
Differential Impact of CD43 and CD28 on T-Cell Differentiation Depending on the Order of Engagement with the TCR
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
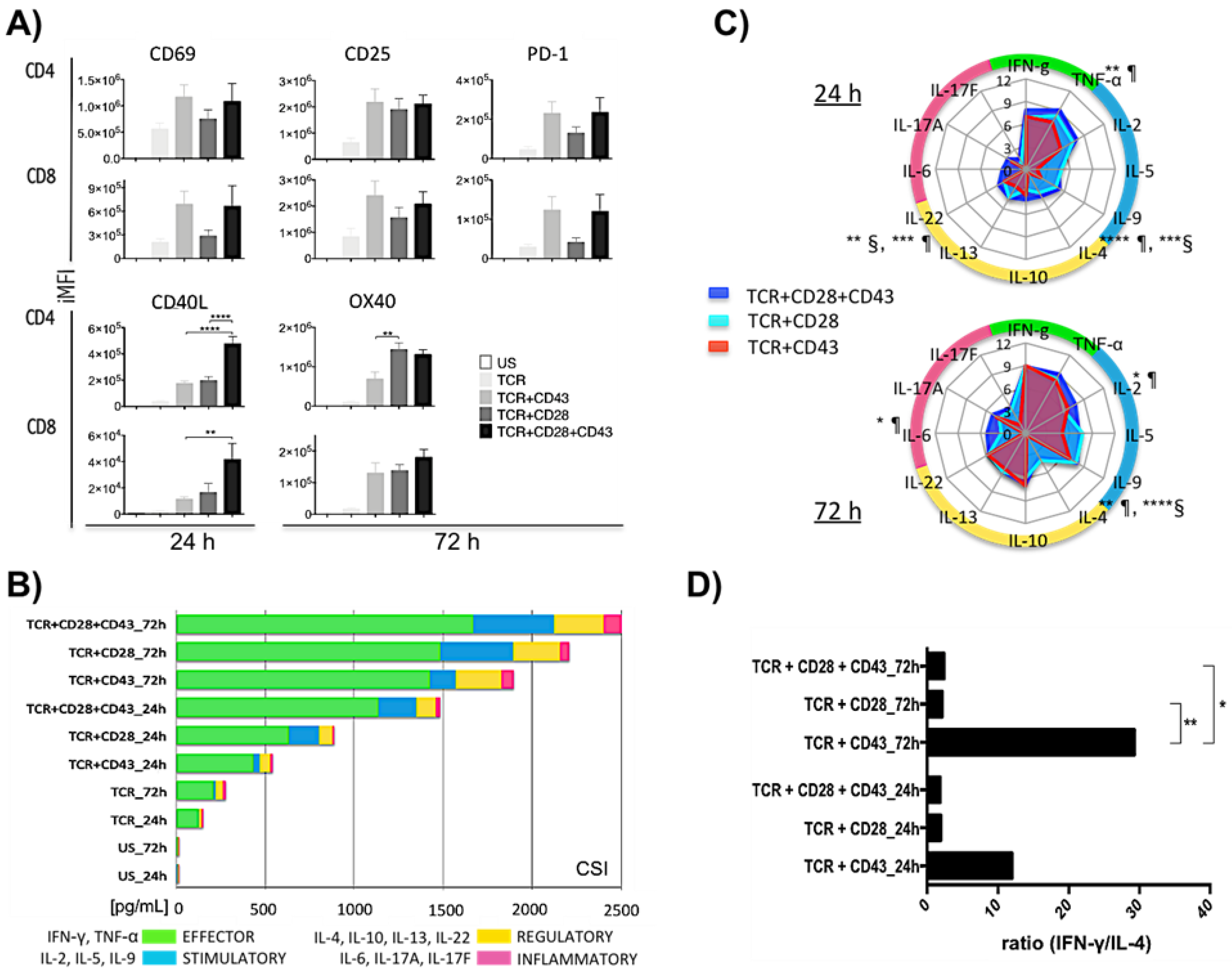
2.1. Engaging either CD28 or CD43 with the TCR Results in Robust T-Cell Activation
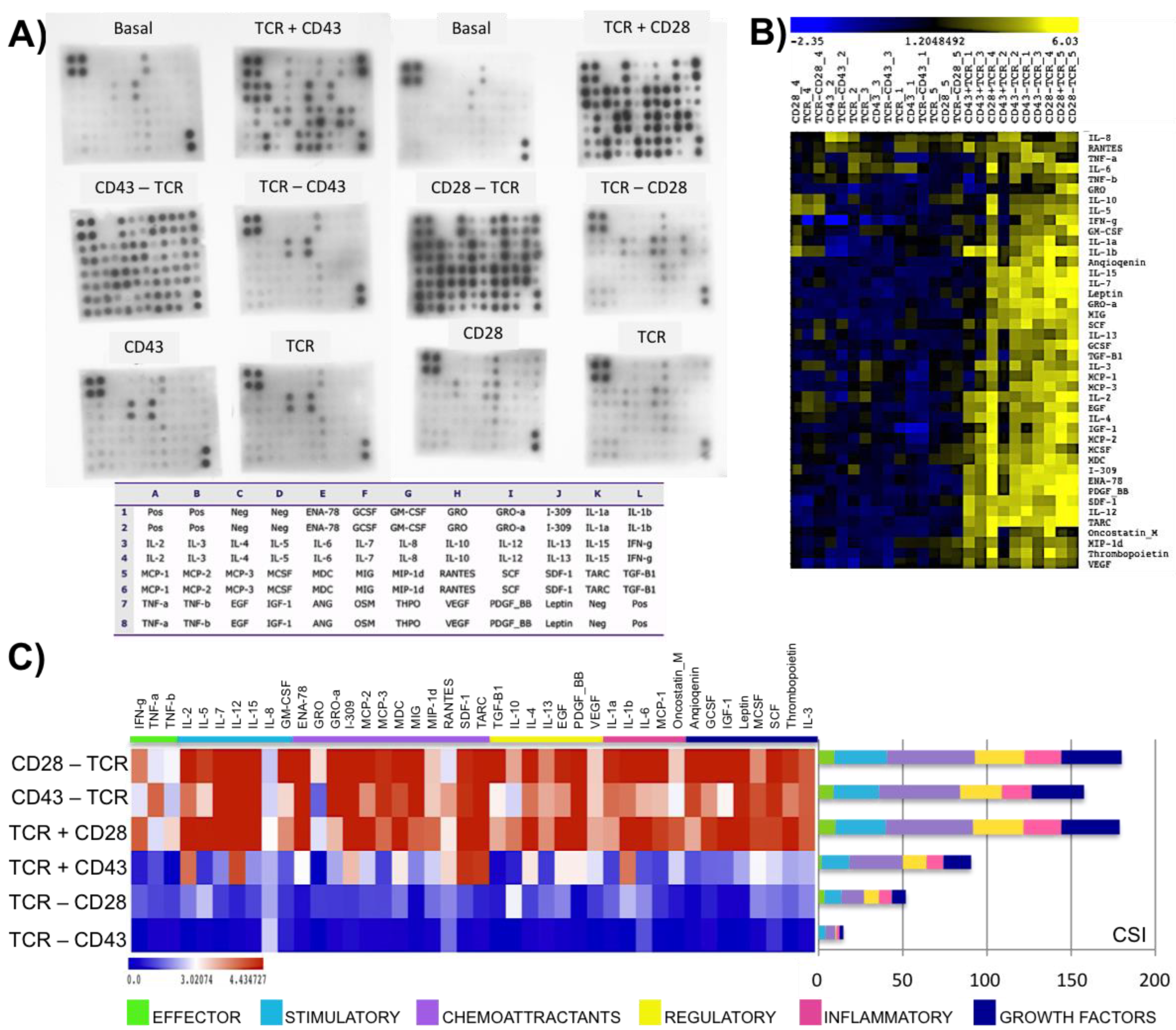
2.2. Engagement of CD43 or CD28 before or after TCR Activation Leads to Different Outcomes
2.3. The Timing of CD28 or CD43 Engagement Relative to the TCR Defines Distinct Activation Patterns
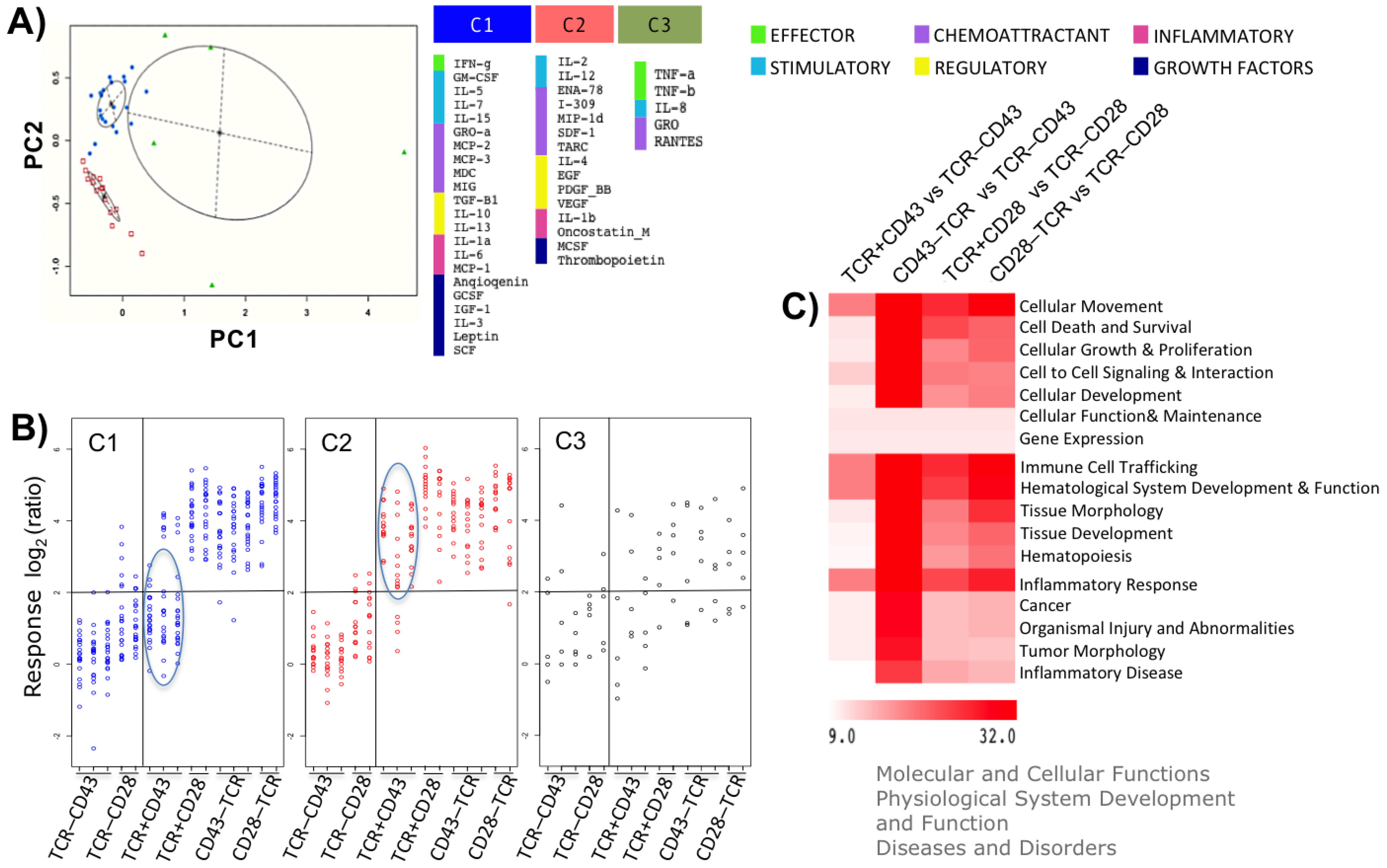
2.4. Different Activation Programs Predict the Usage of Specific Groups of Transcription Regulatory Elements and Different Outcomes
3. Discussion
4. Materials and Methods
4.1. Cell Activation
4.2. Flow Cytometry
4.3. Cytokine Detection
4.4. Image Analysis and Data Normalization
4.5. Correlations, Principal Component Analysis, and Protein Expression Signatures
4.6. Differential Protein Expression Analysis
4.7. Transcription Factors Prediction
4.8. Ingenuity Pathway Analysis
4.9. Clustering Analysis
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bretscher, P.A. A two-step, two-signal model for the primary activation of precursor helper T cells. Proc. Natl. Acad. Sci. USA 1999, 96, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.P.; Taylor, C.; Petryniak, B.; Turka, L.A.; June, C.H.; Thompson, C.B. The genomic organization of the CD28 gene. Implications for the regulation of CD28 mRNA expression and heterogeneity. J. Immunol. 1990, 145, 344–352. [Google Scholar] [CrossRef]
- Venuprasad, K.; Parab, P.; Prasad, D.V.; Sharma, S.; Banerjee, P.R.; Deshpande, M.; Mitra, D.K.; Pal, S.; Bhadra, R.; Mitra, D.; et al. Immunobiology of CD28 expression on human neutrophils. I. CD28 regulates neutrophil migration by modulating CXCR-1 expression. Eur. J. Immunol. 2001, 31, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Woerly, G.; Lacy, P.; Younes, A.B.; Roger, N.; Loiseau, S.; Moqbel, R.; Capron, M. Human eosinophils express and release IL-13 following CD28-dependent activation. J. Leukoc. Biol. 2002, 72, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Boomer, J.S.; Green, J.M. An enigmatic tail of CD28 signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002436. [Google Scholar] [CrossRef] [PubMed]
- Rudd, C.E.; Taylor, A.; Schneider, H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 2009, 229, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Esensten, J.H.; Helou, Y.A.; Chopra, G.; Weiss, A.; Bluestone, J.A. CD28 Costimulation: From Mechanism to Therapy. Immunity 2016, 44, 973–988. [Google Scholar] [CrossRef]
- Sperling, A.I.; Green, J.M.; Mosley, R.L.; Smith, P.L.; DiPaolo, R.J.; Klein, J.R.; Bluestone, J.A.; Thompson, C.B. CD43 is a murine T cell costimulatory receptor that functions independently of CD28. J. Exp. Med. 1995, 182, 139–146. [Google Scholar] [CrossRef]
- Shahinian, A.; Pfeffer, K.; Lee, K.P.; Kundig, T.M.; Kishihara, K.; Wakeham, A.; Kawai, K.; Ohashi, P.S.; Thompson, C.B.; Mak, T.W. Differential T cell costimulatory requirements in CD28-deficient mice. Science 1993, 261, 609–612. [Google Scholar] [CrossRef]
- Cyster, J.G.; Shotton, D.M.; Williams, A.F. The dimensions of the T lymphocyte glycoprotein leukosialin and identification of linear protein epitopes that can be modified by glycosylation. EMBO J. 1991, 10, 893–902. [Google Scholar] [CrossRef]
- Matsumoto, M.; Atarashi, K.; Umemoto, E.; Furukawa, Y.; Shigeta, A.; Miyasaka, M.; Hirata, T. CD43 functions as a ligand for E-Selectin on activated T cells. J. Immunol. 2005, 175, 8042–8050. [Google Scholar] [CrossRef] [PubMed]
- Nonomura, C.; Kikuchi, J.; Kiyokawa, N.; Ozaki, H.; Mitsunaga, K.; Ando, H.; Kanamori, A.; Kannagi, R.; Fujimoto, J.; Muroi, K.; et al. CD43, but not P-selectin glycoprotein ligand-1, functions as an E-selectin counter-receptor in human pre-B-cell leukemia NALL-1. Cancer Res. 2008, 68, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Rosenstein, Y.; Park, J.K.; Hahn, W.C.; Rosen, F.S.; Bierer, B.E.; Burakoff, S.J. CD43, a molecule defective in Wiskott-Aldrich syndrome, binds ICAM-1. Nature 1991, 354, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Stöckl, J.; Majdic, O.; Kohl, P.; Pickl, W.F.; Menzel, J.E.; Knapp, W. Leukosialin (CD43)-major histocompatibility class I molecule interactions involved in spontaneous T cell conjugate formation. J. Exp. Med. 1996, 184, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, T.K.; Nath, D.; Ziltener, H.J.; Vestweber, D.; Fukuda, M.; van Die, I.; Crocker, P.R. Cutting edge: CD43 functions as a T cell counterreceptor for the macrophage adhesion receptor sialoadhesin (Siglec-1). J. Immunol. 2001, 166, 3637–3640. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, F.E.; Anastasiou, M.; Carrillo-Salinas, F.J.; Ngwenyama, N.; Salvador, A.M.; Nevers, T.; Alcaide, P. Sialomucin CD43 regulates T helper type 17 cell intercellular adhesion molecule 1 dependent adhesion, apical migration and transendothelial migration. Immunology 2019, 157, 52–69. [Google Scholar] [CrossRef] [PubMed]
- Velázquez, F.; Grodecki-Pena, A.; Knapp, A.; Salvador, A.M.; Nevers, T.; Croce, K.; Alcaide, P. CD43 Functions as an E-Selectin Ligand for Th17 Cells In Vitro and Is Required for Rolling on the Vascular Endothelium and Th17 Cell Recruitment during Inflammation In Vivo. J. Immunol. 2016, 196, 1305–1316. [Google Scholar] [CrossRef]
- Torres-Huerta, A.; Aleman-Navarro, E.; Bravo-Adame, M.E.; Sandoval-Hernandez, M.A.; Migueles-Lozano, O.A.; Rosenstein, Y. CD43. In Encyclopedia of Signaling Molecules; Choi, S., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 893–905. [Google Scholar] [CrossRef]
- Mattioli, I.; Dittrich-Breiholz, O.; Livingstone, M.; Kracht, M.; Schmitz, M.L. Comparative analysis of T-cell costimulation and CD43 activation reveals novel signaling pathways and target genes. Blood 2004, 104, 3302–3304. [Google Scholar] [CrossRef]
- del Rio, R.; Rincón, M.; Layseca-Espinosa, E.; Fierro, N.A.; Rosenstein, Y.; Pedraza-Alva, G. PKCtheta is required for the activation of human T lymphocytes induced by CD43 engagement. Biochem. Biophys. Res. Commun. 2004, 325, 133–143. [Google Scholar] [CrossRef]
- Montufar-Solis, D.; Garza, T.; Klein, J.R. Selective upregulation of immune regulatory and effector cytokine synthesis by intestinal intraepithelial lymphocytes following CD43 costimulation. Biochem. Biophys. Res. Commun. 2005, 338, 1158–1163. [Google Scholar] [CrossRef]
