
Metabolic Crossroads: Unveiling the Complex Interactions between Obstructive Sleep Apnoea and Metabolic Syndrome

 ,
, 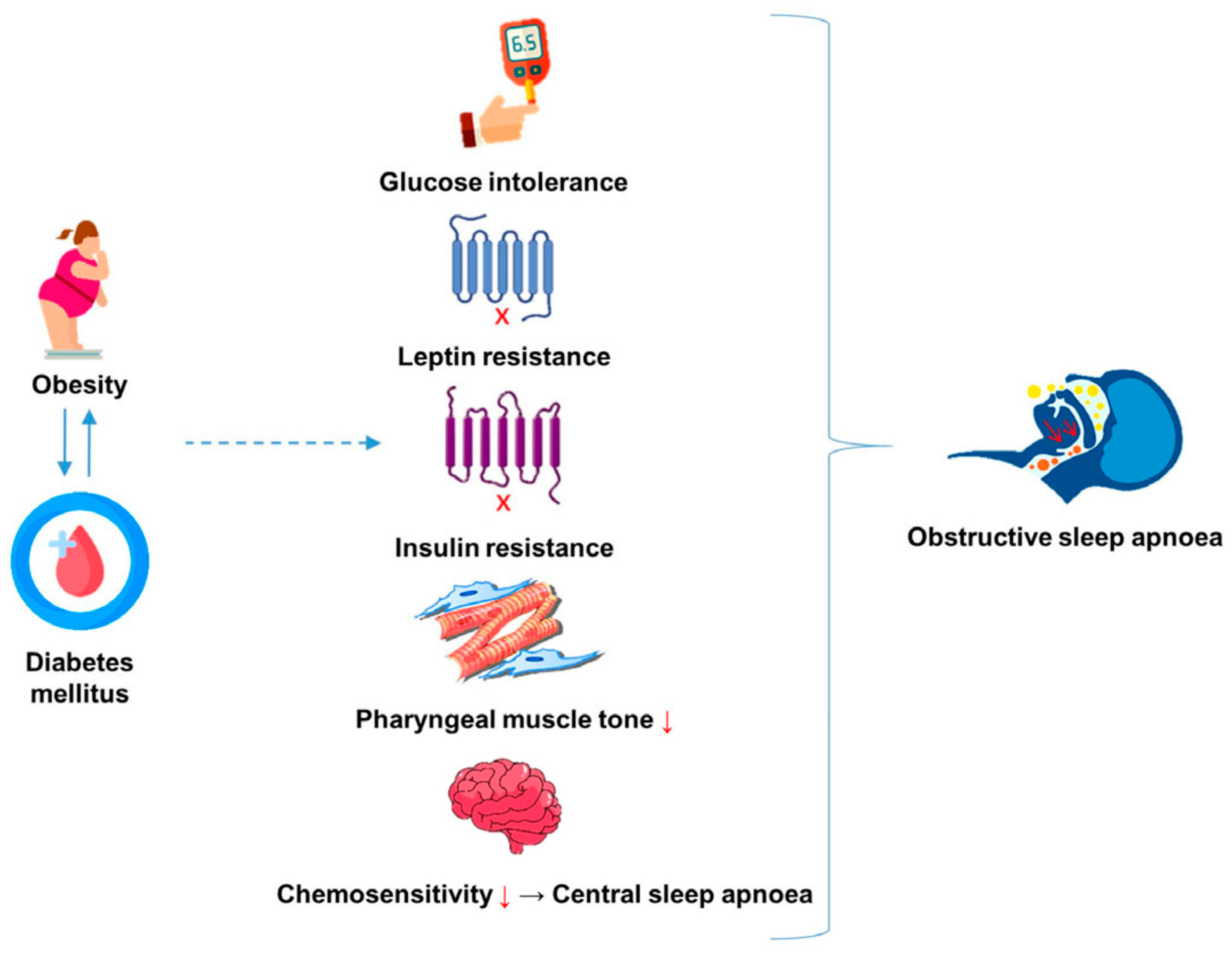{kind=link}
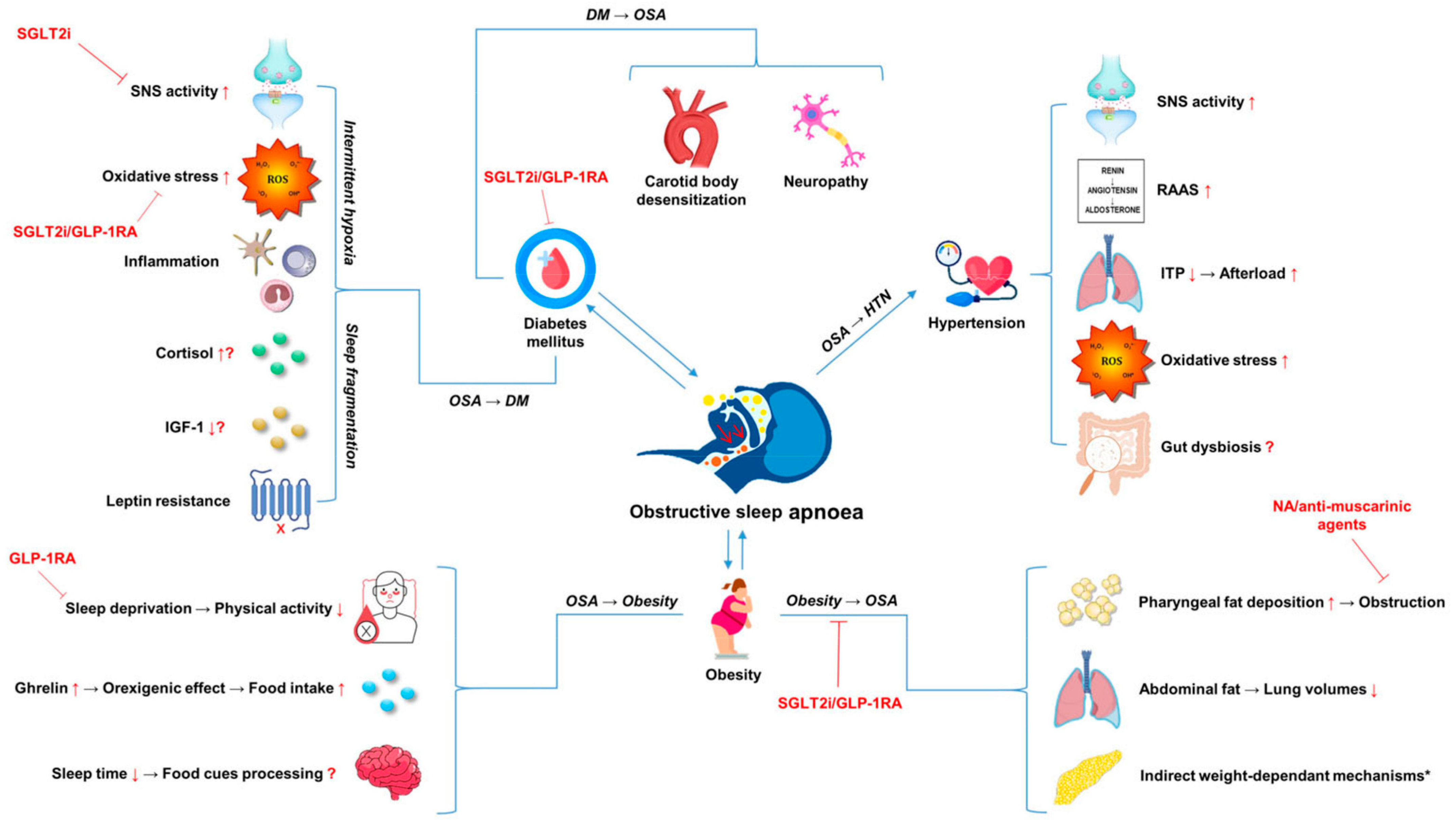{kind=link}
Abstract
:1. Introduction
2. Obesity, OSA and MetS: Cause or Consequence?
3. OSA, Hyperglycaemia and Diabetes Mellitus
4. Leptin
5. Hypertension
6. Dietary and Lifestyle Modification in OSA and MetS
7. Future Directions and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Osman, A.M.; Carter, S.G.; Carberry, J.C.; Eckert, D.J. Obstructive Sleep Apnea: Current Perspectives. Nat. Sci. Sleep 2018, 10, 21–34. [Google Scholar] [CrossRef]
- Franklin, K.A.; Lindberg, E. Obstructive Sleep Apnea Is a Common Disorder in the Population—A Review on the Epidemiology of Sleep Apnea. J. Thorac. Dis. 2015, 7, 1311–1322. [Google Scholar]
- Younas, H.; Gu, C.; Rathore, A.; Jun, J.C.; Polotsky, V.Y. Chapter 8—Metabolic Syndrome and Sleep Apnea: A Bidirectional Relationship. In Mechanisms and Manifestations of Obesity in Lung Disease; Johnston, R.A., Suratt, B.T., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 169–200. ISBN 9780128135532. [Google Scholar]
- Benjafield, A.V.; Ayas, N.T.; Eastwood, P.R.; Heinzer, R.; Ip, M.S.M.; Morrell, M.J.; Nunez, C.M.; Patel, S.R.; Penzel, T.; Pépin, J.-L.; et al. Estimation of the Global Prevalence and Burden of Obstructive Sleep Apnoea: A Literature-Based Analysis. Lancet Respir. Med. 2019, 7, 687–698. [Google Scholar] [CrossRef]
- Song, S.O.; He, K.; Narla, R.R.; Kang, H.G.; Ryu, H.U.; Boyko, E.J. Metabolic Consequences of Obstructive Sleep Apnea Especially Pertaining to Diabetes Mellitus and Insulin Sensitivity. Diabetes Metab. J. 2019, 43, 144–155. [Google Scholar] [CrossRef]
- Bozic, J.; Borovac, J.A.; Galic, T.; Kurir, T.T.; Supe-Domic, D.; Dogas, Z. Adropin and Inflammation Biomarker Levels in Male Patients with Obstructive Sleep Apnea: A Link with Glucose Metabolism and Sleep Parameters. J. Clin. Sleep Med. 2018, 14, 1109–1118. [Google Scholar] [CrossRef]
- Rochlani, Y.; Pothineni, N.V.; Kovelamudi, S.; Mehta, J.L. Metabolic Syndrome: Pathophysiology, Management, and Modulation by Natural Compounds. Ther. Adv. Cardiovasc. Dis. 2017, 11, 215–225. [Google Scholar] [CrossRef]
- Fahed, G.; Aoun, L.; Bou Zerdan, M.; Allam, S.; Bou Zerdan, M.; Bouferraa, Y.; Assi, H.I. Metabolic Syndrome: Updates on Pathophysiology and Management in 2021. Int. J. Mol. Sci. 2022, 23, 786. [Google Scholar] [CrossRef]
- Lam, J.C.M.; Mak, J.C.W.; Ip, M.S.M. Obesity, Obstructive Sleep Apnoea and Metabolic Syndrome. Respirology 2012, 17, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Jha, B.K.; Sherpa, M.L.; Imran, M.; Mohammed, Y.; Jha, L.A.; Paudel, K.R.; Jha, S.K. Progress in Understanding Metabolic Syndrome and Knowledge of Its Complex Pathophysiology. Diabetology 2023, 4, 134–159. [Google Scholar] [CrossRef]
- Wilcox, I.; McNamara, S.G.; Collins, F.L.; Grunstein, R.R.; Sullivan, C.E. “Syndrome Z”: The Interaction of Sleep Apnoea, Vascular Risk Factors and Heart Disease. Thorax 1998, 53 (Suppl. S3), S25–S28. [Google Scholar] [PubMed]
- Xu, S.; Wan, Y.; Xu, M.; Ming, J.; Xing, Y.; An, F.; Ji, Q. The Association between Obstructive Sleep Apnea and Metabolic Syndrome: A Systematic Review and Meta-Analysis. BMC Pulm. Med. 2015, 15, 105. [Google Scholar] [CrossRef]
- Amini, M.; Zayeri, F.; Salehi, M. Trend Analysis of Cardiovascular Disease Mortality, Incidence, and Mortality-to-Incidence Ratio: Results from Global Burden of Disease Study 2017. BMC Public Health 2021, 21, 401. [Google Scholar] [CrossRef]
- Purnell, J.Q. Definitions, Classification, and Epidemiology of Obesity; MDText.com, Inc.: South Dartmouth, MA, USA, 2023. [Google Scholar]
- Lopez, P.P.; Stefan, B.; Schulman, C.I.; Byers, P.M. Prevalence of Sleep Ap nea in Morbidly Obese Patients Who Presented for Weight Loss Surgery Evaluation: More Evidence for Routine Screening for Obstructive Sleep Apnea before Weight Loss Surgery. Am. Surg. 2008, 74, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Garvey, J.F.; Pengo, M.F.; Drakatos, P.; Kent, B.D. Epidemiological Aspects of Obstructive Sleep Apnea. J. Thorac. Dis. 2015, 7, 920–929. [Google Scholar] [PubMed]
- Tom, C.; Roy, B.; Vig, R.; Kang, D.W.; Aysola, R.S.; Woo, M.A.; Harper, R.M.; Kumar, R. Correlations between Waist and Neck Circumferences and Obstructive Sleep Apnea Characteristics. Sleep Vigil. 2018, 2, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Sekizuka, H.; Ono, Y.; Saitoh, T.; Ono, Y. Visceral Fat Area by Abdominal Bioelectrical Impedance Analysis as a Risk of Obstructive Sleep Apnea. Int. Heart J. 2021, 62, 1091–1095. [Google Scholar] [CrossRef] [PubMed]
- Cizza, G.; de Jonge, L.; Piaggi, P.; Mattingly, M.; Zhao, X.; Lucassen, E.; Rother, K.I.; Sumner, A.E.; Csako, G.; NIDDK Sleep Extension Study. Neck Circumference Is a Predictor of Metabolic Syndrome and Obstructive Sleep Apnea in Short-Sleeping Obese Men and Women. Metab. Syndr. Relat. Disord. 2014, 12, 231–241. [Google Scholar] [CrossRef]
- Gaines, J.; Vgontzas, A.N.; Fernandez-Mendoza, J.; Bixler, E.O. Obstructive Sleep Apnea and the Metabolic Syndrome: The Road to Clinically-Meaningful Phenotyping, Improved Prognosis, and Personalized Treatment. Sleep Med. Rev. 2018, 42, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Framnes, S.N.; Arble, D.M. The Bidirectional Relationship between Obstructive Sleep Apnea and Metabolic Disease. Front. Endocrinol. 2018, 9, 440. [Google Scholar] [CrossRef]
- Isono, S. Obesity and Obstructive Sleep Apnoea: Mechanisms for Increased Collapsibility of the Passive Pharyngeal Airway. Respirology 2012, 17, 32–42. [Google Scholar] [CrossRef]
- Genta, P.R.; Schorr, F.; Edwards, B.A.; Wellman, A.; Lorenzi-Filho, G. Discriminating the Severity of Pharyngeal Collapsibility in Men Using Anthropometric and Polysomnographic Indices. J. Clin. Sleep Med. 2020, 16, 1531–1537. [Google Scholar] [CrossRef]
- Genta, P.R.; Schorr, F.; Eckert, D.J.; Gebrim, E.; Kayamori, F.; Moriya, H.T.; Malhotra, A.; Lorenzi-Filho, G. Upper Airway Collapsibility Is Associated with Obesity and Hyoid Position. Sleep 2014, 37, 1673–1678. [Google Scholar] [CrossRef]
- Kazemeini, E.; Van de Perck, E.; Dieltjens, M.; Willemen, M.; Verbraecken, J.; Op de Beeck, S.; Vanderveken, O.M. Critical to Know Pcrit: A Review on Pharyngeal Critical Closing Pressure in Obstructive Sleep Apnea. Front. Neurol. 2022, 13, 775709. [Google Scholar] [CrossRef]
- Martinovic, D.; Tokic, D.; Puizina-Mladinic, E.; Kadic, S.; Lesin, A.; Lupi-Ferandin, S.; Kumric, M.; Bozic, J. Oromaxillofacial Surgery: Both a Treatment and a Possible Cause of Obstructive Sleep Apnea—A Narrative Review. Life 2023, 13, 142. [Google Scholar] [CrossRef]
- Dixon, A.E.; Peters, U. The Effect of Obesity on Lung Function. Expert Rev. Respir. Med. 2018, 12, 755–767. [Google Scholar] [CrossRef]
- Pugliese, G.; Barrea, L.; Laudisio, D.; Salzano, C.; Aprano, S.; Colao, A.; Savastano, S.; Muscogiuri, G. Sleep Apnea, Obesity, and Disturbed Glucose Homeostasis: Epidemiologic Evidence, Biologic Insights, and Therapeutic Strategies. Curr. Obes. Rep. 2020, 9, 30–38. [Google Scholar] [CrossRef]
- Phillips, C.L.; Hoyos, C.M.; Yee, B.J.; Grunstein, R.R. CrossTalk Opposing View: Sleep Apnoea Causes Metabolic Syndrome. J. Physiol. 2016, 594, 4691–4694. [Google Scholar] [CrossRef]
- Shechter, A. Obstructive Sleep Apnea and Energy Balance Regulation: A Systematic Review. Sleep Med. Rev. 2017, 34, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Kline, C.E.; Burke, L.E.; Sereika, S.M.; Imes, C.C.; Rockette-Wagner, B.; Mendez, D.D.; Strollo, P.J.; Zheng, Y.; Rathbun, S.L.; Chasens, E.R. Bidirectional Relationships Between Weight Change and Sleep Apnea in a Behavioral Weight Loss Intervention. Mayo Clin. Proc. 2018, 93, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Tachikawa, R.; Ikeda, K.; Minami, T.; Matsumoto, T.; Hamada, S.; Murase, K.; Tanizawa, K.; Inouchi, M.; Oga, T.; Akamizu, T.; et al. Changes in Energy Metabolism after Continuous Positive Airway Pressure for Obstructive Sleep Apnea. Am. J. Respir. Crit. Care Med. 2016, 194, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Karlsen, T.; Engstrøm, M.; Steinshamn, S.L. Exercise and Obstructive Sleep Apnoea: A 24-Week Follow-up Study. BMJ Open Sport Exerc. Med. 2022, 8, e001366. [Google Scholar] [CrossRef]
- Pardak, P.; Filip, R.; Woliński, J. The Impact of Sleep-Disordered Breathing on Ghrelin, Obestatin, and Leptin Profiles in Patients with Obesity or Overweight. J. Clin. Med. Res. 2022, 11, 2032. [Google Scholar] [CrossRef] [PubMed]
- Demos, K.E.; Sweet, L.H.; Hart, C.N.; McCaffery, J.M.; Williams, S.E.; Mailloux, K.A.; Trautvetter, J.; Owens, M.M.; Wing, R.R. The Effects of Experimental Manipulation of Sleep Duration on Neural Response to Food Cues. Sleep 2017, 40, zsx125. [Google Scholar] [CrossRef] [PubMed]
- St-Onge, M.-P.; Wolfe, S.; Sy, M.; Shechter, A.; Hirsch, J. Sleep Restriction Increases the Neuronal Response to Unhealthy Food in Normal-Weight Individuals. Int. J. Obes. 2014, 38, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Fattal, D.; Hester, S.; Wendt, L. Body Weight and Obstructive Sleep Apnea: A Mathematical Relationship between Body Mass Index and Apnea-Hypopnea Index in Veterans. J. Clin. Sleep Med. 2022, 18, 2723–2729. [Google Scholar] [CrossRef] [PubMed]
- Reutrakul, S.; Mokhlesi, B. Obstructive Sleep Apnea and Diabetes: A State of the Art Review. Chest 2017, 152, 1070–1086. [Google Scholar] [CrossRef] [PubMed]
- Jehan, S.; Myers, A.K.; Zizi, F.; Pandi-Perumal, S.R.; Jean-Louis, G.; McFarlane, S.I. Obesity, Obstructive Sleep Apnea and Type 2 Diabetes Mellitus: Epidemiology and Pathophysiologic Insights. Sleep Med. Disord. 2018, 2, 52–58. [Google Scholar]
- Sajkov, D.; Mupunga, B.; Bowden, J.J.; Langton, C.; Petrovsky, N. Narrative Review: Obesity, Type 2 DM and Obstructive Sleep Apnoea—Common Bedfellows. Diabetol. Metab. Syndr. 2022, 3, 447–459. [Google Scholar] [CrossRef]
- Tan, H.L.; Babwah, F.; Waheed, N.; Butt, M.I. Obstructive Sleep Apnoea and Type 1 Diabetes Mellitus. Br. J. Diabetes 2015, 15, 96–98. [Google Scholar] [CrossRef]
- Gutiérrez-Carrasquilla, L.; López-Cano, C.; Sánchez, E.; Barbé, F.; Dalmases, M.; Hernández, M.; Campos, A.; Gaeta, A.M.; Carmona, P.; Hernández, C.; et al. Effect of Glucose Improvement on Nocturnal Sleep Breathing Parameters in Patients with Type 2 Diabetes: The Candy Dreams Study. J. Clin. Med. Res. 2020, 9, 1022. [Google Scholar] [CrossRef]
- Lecube, A.; Ciudin, A.; Sampol, G.; Valladares, S.; Hernández, C.; Simó, R. Effect of Glycemic Control on Nocturnal Arterial Oxygen Saturation: A Case-Control Study in Type 2 Diabetic Patients. J. Diabetes 2015, 7, 133–138. [Google Scholar] [CrossRef]
- Feldman, E.L.; Callaghan, B.C.; Pop-Busui, R.; Zochodne, D.W.; Wright, D.E.; Bennett, D.L.; Bril, V.; Russell, J.W.; Viswanathan, V. Diabetic Neuropathy. Nat. Rev. Dis. Primers 2019, 5, 41. [Google Scholar] [CrossRef]
- Keller, T.; Hader, C.; De Zeeuw, J.; Rasche, K. Obstructive Sleep Apnea Syndrome: The Effect of Diabetes and Autonomic Neuropathy. J. Physiol. Pharmacol. 2007, 58 (Suppl. S5), 313–318. [Google Scholar]
- Ficker, J.H.; Dertinger, S.H.; Siegfried, W.; König, H.J.; Pentz, M.; Sailer, D.; Katalinic, A.; Hahn, E.G. Obstructive Sleep Apnoea and Diabetes Mellitus: The Role of Cardiovascular Autonomic Neuropathy. Eur. Respir. J. 1998, 11, 14–19. [Google Scholar] [CrossRef]
- Bottini, P.; Redolfi, S.; Dottorini, M.L.; Tantucci, C. Autonomic Neuropathy Increases the Risk of Obstructive Sleep Apnea in Obese Diabetics. Respiration 2008, 75, 265–271. [Google Scholar] [CrossRef]
- Aurora, R.N.; Punjabi, N.M. Obstructive Sleep Apnoea and Type 2 Diabetes Mellitus: A Bidirectional Association. Lancet Respir. Med. 2013, 1, 329–338. [Google Scholar] [CrossRef]
- Neumann, C.; Martinez, D.; Schmid, H. Nocturnal Oxygen Desaturation in Diabetic Patients with Severe Autonomic Neuropathy. Diabetes Res. Clin. Pract. 1995, 28, 97–102. [Google Scholar] [CrossRef]
- Rasche, K.; Keller, T.; Tautz, B.; Hader, C.; Hergenc, G.; Antosiewicz, J.; Di Giulio, C.; Pokorski, M. Obstructive Sleep Apnea and Type 2 Diabetes. Eur. J. Med. Res. 2010, 15 (Suppl. S2), 152–156. [Google Scholar] [CrossRef] [PubMed]
- Badoer, E. The Carotid Body a Common Denominator for Cardiovascular and Metabolic Dysfunction? Front. Physiol. 2020, 11, 1069. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.J.; Polotsky, V.Y. Carotid Body and Metabolic Syndrome: Mechanisms and Potential Therapeutic Targets. Int. J. Mol. Sci. 2020, 21, 5117. [Google Scholar] [CrossRef] [PubMed]
- Ward, D.S.; Voter, W.A.; Karan, S. The Effects of Hypo- and Hyperglycaemia on the Hypoxic Ventilatory Response in Humans. J. Physiol. 2007, 582, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Bin-Jaliah, I.; Maskell, P.D.; Kumar, P. Indirect Sensing of Insulin-Induced Hypoglycaemia by the Carotid Body in the Rat. J. Physiol. 2004, 556, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Bin-Jaliah, I.; Maskell, P.D.; Kumar, P. Carbon Dioxide Sensitivity during Hypoglycaemia-Induced, Elevated Metabolism in the Anaesthetized Rat. J. Physiol. 2005, 563, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Haag, R.; Mayer, T.; Wehrwein, E. Hyperglycemia Blunts Hypoxic Ventilatory Response. Auton. Neurosci. 2015, 192, 68. [Google Scholar] [CrossRef]
- Subramanian, A.; Adderley, N.J.; Tracy, A.; Taverner, T.; Hanif, W.; Toulis, K.A.; Thomas, G.N.; Tahrani, A.A.; Nirantharakumar, K. Risk of Incident Obstructive Sleep Apnea among Patients with Type 2 Diabetes. Diabetes Care 2019, 42, 954–963. [Google Scholar] [CrossRef]
- Liu, C.-L.; Wu, C.-S. Assessing Whether the Association Between Sleep Apnea and Diabetes Is Bidirectional. Can. J. Diabetes 2017, 41, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Punjabi, N.M.; Ahmed, M.M.; Polotsky, V.Y.; Beamer, B.A.; O’Donnell, C.P. Sleep-Disordered Breathing, Glucose Intolerance, and Insulin Resistance. Respir. Physiol. Neurobiol. 2003, 136, 167–178. [Google Scholar] [CrossRef]
- Almendros, I.; García-Río, F. Sleep Apnoea, Insulin Resistance and Diabetes: The First Step Is in the Fat. Eur. Respir. J. 2017, 49, 1700179. [Google Scholar] [CrossRef]
- Pack, A.I. Gut Microbiome: Role in Insulin Resistance in Obstructive Sleep Apnea. EBioMedicine 2021, 65, 103278. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Kim, C.-W.; Lee, K.-C.; Lee, J.-H.; Kang, S.-H.; Li, S.-W.; Kim, K.; Hong, S.-J. Effect of Intermittent Hypoxia on Metabolic Syndrome and Insulin Resistance in the General Male Population. Medicina 2021, 57, 668. [Google Scholar] [CrossRef]
- Briançon-Marjollet, A.; Netchitailo, M.; Fabre, F.; Belaidi, E.; Arnaud, C.; Borel, A.-L.; Levy, P.; Pépin, J.-L.; Tamisier, R. Intermittent Hypoxia Exposure Increases Adipose Insulin Resistance and Lipolysis in Healthy Humans: A Randomized Cross over Trial. Eur. Respir. J. 2023, 62, OA4337. [Google Scholar] [CrossRef]
- Green, M.; Ken-Dror, G.; Fluck, D.; Sada, C.; Sharma, P.; Fry, C.H.; Han, T.S. Meta-Analysis of Changes in the Levels of Catecholamines and Blood Pressure with Continuous Positive Airway Pressure Therapy in Obstructive Sleep Apnea. J. Clin. Hypertens. 2021, 23, 12–20. [Google Scholar] [CrossRef]
- Hoeldtke, R. Primer on the Autonomic Nervous System, 2nd ed.; Robertson, D., Biaggioni, I., Burnstock, G., Low, P.A., Eds.; Academic Press: Cambridge, MA, USA, 2004; ISBN 9780125897624. [Google Scholar]
- Bolli, G.B. Importance of Catecholamines in Defense against Insulin Hypoglycemia in Humans. In Advances in Pharmacology; Goldstein, D.S., Eisenhofer, G., McCarty, R., Eds.; Academic Press: Cambridge, MA, USA, 1997; Volume 42, pp. 627–630. [Google Scholar]
- Ghigo, E.; Porta, M. Diabetes Secondary to Endocrine and Pancreatic Disorders. Front. Diabetes 2014, 22, 44–51. [Google Scholar] [CrossRef]
- Betteridge, D.J. What Is Oxidative Stress? Metabolism 2000, 49, 3–8. [Google Scholar] [CrossRef]
- Peres, B.U.; Allen, A.J.H.; Shah, A.; Fox, N.; Laher, I.; Almeida, F.; Jen, R.; Ayas, N. Obstructive Sleep Apnea and Circulating Biomarkers of Oxidative Stress: A Cross-Sectional Study. Antioxidants 2020, 9, 476. [Google Scholar] [CrossRef]
- Lavie, L. Oxidative Stress in Obstructive Sleep Apnea and Intermittent Hypoxia–Revisited—The Bad Ugly and Good: Implications to the Heart and Brain. Sleep Med. Rev. 2015, 20, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Sathyapalan, T.; Atkin, S.L.; Sahebkar, A. Molecular Mechanisms Linking Oxidative Stress and Diabetes Mellitus. Oxidative Med. Cell. Longev. 2020, 2020, 8609213. [Google Scholar] [CrossRef] [PubMed]
- Kritikou, I.; Basta, M.; Vgontzas, A.N.; Pejovic, S.; Fernandez-Mendoza, J.; Liao, D.; Bixler, E.O.; Gaines, J.; Chrousos, G.P. Sleep Apnoea and the Hypothalamic-Pituitary-Adrenal Axis in Men and Women: Effects of Continuous Positive Airway Pressure. Eur. Respir. J. 2016, 47, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.; McQueen, A.; Chen, T.-C.; Wang, J.-C. Regulation of Glucose Homeostasis by Glucocorticoids. Adv. Exp. Med. Biol. 2015, 872, 99–126. [Google Scholar] [PubMed]
- Vgontzas, A.N.; Pejovic, S.; Zoumakis, E.; Lin, H.-M.; Bentley, C.M.; Bixler, E.O.; Sarrigiannidis, A.; Basta, M.; Chrousos, G.P. Hypothalamic-Pituitary-Adrenal Axis Activity in Obese Men with and without Sleep Apnea: Effects of Continuous Positive Airway Pressure Therapy. J. Clin. Endocrinol. Metab. 2007, 92, 4199–4207. [Google Scholar] [CrossRef] [PubMed]
- Dadoun, F.; Darmon, P.; Achard, V.; Boullu-Ciocca, S.; Philip-Joet, F.; Alessi, M.C.; Rey, M.; Grino, M.; Dutour, A. Effect of Sleep Apnea Syndrome on the Circadian Profile of Cortisol in Obese Men. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E466–E474. [Google Scholar] [CrossRef]
- Carneiro, G.; Togeiro, S.M.; Hayashi, L.F.; Ribeiro-Filho, F.F.; Ribeiro, A.B.; Tufik, S.; Zanella, M.T. Effect of Continuous Positive Airway Pressure Therapy on Hypothalamic-Pituitary-Adrenal Axis Function and 24-H Blood Pressure Profile in Obese Men with Obstructive Sleep Apnea Syndrome. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E380–E384. [Google Scholar] [CrossRef]
- Aguirre, G.A.; De Ita, J.R.; de la Garza, R.G.; Castilla-Cortazar, I. Insulin-like Growth Factor-1 Deficiency and Metabolic Syndrome. J. Transl. Med. 2016, 14, 3. [Google Scholar] [CrossRef]
- Kubo, H.; Sawada, S.; Satoh, M.; Asai, Y.; Kodama, S.; Sato, T.; Tomiyama, S.; Seike, J.; Takahashi, K.; Kaneko, K.; et al. Insulin-like Growth Factor-1 Levels Are Associated with High Comorbidity of Metabolic Disorders in Obese Subjects; a Japanese Single-Center, Retrospective-Study. Sci. Rep. 2022, 12, 20130. [Google Scholar] [CrossRef] [PubMed]
- Izumi, S.; Ribeiro-Filho, F.F.; Carneiro, G.; Togeiro, S.M.; Tufik, S.; Zanella, M.T. IGF-1 Levels Are Inversely Associated with Metabolic Syndrome in Obstructive Sleep Apnea. J. Clin. Sleep Med. 2016, 12, 487–493. [Google Scholar] [CrossRef]
- He, J.; Li, X.; Yu, M. The Correlation of Serum/plasma IGF-1 Concentrations with Obstructive Sleep Apnea Hypopnea Syndrome: A Meta-Analysis and Meta-Regression. Front. Endocrinol. 2022, 13, 922229. [Google Scholar] [CrossRef]
- Oh, J.; Kim, J.-Y.; Park, S.; Youn, J.-C.; Son, N.H.; Shin, D.-J.; Lee, S.-H.; Kang, S.-M.; Jee, S.H.; Jang, Y. The Relationship between Insulin-like Growth Factor-1 and Metabolic Syndrome, Independent of Adiponectin. Clin. Chim. Acta 2012, 413, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Bozic, J.; Galic, T.; Supe-Domic, D.; Ivkovic, N.; Ticinovic Kurir, T.; Valic, Z.; Lesko, J.; Dogas, Z. Morning Cortisol Levels and Glucose Metabolism Parameters in Moderate and Severe Obstructive Sleep Apnea Patients. Endocrine 2016, 53, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Loffler, K.A.; Heeley, E.; Freed, R.; Meng, R.; Bittencourt, L.R.; Gonzaga Carvalho, C.C.; Chen, R.; Hlavac, M.; Liu, Z.; Lorenzi-Filho, G.; et al. Continuous Positive Airway Pressure Treatment, Glycemia, and Diabetes Risk in Obstructive Sleep Apnea and Comorbid Cardiovascular Disease. Diabetes Care 2020, 43, 1859–1867. [Google Scholar] [CrossRef]
- Mokhlesi, B.; Tjaden, A.H.; Temple, K.A.; Edelstein, S.L.; Sam, S.; Nadeau, K.J.; Hannon, T.S.; Manchanda, S.; Mather, K.J.; Kahn, S.E.; et al. Obstructive Sleep Apnea, Glucose Tolerance, and β-Cell Function in Adults with Prediabetes or Untreated Type 2 Diabetes in the Restoring Insulin Secretion (RISE) Study. Diabetes Care 2021, 44, 993–1001. [Google Scholar] [CrossRef]
- Temple, K.A.; Tjaden, A.H.; Atkinson, K.M.; Barengolts, E.; Hannon, T.S.; Mather, K.J.; Utzschneider, K.M.; Edelstein, S.L.; Ehrmann, D.A.; Mokhlesi, B.; et al. Association of Habitual Daily Physical Activity with Glucose Tolerance and β-Cell Function in Adults with Impaired Glucose Tolerance or Recently Diagnosed Type 2 Diabetes from the Restoring Insulin Secretion (RISE) Study. Diabetes Care 2019, 42, 1521–1529. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, J.; Moreau, J.M.; Caverson, M.M.; Moranis, R. Leptin: A Potential Link Between Obstructive Sleep Apnea and Obesity. Front. Physiol. 2021, 12, 767318. [Google Scholar] [CrossRef]
- Meszaros, M.; Bikov, A. Obstructive Sleep Apnoea and Lipid Metabolism: The Summary of Evidence and Future Perspectives in the Pathophysiology of OSA-Associated Dyslipidaemia. Biomedicines 2022, 10, 2754. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.; Polotsky, V.Y. Leptin and Leptin Resistance in the Pathogenesis of Obstructive Sleep Apnea: A Possible Link to Oxidative Stress and Cardiovascular Complications. Oxidative Med. Cell. Longev. 2018, 2018, 5137947. [Google Scholar] [CrossRef] [PubMed]
- Harsch, I.A.; Konturek, P.C.; Koebnick, C.; Kuehnlein, P.P.; Fuchs, F.S.; Pour Schahin, S.; Wiest, G.H.; Hahn, E.G.; Lohmann, T.; Ficker, J.H. Leptin and Ghrelin Levels in Patients with Obstructive Sleep Apnoea: Effect of CPAP Treatment. Eur. Respir. J. 2003, 22, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Imayama, I.; Prasad, B. Role of Leptin in Obstructive Sleep Apnea. Ann. Am. Thorac. Soc. 2017, 14, 1607–1621. [Google Scholar] [CrossRef] [PubMed]
- Patial, K.; Mishra, H.P.; Pal, G.; Suvvari, T.K.; Mahapatra, C.; Amanullah, N.A.; Singh, I.; Gaur, S.N.; Behera, R.K. Assessment of Leptin Levels and Their Correlation with the Severity of Obstructive Sleep Apnea Syndrome: A Case-Control Study. Cureus 2023, 15, e42028. [Google Scholar] [CrossRef]
- Li, X.; He, J. The Association between Serum/Plasma Leptin Levels and Obstructive Sleep Apnea Syndrome: A Meta-Analysis and Meta-Regression. Front. Endocrinol. 2021, 12, 696418. [Google Scholar] [CrossRef]
- Gharraf, H.S.; AlHadidy, A.; AlNehr, I. Study of Leptin and Ghrelin Serum Levels in Patients with Obstructive Sleep Apnea. Egypt. J. Chest Dis. Tuberc. 2019, 68, 567. [Google Scholar] [CrossRef]
- Amorim, M.R.; Aung, O.; Mokhlesi, B.; Polotsky, V.Y. Leptin-Mediated Neural Targets in Obesity Hypoventilation Syndrome. Sleep 2022, 45, zsac153. [Google Scholar] [CrossRef]
- Ghadge, A.A.; Khaire, A.A. Leptin as a Predictive Marker for Metabolic Syndrome. Cytokine 2019, 121, 154735. [Google Scholar] [CrossRef] [PubMed]
- Balcan, B.; Thunström, E.; Yucel-Lindberg, T.; Lindberg, K.; Ay, P.; Peker, Y. Impact of CPAP Treatment on Leptin and Adiponectin in Adults with Coronary Artery Disease and Nonsleepy Obstructive Sleep Apnoea in the RICCADSA Trial. Sleep Med. 2020, 67, 7–14. [Google Scholar] [CrossRef]
- Chin, C.-H.; Lin, P.-W.; Lin, H.-C.; Friedman, M.; Lin, M.-C. Effects of OSA Surgery on Leptin and Metabolic Profiles. Otolaryngol.–Head Neck Surg. 2019, 161, 1048–1055. [Google Scholar] [CrossRef]
- Emara, T.A.; Khazbak, A.O.; Mohammed, O.; Elgaml, M.; Zidan, A.; Hosny, S.M. Changes in Serum Leptin Level after Multilevel Surgery in Patients with Obstructive Sleep Apnea. Laryngoscope 2021, 131, E665–E670. [Google Scholar] [CrossRef]
- Tietjens, J.R.; Claman, D.; Kezirian, E.J.; De Marco, T.; Mirzayan, A.; Sadroonri, B.; Goldberg, A.N.; Long, C.; Gerstenfeld, E.P.; Yeghiazarians, Y. Obstructive Sleep Apnea in Cardiovascular Disease: A Review of the Literature and Proposed Multidisciplinary Clinical Management Strategy. J. Am. Heart Assoc. 2019, 8, e010440. [Google Scholar] [CrossRef]
- Lombardi, C.; Pengo, M.F.; Parati, G. Systemic Hypertension in Obstructive Sleep Apnea. J. Thorac. Dis. 2018, 10, S4231–S4243. [Google Scholar] [CrossRef]
- Cai, A.; Wang, L.; Zhou, Y. Hypertension and Obstructive Sleep Apnea. Hypertens. Res. 2016, 39, 391–395. [Google Scholar] [CrossRef]
- Pedrosa, R.P.; Drager, L.F.; Gonzaga, C.C.; Sousa, M.G.; de Paula, L.K.G.; Amaro, A.C.S.; Amodeo, C.; Bortolotto, L.A.; Krieger, E.M.; Bradley, T.D.; et al. Obstructive Sleep Apnea: The Most Common Secondary Cause of Hypertension Associated with Resistant Hypertension. Hypertension 2011, 58, 811–817. [Google Scholar] [CrossRef]
- Tamisier, R.; Lévy, P. Management of Hypertension in Obstructive Sleep Apnoea: Predicting Blood Pressure Reduction under Continuous Positive Airway Pressure. Eur. Respir. J. 2017, 50, 1701822. [Google Scholar] [CrossRef]
- O’Connor, G.T.; Caffo, B.; Newman, A.B.; Quan, S.F.; Rapoport, D.M.; Redline, S.; Resnick, H.E.; Samet, J.; Shahar, E. Prospective Study of Sleep-Disordered Breathing and Hypertension: The Sleep Heart Health Study. Am. J. Respir. Crit. Care Med. 2009, 179, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Konecny, T.; Kara, T.; Somers, V.K. Obstructive Sleep Apnea and Hypertension: An Update. Hypertension 2014, 63, 203–209. [Google Scholar] [CrossRef]
- Peppard, P.E.; Young, T.; Palta, M.; Skatrud, J. Prospective Study of the Association between Sleep-Disordered Breathing and Hypertension. N. Engl. J. Med. 2000, 342, 1378–1384. [Google Scholar] [CrossRef]
- Hou, H.; Zhao, Y.; Yu, W.; Dong, H.; Xue, X.; Ding, J.; Xing, W.; Wang, W. Association of Obstructive Sleep Apnea with Hypertension: A Systematic Review and Meta-Analysis. J. Glob. Health 2018, 8, 010405. [Google Scholar] [CrossRef]
- Simons, S.; Hovens, M. Sleep-Disordered Breathing and Hypertension. Am. J. Respir. Crit. Care Med. 2009, 180, 582. [Google Scholar] [CrossRef]
- Haas, D.C.; Foster, G.L.; Nieto, F.J.; Redline, S.; Resnick, H.E.; Robbins, J.A.; Young, T.; Pickering, T.G. Age-Dependent Associations between Sleep-Disordered Breathing and Hypertension: Importance of Discriminating between Systolic/Diastolic Hypertension and Isolated Systolic Hypertension in the Sleep Heart Health Study. Circulation 2005, 111, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Mihovilovic, A.; Dogas, Z.; Martinovic, D.; Tokic, D.; Puizina Mladinic, E.; Kumric, M.; Ivkovic, N.; Vilovic, M.; Bozic, J. Serum Urotensin II Levels Are Elevated in Patients with Obstructive Sleep Apnea. Biomolecules 2023, 13, 914. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M.; McNicholas, W.T. Bidirectional Relationships of Comorbidity with Obstructive Sleep Apnoea. Eur. Respir. Rev. 2022, 31, 210256. [Google Scholar] [CrossRef]
- Phillips, C.L.; O’Driscoll, D.M. Hypertension and Obstructive Sleep Apnea. Nat. Sci. Sleep 2013, 5, 43–52. [Google Scholar] [CrossRef]
- Brown, J.; Yazdi, F.; Jodari-Karimi, M.; Owen, J.G.; Reisin, E. Obstructive Sleep Apnea and Hypertension: Updates to a Critical Relationship. Curr. Hypertens. Rep. 2022, 24, 173–184. [Google Scholar] [CrossRef]
- Salman, L.A.; Shulman, R.; Cohen, J.B. Obstructive Sleep Apnea, Hypertension, and Cardiovascular Risk: Epidemiology, Pathophysiology, and Management. Curr. Cardiol. Rep. 2020, 22, 6. [Google Scholar] [CrossRef]
- Altay, S.; Fırat, S.; Peker, Y.; The TURCOSACT Collaborators. A Narrative Review of the Association of Obstructive Sleep Apnea with Hypertension: How to Treat Both When They Coexist? J. Clin. Med. Res. 2023, 12, 4144. [Google Scholar] [CrossRef] [PubMed]
- Krasińska, B.; Miazga, A.; Cofta, S.; Szczepaniak-Chicheł, L.; Trafas, T.; Krasiński, Z.; Pawlaczyk-Gabriel, K.; Tykarski, A. Effect of Eplerenone on the Severity of Obstructive Sleep Apnea and Arterial Stiffness in Patients with Resistant Arterial Hypertension. Pol. Arch. Med. Wewn. 2016, 126, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, H.; Cai, M.; Zou, Y.; Jiang, X.; Song, L.; Liang, E.; Bian, J.; Wu, H.; Hui, R. Effect of Spironolactone on Patients with Resistant Hypertension and Obstructive Sleep Apnea. Clin. Exp. Hypertens. 2016, 38, 464–468. [Google Scholar] [CrossRef]
- Gaddam, K.; Pimenta, E.; Thomas, S.J.; Cofield, S.S.; Oparil, S.; Harding, S.M.; Calhoun, D.A. Spironolactone Reduces Severity of Obstructive Sleep Apnoea in Patients with Resistant Hypertension: A Preliminary Report. J. Hum. Hypertens. 2010, 24, 532–537. [Google Scholar] [CrossRef]
- Garvey, J.F.; Taylor, C.T.; McNicholas, W.T. Cardiovascular Disease in Obstructive Sleep Apnoea Syndrome: The Role of Intermittent Hypoxia and Inflammation. Eur. Respir. J. 2009, 33, 1195–1205. [Google Scholar] [CrossRef]
- Ayyaswamy, S.; Shi, H.; Zhang, B.; Bryan, R.M., Jr.; Durgan, D.J. Obstructive Sleep Apnea-Induced Hypertension Is Associated with Increased Gut and Neuroinflammation. J. Am. Heart Assoc. 2023, 12, e029218. [Google Scholar] [CrossRef]
- Munir, S.S.; Sert Kuniyoshi, F.H.; Singh, P.; Covassin, N. Is the Gut Microbiome Implicated in the Excess Risk of Hypertension Associated with Obstructive Sleep Apnea? A Contemporary Review. Antioxidants 2023, 12, 866. [Google Scholar] [CrossRef]
- Walters, W.A.; Xu, Z.; Knight, R. Meta-Analyses of Human Gut Microbes Associated with Obesity and IBD. FEBS Lett. 2014, 588, 4223–4233. [Google Scholar] [CrossRef]
- Puljiz, Z.; Kumric, M.; Vrdoljak, J.; Martinovic, D.; Ticinovic Kurir, T.; Krnic, M.O.; Urlic, H.; Puljiz, Z.; Zucko, J.; Dumanic, P.; et al. Obesity, Gut Microbiota, and Metabolome: From Pathophysiology to Nutritional Interventions. Nutrients 2023, 15, 2236. [Google Scholar] [CrossRef]
- Nearing, J.T.; Comeau, A.M.; Langille, M.G.I. Identifying Biases and Their Potential Solutions in Human Microbiome Studies. Microbiome 2021, 9, 113. [Google Scholar] [CrossRef]
- Carneiro-Barrera, A.; Amaro-Gahete, F.J.; Guillén-Riquelme, A.; Jurado-Fasoli, L.; Sáez-Roca, G.; Martín-Carrasco, C.; Buela-Casal, G.; Ruiz, J.R. Effect of an Interdisciplinary Weight Loss and Lifestyle Intervention on Obstructive Sleep Apnea Severity: The INTERAPNEA Randomized Clinical Trial. JAMA Netw. Open 2022, 5, e228212. [Google Scholar] [CrossRef]
- Marc-André, C. A Review of Current Guidelines for the Treatment of Obesity. Am. J. Manag. Care 2022, 28 (Suppl. S15), S288–S296. Available online: https://www.ajmc.com/view/review-of-current-guidelines-for-the-treatment-of-obesity (accessed on 5 March 2024).
- Fernandes, J.F.R.; da Silva Araújo, L.; Kaiser, S.E.; Sanjuliani, A.F.; Klein, M.R.S.T. The effects of moderate energy restriction on apnoea severity and CVD risk factors in obese patients with obstructive sleep apnoea. Br. J. Nutr. 2015, 114, 2022–2031. [Google Scholar] [CrossRef]
- Hall, K.D.; Kahan, S. Maintenance of Lost Weight and Long-Term Management of Obesity. Med. Clin. N. Am. 2018, 102, 183–197. [Google Scholar] [CrossRef]
- Dixon, J.B.; Schachter, L.M.; O’Brien, P.E.; Jones, K.; Grima, M.; Lambert, G.; Brown, W.; Bailey, M.; Naughton, M.T. Surgical vs conventional therapy for weight loss treatment of obstructive sleep apnea: A randomized controlled trial. JAMA 2012, 308, 1142–1149. [Google Scholar] [CrossRef]
- Georgoulis, M.; Yiannakouris, N.; Kechribari, I.; Lamprou, K.; Perraki, E.; Vagiakis, E.; Kontogianni, M.D. Dose-response relationship between weight loss and improvements in obstructive sleep apnea severity after a diet/lifestyle interventions: Secondary analyses of the “MIMOSA” randomized clinical trial. J. Clin. Sleep Med. 2022, 18, 1251–1261. [Google Scholar] [CrossRef]
- Georgoulis, M.; Yiannakouris, N.; Tenta, R.; Fragopoulou, E.; Kechribari, I.; Lamprou, K.; Perraki, E.; Vagiakis, E.; Kontogianni, M.D. A weight-loss Mediterranean diet/lifestyle intervention ameliorates inflammation and oxidative stress in patients with obstructive sleep apnea: Results of the “MIMOSA” randomized clinical trial. Eur. J. Nutr. 2021, 60, 3799–3810. [Google Scholar] [CrossRef]
- Georgoulis, M.; Yiannakouris, N.; Kechribari, I.; Lamprou, K.; Perraki, E.; Vagiakis, E.; Kontogianni, M.D. Sustained improvements in the cardiometabolic profile of patients with obstructive sleep apnea after a weight-loss Mediterranean diet/lifestyle intervention: 12-month follow-up (6 months post-intervention) of the “MIMOSA” randomized clinical trial. Nutr. Metab. Cardiovasc. Dis. 2023, 33, 1019–1028. [Google Scholar] [CrossRef]
- Hamilton, G.S.; Edwards, B.A. The Potential Impact of GLP-1 Agonists on Obstructive Sleep Apnoea. Respirology 2023, 28, 824–825. [Google Scholar] [CrossRef]
- Matsumoto, T.; Harada, N.; Azuma, M.; Chihara, Y.; Murase, K.; Tachikawa, R.; Minami, T.; Hamada, S.; Tanizawa, K.; Inouchi, M.; et al. Plasma Incretin Levels and Dipeptidyl Peptidase-4 Activity in Patients with Obstructive Sleep Apnea. Ann. Am. Thorac. Soc. 2016, 13, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.; Barclay, J.L.; Ott, V.; Oster, H.; Hallschmid, M. Acute Sleep Deprivation Delays the Glucagon-Like Peptide 1 Peak Response to Breakfast in Healthy Men. Nutr. Diabetes 2013, 3, e78. [Google Scholar] [CrossRef] [PubMed]
- Blackman, A.; on behalf of the SCALE Study Group; Foster, G.D.; Zammit, G.; Rosenberg, R.; Aronne, L.; Wadden, T.; Claudius, B.; Jensen, C.B.; Mignot, E. Effect of Liraglutide 3.0 mg in Individuals with Obesity and Moderate or Severe Obstructive Sleep Apnea: The SCALE Sleep Apnea Randomized Clinical Trial. Int. J. Obes. 2016, 40, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Lincoff, A.M.; Brown-Frandsen, K.; Colhoun, H.M.; Deanfield, J.; Emerson, S.S.; Esbjerg, S.; Hardt-Lindberg, S.; Hovingh, G.K.; Kahn, S.E.; Kushner, R.F.; et al. SELECT Trial Investigators. Semaglutide and Cardiovascular Outcomes in Obesity without Diabetes. N. Engl. J. Med. 2023, 389, 2221–2232. [Google Scholar] [CrossRef]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef]
- Holman, R.R.; Bethel, M.A.; Mentz, R.J.; Thompson, V.P.; Lokhnygina, Y.; Buse, J.B.; Chan, J.C.; Choi, J.; Gustavson, S.M.; Iqbal, N.; et al. Effects of Once-Weekly Exenatide on Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 1228–1239. [Google Scholar] [CrossRef]
- Wilding, J.P.; Batterham, R.L.; Davies, M.; Van Gaal, L.F.; Kandler, K.; Konakli, K.; Lingvay, I.; McGowan, B.M.; Oral, T.K.; Rosenstock, J.; et al. Weight Regain and Cardiometabolic Effects after Withdrawal of Semaglutide: The STEP 1 Trial Extension. Diabetes Obes. Metab. 2022, 24, 1553–1564. [Google Scholar] [CrossRef]
- Filippatos, T.D.; Panagiotopoulou, T.V.; Elisaf, M.S. Adverse Effects of GLP-1 Receptor Agonists. Rev. Diabet. Stud. 2014, 11, 202–230. [Google Scholar] [CrossRef]
- Wojeck, B.S.; Inzucchi, S.E.; Neeland, I.J.; Mancuso, J.P.; Frederich, R.; Masiukiewicz, U.; Cater, N.B.; McGuire, D.K.; Cannon, C.P.; Yaggi, H.K. Ertugliflozin and Incident Obstructive Sleep Apnea: An Analysis from the VERTIS CV Trial. Sleep Breath. 2022, 27, 669–672. [Google Scholar] [CrossRef]
- Furukawa, S.; Miyake, T.; Senba, H.; Sakai, T.; Furukawa, E.; Yamamoto, S.; Niiya, T.; Matsuura, B.; Hiasa, Y. The Effectiveness of Dapagliflozin for Sleep-Disordered Breathing among Japanese Patients with Obesity and Type 2 Diabetes Mellitus. Endocr. J. 2018, 65, 953–961. [Google Scholar] [CrossRef]
- Kusunoki, M.; Hisano, F.; Wakazono, N.; Tsutsumi, K.; Oshida, Y.; Miyata, T. Effect of Treatment with Sodium-Glucose Cotransporter 2 Inhibitor on the Initiation of Continuous Positive Airway Pressure Therapy in Type 2 Diabetic Patients with Obstructive Sleep Apnea Syndrome. J. Clin. Med. Res. 2021, 13, 497–501. [Google Scholar] [CrossRef]
- Kasai, T. Fluid Retention and Rostral Fluid Shift in Sleep-Disordered Breathing. Curr. Hypertens. Rev. 2016, 12, 32–42. [Google Scholar] [CrossRef]
- Alzoubaidi, M.; Mokhlesi, B. Obstructive Sleep Apnea during Rapid Eye Movement Sleep: Clinical Relevance and Therapeutic Implications. Curr. Opin. Pulm. Med. 2016, 22, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Brikman, S.; Dori, G. Sodium Glucose Cotransporter2 Inhibitor-Possible Treatment for Patients with Diabetes, Pulmonary Disease and CO2 Retention. Med. Hypotheses 2020, 139, 109631. [Google Scholar] [CrossRef] [PubMed]
- Neeland, I.J.; Eliasson, B.; Kasai, T.; Marx, N.; Zinman, B.; Inzucchi, S.E.; Wanner, C.; Zwiener, I.; Wojeck, B.S.; Yaggi, H.K.; et al. The Impact of Empagliflozin on Obstructive Sleep Apnea and Cardiovascular and Renal Outcomes: An Exploratory Analysis of the EMPA-REG OUTCOME Trial. Diabetes Care 2020, 43, 3007–3301, Erratum in Diabetes Care 2021, 44, e136. [Google Scholar] [CrossRef]
- Lee, Y.-C.; Lu, C.-T.; Chuang, L.-P.; Lee, L.-A.; Fang, T.-J.; Cheng, W.-N.; Li, H.-Y. Pharmacotherapy for obstructive sleep apnea—A systematic review and meta-analysis of randomized controlled trials. Sleep Med. Rev. 2023, 70, 101809. [Google Scholar] [CrossRef]
- Toh, S.T. Pharmacotherapy for obstructive sleep apnea: Reality or Pipe Dream? Sleep Med. Rev. 2023, 70, 101817. [Google Scholar] [CrossRef]
- Bonsignore, M.R.; Saaresranta, T.; Riha, R.L. Sex differences in obstructive sleep apnoea. Eur. Respir. Rev. 2019, 28, 190030. [Google Scholar] [CrossRef]
- Martins, F.O.; Conde, S.V. Gender Differences in the Context of Obstructive Sleep Apnea and Metabolic Diseases. Front. Physiol. 2021, 12, 792633. [Google Scholar] [CrossRef]
- Chaudhary, P.; Goyal, A.; Goel, S.; Kumar, A.; Chaudhary, S.; Keshri, S.K.; Subhedar, R.P. Women with OSA have higher chances of having metabolic syndrome than men: Effect of gender on syndrome Z in cross sectional study. Sleep Med. 2021, 79, 83–87. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heffernan, A.; Duplancic, D.; Kumric, M.; Ticinovic Kurir, T.; Bozic, J. Metabolic Crossroads: Unveiling the Complex Interactions between Obstructive Sleep Apnoea and Metabolic Syndrome. Int. J. Mol. Sci. 2024, 25, 3243. https://doi.org/10.3390/ijms25063243
Heffernan A, Duplancic D, Kumric M, Ticinovic Kurir T, Bozic J. Metabolic Crossroads: Unveiling the Complex Interactions between Obstructive Sleep Apnoea and Metabolic Syndrome. International Journal of Molecular Sciences. 2024; 25(6):3243. https://doi.org/10.3390/ijms25063243
Chicago/Turabian StyleHeffernan, Aisling, Darko Duplancic, Marko Kumric, Tina Ticinovic Kurir, and Josko Bozic. 2024. "Metabolic Crossroads: Unveiling the Complex Interactions between Obstructive Sleep Apnoea and Metabolic Syndrome" International Journal of Molecular Sciences 25, no. 6: 3243. https://doi.org/10.3390/ijms25063243