
Metformin in Esophageal Carcinoma: Exploring Molecular Mechanisms and Therapeutic Insights

 ,
,  , , , , and
, , , , and
Abstract
:1. Introduction
1.1. Exploring Esophageal Cancer: Trends, Treatments, and Future Horizons
1.2. Understanding the Mechanisms and Pathophysiology of Esophageal Carcinoma
1.3. Metformin: Unraveling Physiological Functions and Anti-Cancer Mechanisms
2. Metformin in Esophageal Cancer: Exploring Its Clinical Significance and Therapeutic Implications
2.1. Metformin for Esophageal Cancer Risk Reduction
2.2. Metformin’s Impact on Esophageal Cancer Survival
2.3. Metformin as a Chemopreventive Agent
3. Understanding Metformin’s Molecular Mechanisms in EC
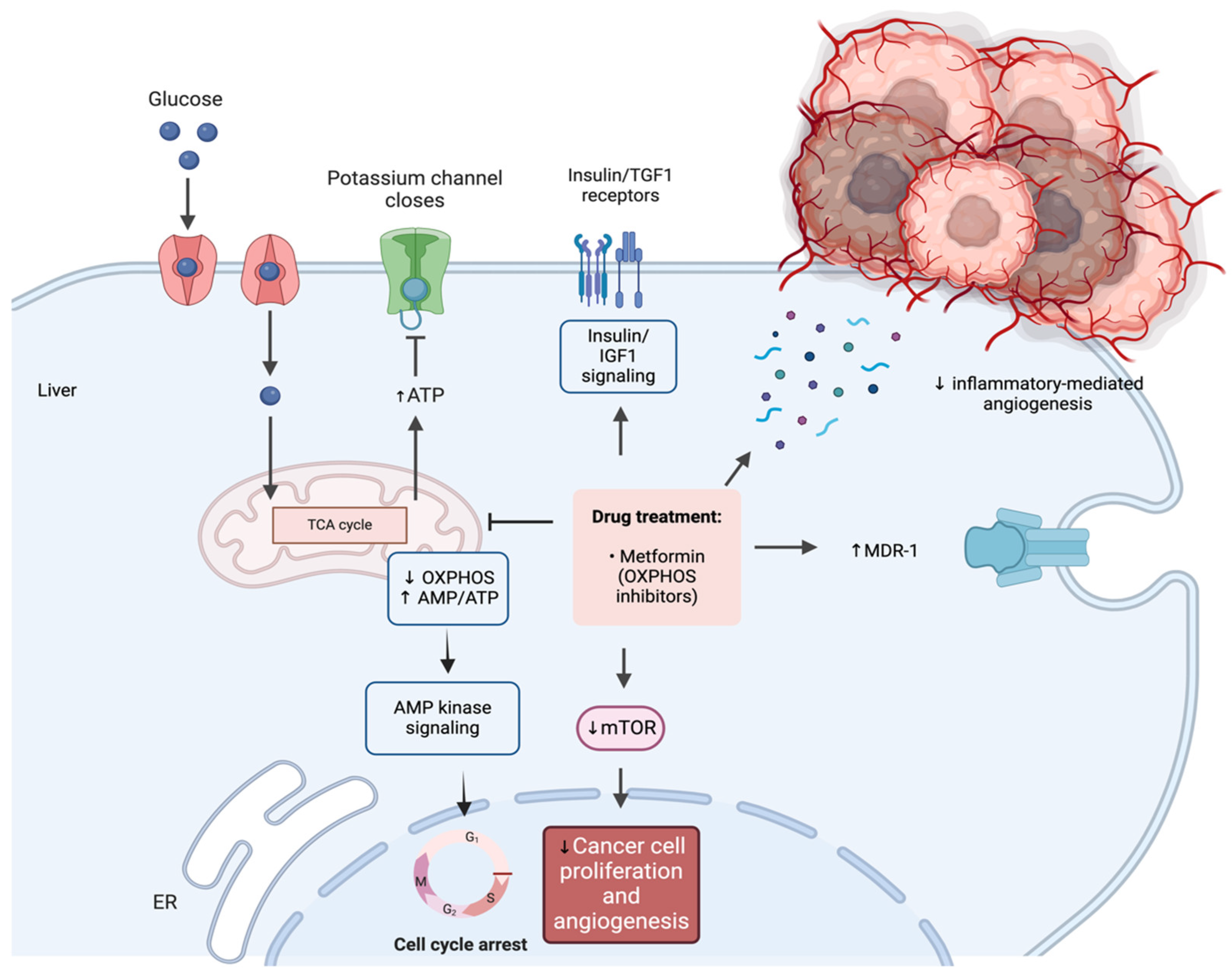
3.1. The Regulatory Effects of Metformin on EC Cellular Proliferation
3.2. Deciphering the Metformin’s Influence on Apoptosis in EC
3.3. Metformin’s Effects on Angiogenesis, Invasion, and Metastasis
3.4. Metformin-Mediated Epigenetic Changes: Understanding the Molecular Mechanisms
3.5. Enhancing Chemotherapeutic Efficacy: Synergies of Metformin in Combination Therapies
3.5.1. Metformin and Cisplatin
3.5.2. Metformin and 5-Fluorouracil
3.5.3. Metformin and Ionizing Radiation
3.5.4. Metformin and Disulfiram
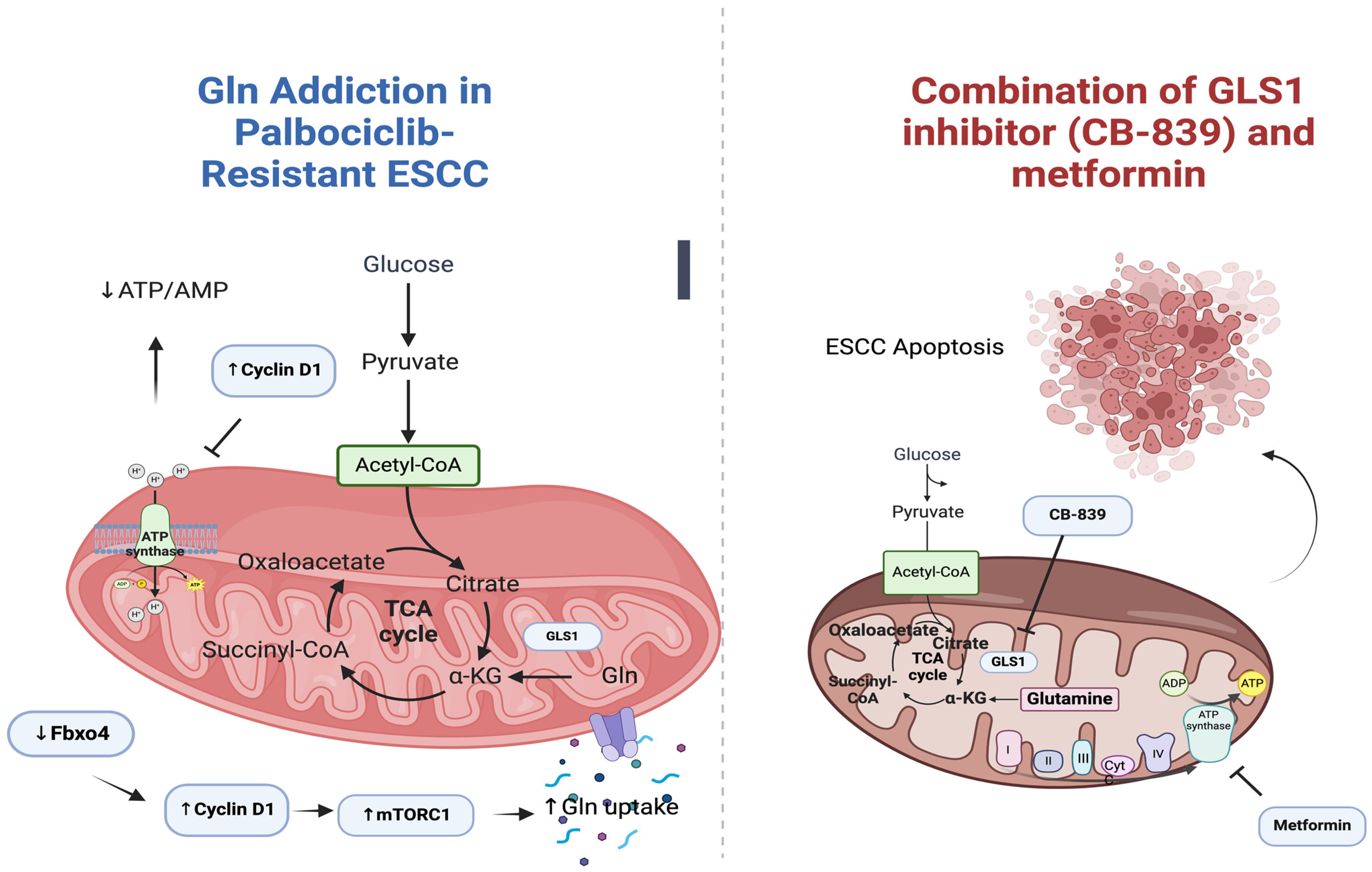
3.5.5. Metformin and CB-839 (Glutaminase 1 Inhibitor)
3.5.6. Metformin and 2-Deoxy-d-glucose (2DG)
3.5.7. 3-Aminobenzamide (3-ABA) Combined with Cisplatin or Metformin
4. Understanding Metformin’s Impact on Immune Responses in EC
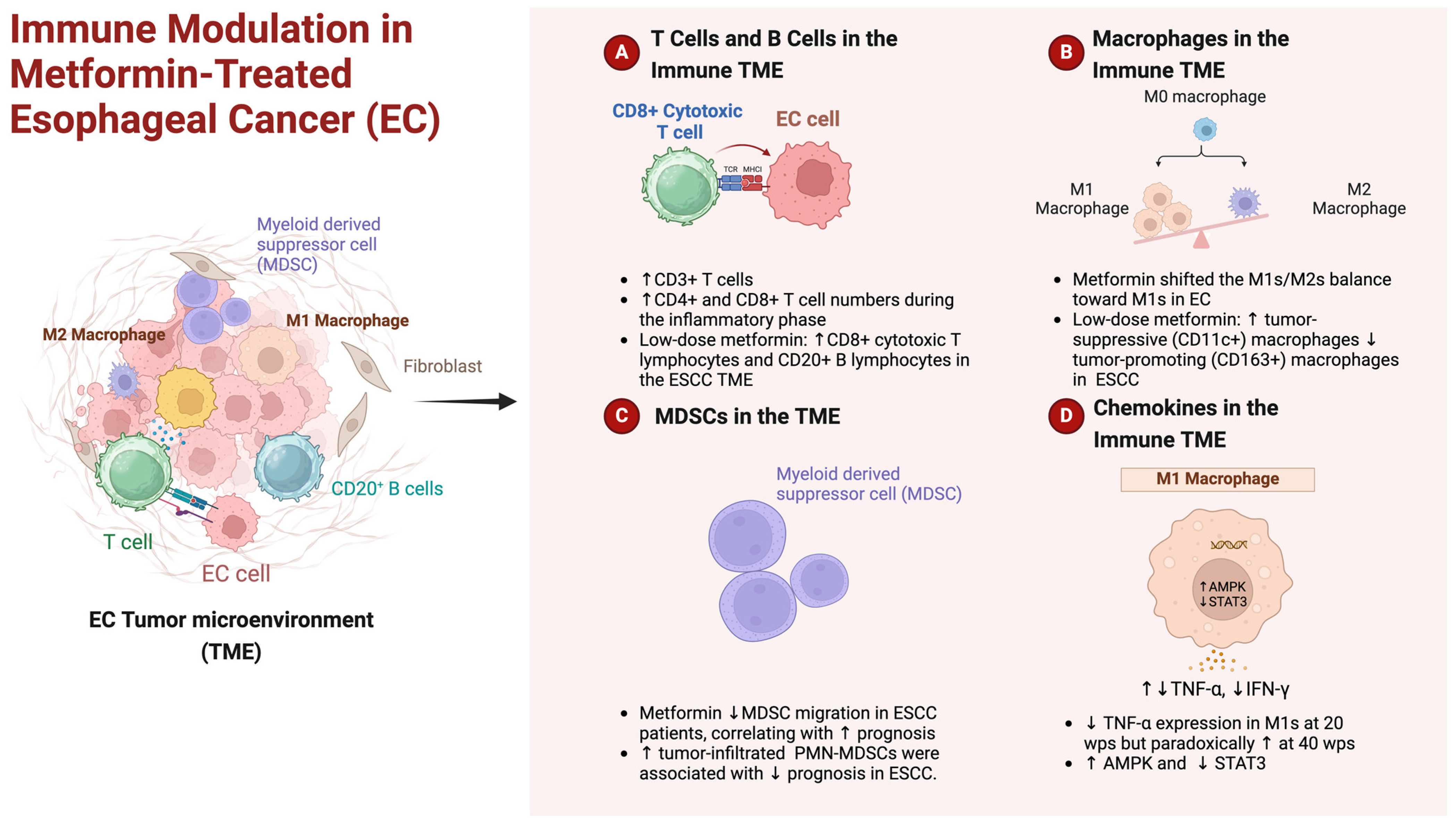
4.1. Navigating Immune TME: Metformin’s Role in Esophageal Cancer
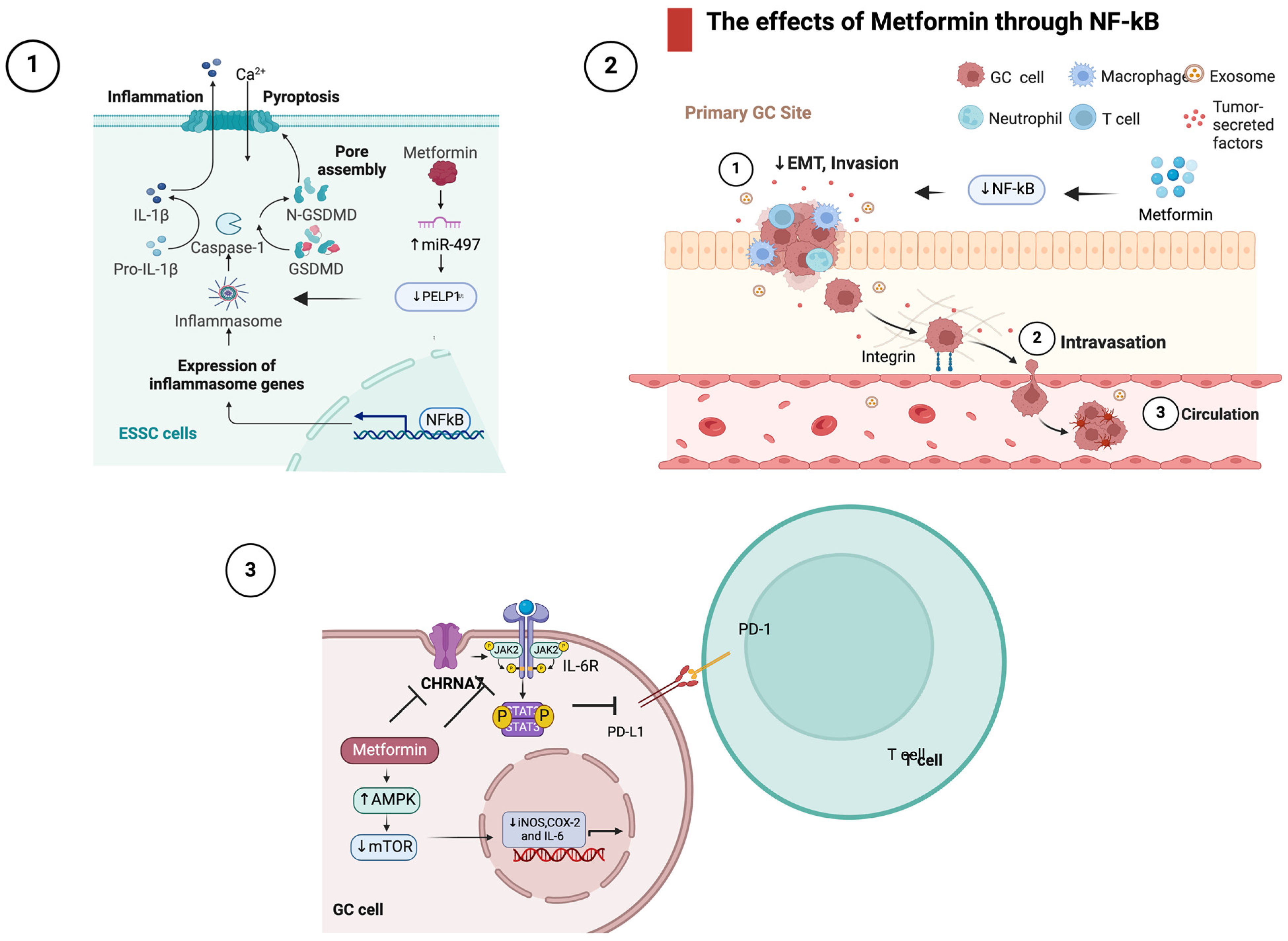
4.2. The Effects of Metformin in Inflammatory Signaling
5. Discussion
6. Conclusions—Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lagergren, J.; Smyth, E.; Cunningham, D.; Lagergren, P. Oesophageal Cancer. Lancet 2017, 390, 2383–2396. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Thrumurthy, S.G.; Chaudry, M.A.; Thrumurthy, S.S.D.; Mughal, M. Oesophageal Cancer: Risks, Prevention, and Diagnosis. BMJ 2019, 366, l4373. [Google Scholar] [CrossRef]
- Kamangar, F.; Nasrollahzadeh, D.; Safiri, S.; Sepanlou, S.G.; Fitzmaurice, C.; Ikuta, K.S.; Bisignano, C.; Islami, F.; Roshandel, G.; Lim, S.S.; et al. The Global, Regional, and National Burden of Oesophageal Cancer and Its Attributable Risk Factors in 195 Countries and Territories, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 582–597. [Google Scholar] [CrossRef]
- Santucci, C.; Mignozzi, S.; Malvezzi, M.; Collatuzzo, G.; Levi, F.; La Vecchia, C.; Negri, E. Global Trends in Esophageal Cancer Mortality with Predictions to 2025, and in Incidence by Histotype. Cancer Epidemiol. 2023, 87, 102486. [Google Scholar] [CrossRef]
- Bolger, J.C.; Donohoe, C.L.; Lowery, M.; Reynolds, J.V. Advances in the Curative Management of Oesophageal Cancer. Br. J. Cancer 2022, 126, 706–717. [Google Scholar] [CrossRef]
- Pennathur, A.; Godfrey, T.E.; Luketich, J.D. The Molecular Biologic Basis of Esophageal and Gastric Cancers. Surg. Clin. N. Am. 2019, 99, 403–418. [Google Scholar] [CrossRef]
- Iwaya, T.; Endo, F.; Takahashi, F.; Tokino, T.; Sasaki, Y.; Nishizuka, S.S. Frequent Tumor Burden Monitoring of Esophageal Squamous Cell Carcinoma with Circulating Tumor DNA Using Individually Designed Digital Polymerase Chain Reaction. Gastroenterology 2021, 160, 463–465.e4. [Google Scholar] [CrossRef]
- Papadakos, S.P.; Ferraro, D.; Carbone, G.; Frampton, A.E.; Vennarecci, G.; Kykalos, S.; Schizas, D.; Theocharis, S.; Machairas, N. The Emerging Role of Metformin in the Treatment of Hepatocellular Carcinoma: Is There Any Value in Repurposing Metformin for HCC Immunotherapy? Cancers 2023, 15, 3161. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, M.; Ying, S.; Zhang, C.; Lin, R.; Zheng, J.; Zhang, G.; Tian, D.; Guo, Y.; Du, C.; et al. Genetic Alterations in Esophageal Tissues from Squamous Dysplasia to Carcinoma. Gastroenterology 2017, 153, 166–177. [Google Scholar] [CrossRef]
- Wu, C.; Wang, Z.; Song, X.; Feng, X.-S.; Abnet, C.C.; He, J.; Hu, N.; Zuo, X.-B.; Tan, W.; Zhan, Q.; et al. Joint Analysis of Three Genome-Wide Association Studies of Esophageal Squamous Cell Carcinoma in Chinese Populations. Nat. Genet. 2014, 46, 1001–1006. [Google Scholar] [CrossRef]
- Cui, R.; Kamatani, Y.; Takahashi, A.; Usami, M.; Hosono, N.; Kawaguchi, T.; Tsunoda, T.; Kamatani, N.; Kubo, M.; Nakamura, Y.; et al. Functional Variants in ADH1B and ALDH2 Coupled with Alcohol and Smoking Synergistically Enhance Esophageal Cancer Risk. Gastroenterology 2009, 137, 1768–1775. [Google Scholar] [CrossRef]
- Gao, Y.-B.; Chen, Z.-L.; Li, J.-G.; Hu, X.-D.; Shi, X.-J.; Sun, Z.-M.; Zhang, F.; Zhao, Z.-R.; Li, Z.-T.; Liu, Z.-Y.; et al. Genetic Landscape of Esophageal Squamous Cell Carcinoma. Nat. Genet. 2014, 46, 1097–1102. [Google Scholar] [CrossRef]
- Kim, J.; Bowlby, R.; Mungall, A.J.; Robertson, A.G.; Odze, R.D.; Cherniack, A.D.; Shih, J.; Pedamallu, C.S.; Cibulskis, C.; Dunford, A.; et al. Integrated Genomic Characterization of Oesophageal Carcinoma. Nature 2017, 541, 169–174. [Google Scholar] [CrossRef]
- Dulak, A.M.; Stojanov, P.; Peng, S.; Lawrence, M.S.; Fox, C.; Stewart, C.; Bandla, S.; Imamura, Y.; Schumacher, S.E.; Shefler, E.; et al. Exome and Whole-Genome Sequencing of Esophageal Adenocarcinoma Identifies Recurrent Driver Events and Mutational Complexity. Nat. Genet. 2013, 45, 478–486. [Google Scholar] [CrossRef]
- Ross-Innes, C.S.; Becq, J.; Warren, A.; Cheetham, R.K.; Northen, H.; O’Donovan, M.; Malhotra, S.; di Pietro, M.; Ivakhno, S.; He, M.; et al. Whole-Genome Sequencing Provides New Insights into the Clonal Architecture of Barrett’s Esophagus and Esophageal Adenocarcinoma. Nat. Genet. 2015, 47, 1038–1046. [Google Scholar] [CrossRef]
- Thrift, A.P.; Pandeya, N.; Smith, K.J.; Green, A.C.; Hayward, N.K.; Webb, P.M.; Whiteman, D.C. Helicobacter Pylori Infection and the Risks of Barrett’s Oesophagus: A Population-Based Case-Control Study. Int. J. Cancer 2012, 130, 2407–2416. [Google Scholar] [CrossRef]
- Mallik, R.; Chowdhury, T.A. Metformin in Cancer. Diabetes Res. Clin. Pract. 2018, 143, 409–419. [Google Scholar] [CrossRef]
- Viollet, B.; Guigas, B.; Garcia, N.S.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and Molecular Mechanisms of Metformin: An Overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef]
- Maida, A.; Lamont, B.J.; Cao, X.; Drucker, D.J. Metformin Regulates the Incretin Receptor Axis via a Pathway Dependent on Peroxisome Proliferator-Activated Receptor-α in Mice. Diabetologia 2011, 54, 339–349. [Google Scholar] [CrossRef]
- Shu, Y.; Sheardown, S.A.; Brown, C.; Owen, R.P.; Zhang, S.; Castro, R.A.; Ianculescu, A.G.; Yue, L.; Lo, J.C.; Burchard, E.G.; et al. Effect of Genetic Variation in the Organic Cation Transporter 1 (OCT1) on Metformin Action. J. Clin. Investig. 2007, 117, 1422–1431. [Google Scholar] [CrossRef]
- Faubert, B.; Vincent, E.E.; Poffenberger, M.C.; Jones, R.G. The AMP-Activated Protein Kinase (AMPK) and Cancer: Many Faces of a Metabolic Regulator. Cancer Lett. 2015, 356, 165–170. [Google Scholar] [CrossRef]
- Bridges, H.R.; Jones, A.J.Y.; Pollak, M.N.; Hirst, J. Effects of Metformin and Other Biguanides on Oxidative Phosphorylation in Mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef]
- Vakana, E.; Altman, J.K.; Glaser, H.; Donato, N.J.; Platanias, L.C. Antileukemic Effects of AMPK Activators on BCR-ABL–Expressing Cells. Blood 2011, 118, 6399–6402. [Google Scholar] [CrossRef]
- Kim, H.G.; Hien, T.T.; Han, E.H.; Hwang, Y.P.; Choi, J.H.; Kang, K.W.; Kwon, K.; Kim, B.-H.; Kim, S.K.; Song, G.Y.; et al. Metformin Inhibits P-Glycoprotein Expression via the NF-ΚB Pathway and CRE Transcriptional Activity through AMPK Activation. Br. J. Pharmacol. 2011, 162, 1096–1108. [Google Scholar] [CrossRef]
- Rocha, G.Z.; Dias, M.M.; Ropelle, E.R.; Osório-Costa, F.; Rossato, F.A.; Vercesi, A.E.; Saad, M.J.A.; Carvalheira, J.B.C. Metformin Amplifies Chemotherapy-Induced AMPK Activation and Antitumoral Growth. Clin. Cancer Res. 2011, 17, 3993–4005. [Google Scholar] [CrossRef]
- Abo-Elmatty, D.M.; Ahmed, E.A.; Tawfik, M.K.; Helmy, S.A. Metformin Enhancing the Antitumor Efficacy of Carboplatin against Ehrlich Solid Carcinoma Grown in Diabetic Mice: Effect on IGF-1 and Tumoral Expression of IGF-1 Receptors. Int. Immunopharmacol. 2017, 44, 72–86. [Google Scholar] [CrossRef]
- Wu, N.; Gu, C.; Gu, H.; Hu, H.; Han, Y.; Li, Q. Metformin Induces Apoptosis of Lung Cancer Cells through Activating JNK/P38 MAPK Pathway and GADD153. Neoplasma 2011, 58, 482–490. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Cusack, J.C.; Liu, R.; Baldwin, A.S. Control of Inducible Chemoresistance: Enhanced Anti-Tumor Therapy through Increased Apoptosis by Inhibition of NF-ΚB. Nat. Med. 1999, 5, 412–417. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Barco, S.D.; Martin-Castillo, B.; Menendez, J.A. The Anti-Diabetic Drug Metformin Suppresses Self-Renewal and Proliferation of Trastuzumab-Resistant Tumor-Initiating Breast Cancer Stem Cells. Breast Cancer Res. Treat. 2011, 126, 355–364. [Google Scholar] [CrossRef]
- Xavier, D.O.; Amaral, L.S.; Gomes, M.A.; Rocha, M.A.; Campos, P.R.; Cota, B.D.C.V.; Tafuri, L.S.A.; Paiva, A.M.R.; Silva, J.H.; Andrade, S.P.; et al. Metformin Inhibits Inflammatory Angiogenesis in a Murine Sponge Model. Biomed. Pharmacother. 2010, 64, 220–225. [Google Scholar] [CrossRef]
- Wang, S.; Lin, Y.; Xiong, X.; Wang, L.; Guo, Y.; Chen, Y.; Chen, S.; Wang, G.; Lin, P.; Chen, H.; et al. Low-Dose Metformin Reprograms the Tumor Immune Microenvironment in Human Esophageal Cancer: Results of a Phase II Clinical Trial. Clin. Cancer Res. 2020, 26, 4921–4932. [Google Scholar] [CrossRef]
- Lee, M.; Hsu, C.; Wahlqvist, M.L.; Tsai, H.; Chang, Y.; Huang, Y. Type 2 Diabetes Increases and Metformin Reduces Total, Colorectal, Liver and Pancreatic Cancer Incidences in Taiwanese: A Representative Population Prospective Cohort Study of 800,000 Individuals. BMC Cancer 2011, 11, 20. [Google Scholar] [CrossRef]
- Tseng, C. Metformin and Esophageal Cancer Risk in Taiwanese Patients with Type 2 Diabetes Mellitus. Oncotarget 2017, 8, 18802–18810. [Google Scholar] [CrossRef]
- Becker, C.; Meier, C.R.; Jick, S.S.; Bodmer, M. Case-Control Analysis on Metformin and Cancer of the Esophagus. Cancer Causes Control 2013, 24, 1763–1770. [Google Scholar] [CrossRef]
- Wang, Q.L.; Santoni, G.; Ness-Jensen, E.; Lagergren, J.; Xie, S.H. Association between Metformin Use and Risk of Esophageal Squamous Cell Carcinoma in a Population-Based Cohort Study. Am. J. Gastroenterol. 2020, 115, 73–78. [Google Scholar] [CrossRef]
- Loomans-Kropp, H.A.; Chaloux, M.; Richmond, E.; Umar, A. Association of Common Use Pharmaceuticals in Reducing Risk of Esophageal Adenocarcinoma: A SEER-Medicare Analysis. Cancer Prev. Res. 2021, 14, 195–204. [Google Scholar] [CrossRef]
- Chen, Y.; Cheng, X.; Sun, C.; Kim, N.H.; Kailas, S.; Qureshi, F.; Karadsheh, Z.; Wu, Y.; Hu, L.; Zhou, Z.; et al. Is Metformin Use Associated with a Reduced Risk of Oesophageal Cancer? A Systematic Review and Meta-Analysis. Postgrad. Med. J. 2022, 98, 866–870. [Google Scholar] [CrossRef]
- Wu, H.D.; Zhang, J.J.; Zhou, B.J. The Effect of Metformin on Esophageal Cancer Risk in Patients with Type 2 Diabetes Mellitus: A Systematic Review and Meta-analysis. Clin. Transl. Oncol. 2021, 23, 275–282. [Google Scholar] [CrossRef]
- Wang, Q.L.; Santoni, G.; Lagergren, J. Diabetes, Metformin Use, and Survival in Esophageal Cancer: A Population-Based Cohort Study. JNCI Cancer Spectr. 2023, 7, pkad043. [Google Scholar] [CrossRef]
- Skinner, H.D.; McCurdy, M.R.; Echeverria, A.E.; Lin, S.H.; Welsh, J.W.; O’Reilly, M.S.; Hofstetter, W.L.; Ajani, J.A.; Komaki, R.; Cox, J.D.; et al. Metformin Use and Improved Response to Therapy in Esophageal Adenocarcinoma. Acta Oncol. 2013, 52, 1002–1009. [Google Scholar] [CrossRef]
- Spierings, L.E.A.M.M.; Lagarde, S.M.; van Oijen, M.G.H.; Gisbertz, S.S.; Wilmink, J.W.; Hulshof, M.C.C.M.; Meijer, S.L.; Anderegg, M.C.; van Berge Henegouwen, M.I.; van Laarhoven, H.W.M. Metformin Use During Treatment of Potentially Curable Esophageal Cancer Patients Is Not Associated with Better Outcomes. Ann. Surg. Oncol. 2015, 22, 766–771. [Google Scholar] [CrossRef]
- Van De Voorde, L.; Janssen, L.; Larue, R.; Houben, R.; Buijsen, J.; Sosef, M.; Vanneste, B.; Schraepen, M.C.; Berbée, M.; Lambin, P. Can Metformin Improve “the Tomorrow” of Patients Treated for Oesophageal Cancer? Eur. J. Surg. Oncol. 2015, 41, 1333–1339. [Google Scholar] [CrossRef]
- Sakamoto, K.; Okabayashi, K.; Matsui, S.; Seishima, R.; Shigeta, K.; Kitagawa, Y. Association of Tumor Pathological Response with the Use of Metformin During Neoadjuvant Chemoradiotherapy in Rectal and Esophageal/Gastroesophageal Cancer Patients: A Systematic Review and Meta-Analysis. J. Gastrointest. Surg. 2022, 26, 2227–2236. [Google Scholar] [CrossRef]
- Arai, J.; Niikura, R.; Hayakawa, Y.; Kawahara, T.; Honda, T.; Hasatani, K.; Yoshida, N.; Nishida, T.; Sumiyoshi, T.; Kiyotoki, S.; et al. Chemoprevention of Oesophageal Squamous-Cell Carcinoma and Adenocarcinoma: A Multicentre Retrospective Cohort Study. Digestion 2022, 103, 192–204. [Google Scholar] [CrossRef]
- Chak, A.; Buttar, N.S.; Foster, N.R.; Seisler, D.K.; Marcon, N.E.; Schoen, R.; Cruz-Correa, M.R.; Falk, G.W.; Sharma, P.; Hur, C.; et al. Metformin Does Not Reduce Markers of Cell Proliferation in Esophageal Tissues of Patients with Barrett’s Esophagus. Clin. Gastroenterol. Hepatol. 2015, 13, 665–672. [Google Scholar] [CrossRef]
- Agrawal, S.; Patel, P.; Agrawal, A.; Makhijani, N.; Markert, R.; Deidrich, W. Metformin Use and the Risk of Esophageal Cancer in Barrett Esophagus. South Med. J. 2014, 107, 774–779. [Google Scholar] [CrossRef]
- Antonowicz, S.; Bodai, Z.; Wiggins, T.; Markar, S.R.; Boshier, P.R.; Goh, Y.M.; Adam, M.E.; Lu, H.; Kudo, H.; Rosini, F.; et al. Endogenous Aldehyde Accumulation Generates Genotoxicity and Exhaled Biomarkers in Esophageal Adenocarcinoma. Nat. Commun. 2021, 12, 1454. [Google Scholar] [CrossRef]
- Xu, Y.; Lu, S. Metformin Inhibits Esophagus Cancer Proliferation through Upregulation of USP7. Cell. Physiol. Biochem. 2013, 32, 1178–1186. [Google Scholar] [CrossRef]
- Kobayashi, M.; Kato, K.; Iwama, H.; Fujihara, S.; Nishiyama, N.; Mimura, S.; Toyota, Y.; Nomura, T.; Nomura, K.; Tani, J.; et al. Antitumor Effect of Metformin in Esophageal Cancer: In Vitro Study. Int. J. Oncol. 2013, 42, 517–524. [Google Scholar] [CrossRef]
- Damelin, L.H.; Jivan, R.; Veale, R.B.; Rousseau, A.L.; Mavri-Damelin, D. Metformin Induces an Intracellular Reductive State That Protects Oesophageal Squamous Cell Carcinoma Cells against Cisplatin but Not Copper-Bis(Thiosemicarbazones). BMC Cancer 2014, 14, 314. [Google Scholar] [CrossRef]
- Cai, X.; Hu, X.; Tan, X.; Cheng, W.; Wang, Q.; Chen, X.; Guan, Y.; Chen, C.; Jing, X. Metformin Induced AMPK Activation, G0/G1 Phase Cell Cycle Arrest and the Inhibition of Growth of Esophageal Squamous Cell Carcinomas In Vitro and In Vivo. PLoS ONE 2015, 10, e0133349. [Google Scholar] [CrossRef]
- Fujihara, S.; Kato, K.; Morishita, A.; Iwama, H.; Nishioka, T.; Chiyo, T.; Nishiyama, N.; Miyoshi, H.; Kobayashi, M.; Kobara, H.; et al. Antidiabetic Drug Metformin Inhibits Esophageal Adenocarcinoma Cell Proliferation In Vitro and In Vivo. Int. J. Oncol. 2015, 46, 2172–2180. [Google Scholar] [CrossRef]
- Feng, Y.; Ke, C.; Tang, Q.; Dong, H.; Zheng, X.; Lin, W.; Ke, J.; Huang, J.; Yeung, S.-C.; Zhang, H. Metformin Promotes Autophagy and Apoptosis in Esophageal Squamous Cell Carcinoma by Downregulating Stat3 Signaling. Cell Death Dis. 2014, 5, e1088. [Google Scholar] [CrossRef]
- Peng, J.; Jing, X.; Wu, J.; Hong, D.; Hu, X.; Wang, Q.; Hu, H.; Cai, X. Metformin’s Effects on Apoptosis of Esophageal Carcinoma Cells and Normal Esophageal Epithelial Cells: An In Vitro Comparative Study. Biomed. Res. Int. 2020, 2020, 1068671. [Google Scholar] [CrossRef]
- Shafaee, A.; Pirayesh Islamian, J.; Zarei, D.; Mohammadi, M.; Nejati-Koshki, K.; Farajollahi, A.; Aghamiri, S.M.R.; Rahmati Yamchi, M.; Baradaran, B.; Asghari Jafarabadi, M. Induction of Apoptosis by a Combination of 2-Deoxyglucose and Metformin in Esophageal Squamous Cell Carcinoma by Targeting Cancer Cell Metabolism. Iran. J. Med. Sci. 2019, 44, 99–107. [Google Scholar]
- Tang, J.-C.; An, R.; Jiang, Y.-Q.; Yang, J. Effects and Mechanisms of Metformin on the Proliferation of Esophageal Cancer Cells In Vitro and In Vivo. Cancer Res. Treat. 2017, 49, 778–789. [Google Scholar] [CrossRef]
- Wang, F.; Ding, X.; Wang, T.; Shan, Z.; Wang, J.; Wu, S.; Chi, Y.; Zhang, Y.; Lv, Z.; Wang, L.; et al. Metformin Inhibited Esophageal Squamous Cell Carcinoma Proliferation in Vitro and in Vivo and Enhanced the Anti-Cancer Effect of Cisplatin. PLoS ONE 2017, 12, e0174276. [Google Scholar] [CrossRef]
- Li, P.D.; Liu, Z.; Cheng, T.T.; Luo, W.G.; Yao, J.; Chen, J.; Zou, Z.W.; Chen, L.L.; Ma, C.; Dai, X.F. Redox-Dependent Modulation of Metformin Contributes to Enhanced Sensitivity of Esophageal Squamous Cell Carcinoma to Cisplatin. Oncotarget 2017, 8, 62057–62068. [Google Scholar] [CrossRef]
- Hong, J.; Maacha, S.; Pidkovka, N.; Bates, A.; Salaria, S.N.; Washington, M.K.; Belkhiri, A. AXL Promotes Metformin-Induced Apoptosis Through Mediation of Autophagy by Activating ROS-AMPK-ULK1 Signaling in Human Esophageal Adenocarcinoma. Front. Oncol. 2022, 12, 903874. [Google Scholar] [CrossRef]
- Papadakos, S.P.; Tsagkaris, C.; Papadakis, M.; Papazoglou, A.S.; Moysidis, D.V.; Zografos, C.G.; Theocharis, S. Angiogenesis in Gastrointestinal Stromal Tumors: From Bench to Bedside. World J. Gastrointest. Oncol. 2022, 14, 1469–1477. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, D.; Li, J.; Deng, X.; Liang, G.; Long, Y.; He, X.; Dai, T.; Ren, D. MACC1 Induces Autophagy to Regulate Proliferation, Apoptosis, Migration and Invasion of Squamous Cell Carcinoma. Oncol. Rep. 2017, 38, 2369–2377. [Google Scholar] [CrossRef]
- Yang, Y.; Jin, G.; Liu, H.; Liu, K.; Zhao, J.; Chen, X.; Wang, D.; Bai, R.; Li, X.; Jang, Y.; et al. Metformin Inhibits Esophageal Squamous Cell Carcinoma-Induced Angiogenesis by Suppressing JAK/STAT3 Signaling Pathway. Oncotarget 2017, 8, 74673–74687. [Google Scholar] [CrossRef]
- He, Y.; Tan, X.; Hu, H.; Wang, Q.; Hu, X.; Cai, X.; Guan, Y.; Chen, B.; Jing, X. Metformin Inhibits the Migration and Invasion of Esophageal Squamous Cell Carcinoma Cells by Downregulating the Protein Kinase B Signaling Pathway. Oncol. Lett. 2018, 15, 2939–2945. [Google Scholar] [CrossRef]
- Liang, F.; Wang, Y.-G.; Wang, C. Metformin Inhibited Growth, Invasion and Metastasis of Esophageal Squamous Cell Carcinoma in Vitro and in Vivo. Cell Physiol. Biochem. 2018, 51, 1276–1286. [Google Scholar] [CrossRef]
- Fan, H.; Yu, X.; Zou, Z.; Zheng, W.; Deng, X.; Guo, L.; Jiang, W.; Zhan, Q.; Lu, S.-H. Metformin Suppresses the Esophageal Carcinogenesis in Rats Treated with NMBzA through Inhibiting AMPK/MTOR Signaling Pathway. Carcinogenesis 2019, 40, 669–679. [Google Scholar] [CrossRef]
- Isozaki, Y.; Hoshino, I.; Akutsu, Y.; Hanari, N.; Mori, M.; Nishimori, T.; Murakami, K.; Akanuma, N.; Toyozumi, T.; Takahashi, M.; et al. Screening of Alternative Drugs to the Tumor Suppressor MiR-375 in Esophageal Squamous Cell Carcinoma Using the Connectivity Map. Oncology 2014, 87, 351–363. [Google Scholar] [CrossRef]
- Chen, F.; Xu, B.; Li, J.; Yang, X.; Gu, J.; Yao, X.; Sun, X. Hypoxic Tumour Cell-Derived Exosomal MiR-340-5p Promotes Radioresistance of Oesophageal Squamous Cell Carcinoma via KLF10. J. Exp. Clin. Cancer Res. 2021, 40, 38. [Google Scholar] [CrossRef]
- Wang, L.; Li, K.; Lin, X.; Yao, Z.; Wang, S.; Xiong, X.; Ning, Z.; Wang, J.; Xu, X.; Jiang, Y.; et al. Metformin Induces Human Esophageal Carcinoma Cell Pyroptosis by Targeting the MiR-497/PELP1 Axis. Cancer Lett. 2019, 450, 22–31. [Google Scholar] [CrossRef]
- Liu, C.; Chou, K.; Hsu, J.; Lin, J.; Hsu, T.; Yen, D.H.; Hung, S.; Hsu, H. High Metabolic Rate and Stem Cell Characteristics of Esophageal Cancer Stem-like Cells Depend on the Hsp27–AKT–HK2 Pathway. Int. J. Cancer 2019, 145, 2144–2156. [Google Scholar] [CrossRef]
- Rustgi, A.K.; El-Serag, H.B. Esophageal Carcinoma. N. Engl. J. Med. 2014, 371, 2499–2509. [Google Scholar] [CrossRef]
- Doki, Y.; Ajani, J.A.; Kato, K.; Xu, J.; Wyrwicz, L.; Motoyama, S.; Ogata, T.; Kawakami, H.; Hsu, C.-H.; Adenis, A.; et al. Nivolumab Combination Therapy in Advanced Esophageal Squamous-Cell Carcinoma. N. Engl. J. Med. 2022, 386, 449–462. [Google Scholar] [CrossRef]
- Rattan, R.; Graham, R.P.; Maguire, J.L.; Giri, S.; Shridhar, V. Metformin Suppresses Ovarian Cancer Growth and Metastasis with Enhancement of Cisplatin Cytotoxicity in Vivo. Neoplasia 2011, 13, 483–491. [Google Scholar] [CrossRef]
- Janjetovic, K.; Vucicevic, L.; Misirkic, M.; Vilimanovich, U.; Tovilovic, G.; Zogovic, N.; Nikolic, Z.; Jovanovic, S.; Bumbasirevic, V.; Trajkovic, V.; et al. Metformin Reduces Cisplatin-Mediated Apoptotic Death of Cancer Cells through AMPK-Independent Activation of Akt. Eur. J. Pharmacol. 2011, 651, 41–50. [Google Scholar] [CrossRef]
- Tchounwou, P.B.; Dasari, S.; Noubissi, F.K.; Ray, P.; Kumar, S. Advances in Our Understanding of the Molecular Mechanisms of Action of Cisplatin in Cancer Therapy. J. Exp. Pharmacol. 2021, 13, 303–328. [Google Scholar] [CrossRef]
- Yu, H.; Bian, X.; Gu, D.; He, X. Metformin Synergistically Enhances Cisplatin-Induced Cytotoxicity in Esophageal Squamous Cancer Cells under Glucose-Deprivation Conditions. Biomed. Res. Int. 2016, 2016, 8678634. [Google Scholar] [CrossRef]
- Honjo, S.; Ajani, J.A.; Scott, A.W.; Chen, Q.; Skinner, H.D.; Stroehlein, J.; Johnson, R.L.; Song, S. Metformin Sensitizes Chemotherapy by Targeting Cancer Stem Cells and the MTOR Pathway in Esophageal Cancer. Int. J. Oncol. 2014, 45, 567–574. [Google Scholar] [CrossRef]
- Feng, T.; Li, L.; Ling, S.; Fan, N.; Fang, M.; Zhang, H.; Fang, X.; Lan, W.; Hou, Z.; Meng, Q.; et al. Metformin Enhances Radiation Response of ECa109 Cells through Activation of ATM and AMPK. Biomed. Pharmacother. 2015, 69, 260–266. [Google Scholar] [CrossRef]
- Nakayama, A.; Ninomiya, I.; Harada, S.; Tsukada, T.; Okamoto, K.; Nakanuma, S.; Sakai, S.; Makino, I.; Kinoshita, J.; Hayashi, H.; et al. Metformin Inhibits the Radiation-Induced Invasive Phenotype of Esophageal Squamous Cell Carcinoma. Int. J. Oncol. 2016, 49, 1890–1898. [Google Scholar] [CrossRef]
- Jivan, R.; Damelin, L.H.; Birkhead, M.; Rousseau, A.L.; Veale, R.B.; Mavri-Damelin, D. Disulfiram/Copper-disulfiram Damages Multiple Protein Degradation and Turnover Pathways and Cytotoxicity Is Enhanced by Metformin in Oesophageal Squamous Cell Carcinoma Cell Lines. J. Cell Biochem. 2015, 116, 2334–2343. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Deberardinis, R.J.; Cheng, T. Q’s next: The Diverse Functions of Glutamine in Metabolism, Cell Biology and Cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Ganapathy, V. Amino Acid Transporters in Cancer and Their Relevance to “Glutamine Addiction”: Novel Targets for the Design of a New Class of Anticancer Drugs. Cancer Res. 2015, 75, 1782–1788. [Google Scholar] [CrossRef]
- Schmidt, M.; Sebastian, M. Palbociclib-The First of a New Class of Cell Cycle Inhibitors. Recent. Results Cancer Res. 2018, 211, 153–175. [Google Scholar] [CrossRef]
- Qie, S.; Yoshida, A.; Parnham, S.; Oleinik, N.; Beeson, G.C.; Beeson, C.C.; Ogretmen, B.; Bass, A.J.; Wong, K.-K.; Rustgi, A.K.; et al. Targeting Glutamine-Addiction and Overcoming CDK4/6 Inhibitor Resistance in Human Esophageal Squamous Cell Carcinoma. Nat. Commun. 2019, 10, 1296. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, J.; Zhao, H.; Zhao, T.; Chen, Y.; Jiang, M.; Zhang, S.; Wei, Y.; Zhang, J.; Zhou, Y.; et al. Targeting the SOX2/PARP1 Complex to Intervene in the Growth of Esophageal Squamous Cell Carcinoma. Biomed. Pharmacother. 2022, 153, 113309. [Google Scholar] [CrossRef]
- Takei, R.; Miyashita, T.; Takada, S.; Tajima, H.; Ninomiya, I.; Takamura, H.; Fushida, S.; Harashima, A.; Munesue, S.; Yagi, S.; et al. Dynamic Switch of Immunity and Antitumor Effects of Metformin in Rat Spontaneous Esophageal Carcinogenesis. Cancer Immunol. Immunother. 2022, 71, 777–789. [Google Scholar] [CrossRef]
- Qin, G.; Lian, J.; Huang, L.; Zhao, Q.; Liu, S.; Zhang, Z.; Chen, X.; Yue, D.; Li, L.; Li, F.; et al. Metformin Blocks Myeloid-Derived Suppressor Cell Accumulation through AMPK-DACH1-CXCL1 Axis. Oncoimmunology 2018, 7, e1442167. [Google Scholar] [CrossRef]
- Sekino, N.; Kano, M.; Matsumoto, Y.; Sakata, H.; Akutsu, Y.; Hanari, N.; Murakami, K.; Toyozumi, T.; Takahashi, M.; Otsuka, R.; et al. Antitumor Effects of Metformin Are a Result of Inhibiting Nuclear Factor Kappa B Nuclear Translocation in Esophageal Squamous Cell Carcinoma. Cancer Sci. 2018, 109, 1066–1074. [Google Scholar] [CrossRef]
- Sekino, N.; Kano, M.; Kobayashi, S.; Murakami, K.; Sakata, H.; Toyozumi, T.; Endo, S.; Matsumoto, Y.; Suito, H.; Takahashi, M.; et al. Metformin-Induced Heat Shock Protein Family A Member 6 Is a Promising Biomarker of Esophageal Squamous Cell Carcinoma. Oncology 2022, 100, 267–277. [Google Scholar] [CrossRef]
- Wang, L.; Liang, D.; Xiong, X.; Lin, Y.; Zhu, J.; Yao, Z.; Wang, S.; Guo, Y.; Chen, Y.; Geary, K.; et al. Repurposing Dextromethorphan and Metformin for Treating Nicotine-Induced Cancer by Directly Targeting CHRNA7 to Inhibit JAK2/STAT3/SOX2 Signaling. Oncogene 2021, 40, 1974–1987. [Google Scholar] [CrossRef]
- Lu, Y.; Xin, D.; Guan, L.; Xu, M.; Yang, Y.; Chen, Y.; Yang, Y.; Wang-Gillam, A.; Wang, L.; Zong, S.; et al. Metformin Downregulates PD-L1 Expression in Esophageal Squamous Cell Catrcinoma by Inhibiting IL-6 Signaling Pathway. Front. Oncol. 2021, 11, 762523. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Z.; Wang, Q.; Yi, Y.; Li, B. Integrated Cohort of Esophageal Squamous Cell Cancer Reveals Genomic Features Underlying Clinical Characteristics. Nat. Commun. 2022, 13, 5268. [Google Scholar] [CrossRef]
- Bao, C.; Tourdot, R.W.; Brunette, G.J.; Stewart, C.; Sun, L.; Baba, H.; Watanabe, M.; Agoston, A.T.; Jajoo, K.; Davison, J.M.; et al. Genomic Signatures of Past and Present Chromosomal Instability in Barrett’s Esophagus and Early Esophageal Adenocarcinoma. Nat. Commun. 2023, 14, 6203. [Google Scholar] [CrossRef]
- Abbas, S.; Pich, O.; Devonshire, G.; Zamani, S.A.; Katz-Summercorn, A.; Killcoyne, S.; Cheah, C.; Nutzinger, B.; Grehan, N.; Lopez-Bigas, N.; et al. Mutational Signature Dynamics Shaping the Evolution of Oesophageal Adenocarcinoma. Nat. Commun. 2023, 14, 4239. [Google Scholar] [CrossRef]
- Zheng, Y.; Chen, Z.; Han, Y.; Han, L.; Zou, X.; Zhou, B.; Hu, R.; Hao, J.; Bai, S.; Xiao, H.; et al. Immune Suppressive Landscape in the Human Esophageal Squamous Cell Carcinoma Microenvironment. Nat. Commun. 2020, 11, 6268. [Google Scholar] [CrossRef]
- Kato, K.; Cho, B.C.; Takahashi, M.; Okada, M.; Lin, C.-Y.; Chin, K.; Kadowaki, S.; Ahn, M.-J.; Hamamoto, Y.; Doki, Y.; et al. Nivolumab versus Chemotherapy in Patients with Advanced Oesophageal Squamous Cell Carcinoma Refractory or Intolerant to Previous Chemotherapy (ATTRACTION-3): A Multicentre, Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2019, 20, 1506–1517. [Google Scholar] [CrossRef]
- Li, S.; Hoefnagel, S.J.M.; Krishnadath, K.K. Molecular Biology and Clinical Management of Esophageal Adenocarcinoma. Cancers 2023, 15, 5410. [Google Scholar] [CrossRef]
- Baretton, G.B.; Lordick, F.; Gaiser, T.; Hofheinz, R.; Horst, D.; Lorenzen, S.; Moehler, M.; Röcken, C.; Schirmacher, P.; Stahl, M.; et al. Standardized and Quality-Assured Predictive PD-L1 Testing in the Upper Gastrointestinal Tract. J. Cancer Res. Clin. Oncol. 2023, 149, 16231–16238. [Google Scholar] [CrossRef]
- Zhao, N.; Zhang, Z.; Wang, Q.; Li, L.; Wei, Z.; Chen, H.; Zhou, M.; Liu, Z.; Su, J. DNA Damage Repair Profiling of Esophageal Squamous Cell Carcinoma Uncovers Clinically Relevant Molecular Subtypes with Distinct Prognoses and Therapeutic Vulnerabilities. EBioMedicine 2023, 96, 104801. [Google Scholar] [CrossRef]
- Reynolds, J.V.; Preston, S.R.; O’Neill, B.; Lowery, M.A.; