.; Zeng, Z. WWOX activates autophagy to alleviate lipopolysaccharide-induced acute lung injury by regulating mTOR. Int. Immunopharmacol. 2023, 115, 109671. [Google Scholar] [CrossRef]
- Shin, M.J.; Kim, H.S.; Lee, P.; Yang, N.G.; Kim, J.Y.; Eun, Y.S.; Lee, W.; Kim, D.; Lee, Y.; Jung, K.E.; et al. Mechanistic Investigation of WWOX Function in NF-kB-Induced Skin Inflammation in Psoriasis. Int. J. Mol. Sci. 2023, 25, 167. [Google Scholar] [CrossRef] [PubMed]
- Kaluzinska-Kolat, Z.; Kolat, D.; Kosla, K.; Pluciennik, E.; Bednarek, A.K. Molecular landscapes of glioblastoma cell lines revealed a group of patients that do not benefit from WWOX tumor suppressor expression. Front. Neurosci. 2023, 17, 1260409. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics, C.; Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Jakel, S.; Agirre, E.; Mendanha Falcao, A.; van Bruggen, D.; Lee, K.W.; Knuesel, I.; Malhotra, D.; Ffrench-Constant, C.; Williams, A.; Castelo-Branco, G. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 2019, 566, 543–547. [Google Scholar] [CrossRef]
- Park, Y.J.; Yoo, S.A.; Kim, M.; Kim, W.U. The Role of Calcium-Calcineurin-NFAT Signaling Pathway in Health and Autoimmune Diseases. Front. Immunol. 2020, 11, 195. [Google Scholar] [CrossRef] [PubMed]
- Macian, F. NFAT proteins: Key regulators of T-cell development and function. Nat. Reviews. Immunol. 2005, 5, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Kook, S.Y.; Hong, H.S.; Moon, M.; Ha, C.M.; Chang, S.; Mook-Jung, I. Abeta(1)(-)(4)(2)-RAGE interaction disrupts tight junctions of the blood-brain barrier via Ca(2)(+)-calcineurin signaling. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 8845–8854. [Google Scholar] [CrossRef] [PubMed]
- Stergioti, E.M.; Manolakou, T.; Boumpas, D.T.; Banos, A. Antiviral Innate Immune Responses in Autoimmunity: Receptors, Pathways, and Therapeutic Targeting. Biomedicines 2022, 10, 2820. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, H.; Kapushesky, M.; Shojatalab, M.; Abeygunawardena, N.; Coulson, R.; Farne, A.; Holloway, E.; Kolesnykov, N.; Lilja, P.; Lukk, M.; et al. ArrayExpress--a public database of microarray experiments and gene expression profiles. Nucleic Acids Res. 2007, 35, D747–D750. [Google Scholar] [CrossRef]
- Gandhi, K.S.; McKay, F.C.; Cox, M.; Riveros, C.; Armstrong, N.; Heard, R.N.; Vucic, S.; Williams, D.W.; Stankovich, J.; Brown, M.; et al. The multiple sclerosis whole blood mRNA transcriptome and genetic associations indicate dysregulation of specific T cell pathways in pathogenesis. Hum. Mol. Genet. 2010, 19, 2134–2143. [Google Scholar] [CrossRef]
- Visentin, L.; Scarpellino, G.; Chinigo, G.; Munaron, L.; Ruffinatti, F.A. BioTEA: Containerized Methods of Analysis for Microarray-Based Transcriptomics Data. Biology 2022, 11, 1346. [Google Scholar] [CrossRef] [PubMed]
- Del Carratore, F.; Jankevics, A.; Eisinga, R.; Heskes, T.; Hong, F.; Breitling, R. RankProd 2.0: A refactored bioconductor package for detecting differentially expressed features in molecular profiling datasets. Bioinformatics 2017, 33, 2774–2775. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | Overlap | p-Value | q-Value | Genes |
|---|---|---|---|---|
| Negative regulation of calcineurin–NFAT signaling cascade (GO:0070885) | 3/11 | 5.32 × 10−5 | 3.51 × 10−2 | HOMER2; FHL2; HOMER3 |
| Negative regulation of calcineurin-mediated signaling (GO:0106057) | 3/11 | 5.32 × 10−5 | 3.51 × 10−2 | HOMER2; FHL2; HOMER3 |
| Regulation of defense response to virus by host (GO:0050691) | 4/37 | 1.27 × 10−4 | 4.19 × 10−2 | DHX9; CGAS; TARBP2; MICB |
| Dataset | Tissue | Secondary Progressive (SPMS) | Relapsing–Remitting (RRMS) | Healthy Controls (HCs) | References | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n | % Females | Mean Age | n | % Females | Mean Age | n | % Females | Mean Age | |||
| GSE126802 | Brain | 18 | 100.0% | 61.3 ± 13.4 | - | - | - | 9 | 100.0% | 61.7 ± 10.1 | [16] |
| GSE131282 | Brain | 23 | 73.9% | 75.1 ± 14.8 | - | - | - | 14 | 28.6% | 58.5 ± 14.5 | [17] |
| GSE17048 | Blood | 20 | 75.0% | 57.5 ± 9.8 | 36 | 80.6% | 48.5 ± 9.0 | 45 | 64.4% | 48.5 ± 13.5 | [18,110] |
| E-MTAB-4890 | Blood | 21 | 52.4% | 54.3 ± 11.1 | 52 | 61.5% | 37.4 ± 10.2 | 40 | 50.0% | 33.3 ± 10.4 | [20] |
| E-MTAB-5151 | Blood | 13 | 69.2% | 41.8 ± 9.3 | 21 | 57.1% | 41.2 ± 8.1 | 27 | 48.2% | 42.4 ± 9.1 | [21] |
| E-MTAB-11415 | Blood | 21 | 47.6% | 54.4 ± 11.1 | 53 | 60.4% | 37.4 ± 10.1 | 40 | 50.0% | 33.4 ± 10.5 | [19] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calabrò, M.; Lui, M.; Mazzon, E.; D’Angiolini, S. In Silico Analysis Highlights Potential Predictive Indicators Associated with Secondary Progressive Multiple Sclerosis. Int. J. Mol. Sci. 2024, 25, 3374. https://doi.org/10.3390/ijms25063374
Calabrò M, Lui M, Mazzon E, D’Angiolini S. In Silico Analysis Highlights Potential Predictive Indicators Associated with Secondary Progressive Multiple Sclerosis. International Journal of Molecular Sciences. 2024; 25(6):3374. https://doi.org/10.3390/ijms25063374
Chicago/Turabian StyleCalabrò, Marco, Maria Lui, Emanuela Mazzon, and Simone D’Angiolini. 2024. "In Silico Analysis Highlights Potential Predictive Indicators Associated with Secondary Progressive Multiple Sclerosis" International Journal of Molecular Sciences 25, no. 6: 3374. https://doi.org/10.3390/ijms25063374