- Santana, M.A.; Pedraza-Alva, G.; Olivares-Zavaleta, N.; Madrid-Marina, V.; Horejsi, V.; Burakoff, S.J.; Rosenstein, Y. CD43-mediated signals induce DNA binding activity of AP-1, NF-AT, and NFkappa B transcription factors in human T lymphocytes. J. Biol. Chem. 2000, 275, 31460–31468. [Google Scholar] [CrossRef] [PubMed]
- Fierro, N.A.; Pedraza-Alva, G.; Rosenstein, Y. TCR-dependent cell response is modulated by the timing of CD43 engagement. J. Immunol. 2006, 176, 7346–7353. [Google Scholar] [CrossRef] [PubMed]
- Pedraza-Alva, G.; Merida, L.B.; del Rio, R.; Fierro, N.A.; Cruz-Munoz, M.E.; Olivares, N.; Melchy, E.; Igras, V.; Hollander, G.A.; Burakoff, S.J.; et al. CD43 regulates the threshold for T cell activation by targeting Cbl functions. IUBMB Life 2011, 63, 940–948. [Google Scholar] [CrossRef]
- An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [CrossRef] [PubMed]
- Cheng, C.; Alexander, R.; Min, R.; Leng, J.; Yip, K.Y.; Rozowsky, J.; Yan, K.K.; Dong, X.; Djebali, S.; Ruan, Y.; et al. Understanding transcriptional regulation by integrative analysis of transcription factor binding data. Genome Res. 2012, 22, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Epstein, C.B.; Shoresh, N.; Adrian, J.; Kawli, T.; Davis, C.A.; Dobin, A.; Kaul, R.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef]
- Cibrián, D.; Sánchez-Madrid, F. CD69: From activation marker to metabolic gatekeeper. Eur. J. Immunol. 2017, 47, 946–953. [Google Scholar] [CrossRef]
- Karnell, J.L.; Rieder, S.A.; Ettinger, R.; Kolbeck, R. Targeting the CD40-CD40L pathway in autoimmune diseases: Humoral immunity and beyond. Adv. Drug Deliv. Rev. 2019, 141, 92–103. [Google Scholar] [CrossRef]
- Létourneau, S.; Krieg, C.; Pantaleo, G.; Boyman, O. IL-2- and CD25-dependent immunoregulatory mechanisms in the homeostasis of T-cell subsets. J. Allergy Clin. Immunol. 2009, 123, 758–762. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Webb, G.J.; Hirschfield, G.M.; Lane, P.J. OX40, OX40L and Autoimmunity: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2016, 50, 312–332. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, M.B.; Kundaje, A.; Hariharan, M.; Landt, S.G.; Yan, K.K.; Cheng, C.; Mu, X.J.; Khurana, E.; Rozowsky, J.; Alexander, R.; et al. Architecture of the human regulatory network derived from ENCODE data. Nature 2012, 489, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Remold-O’Donnell, E.; Davis, A.E., 3rd; Kenney, D.; Bhaskar, K.R.; Rosen, F.S. Purification and chemical composition of gpL115, the human lymphocyte surface sialoglycoprotein that is defective in Wiskott-Aldrich syndrome. J. Biol. Chem. 1986, 261, 7526–7530. [Google Scholar] [CrossRef] [PubMed]
- Shooshtari, P.; Fortuno, E.S., 3rd; Blimkie, D.; Yu, M.; Gupta, A.; Kollmann, T.R.; Brinkman, R.R. Correlation analysis of intracellular and secreted cytokines via the generalized integrated mean fluorescence intensity. Cytometry. Part A J. Int. Soc. Anal. Cytol. 2010, 77, 873–880. [Google Scholar] [CrossRef]
- Rossi, J.; Paczkowski, P.; Shen, Y.W.; Morse, K.; Flynn, B.; Kaiser, A.; Ng, C.; Gallatin, K.; Cain, T.; Fan, R.; et al. Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood 2018, 132, 804–814. [Google Scholar] [CrossRef]
- Xue, Q.; Bettini, E.; Paczkowski, P.; Ng, C.; Kaiser, A.; McConnell, T.; Kodrasi, O.; Quigley, M.F.; Heath, J.; Fan, R.; et al. Single-cell multiplexed cytokine profiling of CD19 CAR-T cells reveals a diverse landscape of polyfunctional antigen-specific response. J. Immunother. Cancer 2017, 5, 85. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Chen, D.; Yuan, D.; Lausted, C.; Choi, J.; Dai, C.L.; Voillet, V.; Duvvuri, V.R.; Scherler, K.; Troisch, P.; et al. Multi-Omics Resolves a Sharp Disease-State Shift between Mild and Moderate COVID-19. Cell 2020, 183, 1479–1495. [Google Scholar] [CrossRef]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef]
- Yeung, K.Y.; Fraley, C.; Murua, A.; Raftery, A.E.; Ruzzo, W.L. Model-based clustering and data transformations for gene expression data. Bioinformatics 2001, 17, 977–987. [Google Scholar] [CrossRef]
- Scrucca, L.; Fop, M.; Murphy, T.B.; Raftery, A.E. mclust 5: Clustering, Classification and Density Estimation Using Gaussian Finite Mixture Models. R J. 2016, 8, 289–317. [Google Scholar] [CrossRef]
- Smyth, G.K.; Michaud, J.; Scott, H.S. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 2005, 21, 2067–2075. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Abdi, H. The Bonferonni and Šidák Corrections for Multiple Comparisons. In Encyclopedia of Measurement and Statistics; SAGE: Thousand Oaks, CA, USA, 2007; Volume 3. [Google Scholar]
- Matys, V.; Kel-Margoulis, O.V.; Fricke, E.; Liebich, I.; Land, S.; Barre-Dirrie, A.; Reuter, I.; Chekmenev, D.; Krull, M.; Hornischer, K.; et al. TRANSFAC and its module TRANSCompel: Transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006, 34, D108–D110. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandoval-Hernández, M.A.; Fierro, N.A.; Veytia-Bucheli, J.I.; Alvarado-Velázquez, D.A.; Alemán-Navarro, E.; Melchy-Pérez, E.; Auvynet, C.; Imaz-Rosshandler, I.; Carneiro, J.; Perez-Rueda, E.; et al. Differential Impact of CD43 and CD28 on T-Cell Differentiation Depending on the Order of Engagement with the TCR. Int. J. Mol. Sci. 2024, 25, 3135. https://doi.org/10.3390/ijms25063135
Sandoval-Hernández MA, Fierro NA, Veytia-Bucheli JI, Alvarado-Velázquez DA, Alemán-Navarro E, Melchy-Pérez E, Auvynet C, Imaz-Rosshandler I, Carneiro J, Perez-Rueda E, et al. Differential Impact of CD43 and CD28 on T-Cell Differentiation Depending on the Order of Engagement with the TCR. International Journal of Molecular Sciences. 2024; 25(6):3135. https://doi.org/10.3390/ijms25063135
Chicago/Turabian StyleSandoval-Hernández, Monserrat Alba, Nora Alma Fierro, José Ignacio Veytia-Bucheli, Den Alejandro Alvarado-Velázquez, Estefanía Alemán-Navarro, Erika Melchy-Pérez, Constance Auvynet, Iván Imaz-Rosshandler, Jorge Carneiro, Ernesto Perez-Rueda, and et al. 2024. "Differential Impact of CD43 and CD28 on T-Cell Differentiation Depending on the Order of Engagement with the TCR" International Journal of Molecular Sciences 25, no. 6: 3135. https://doi.org/10.3390/ijms25063135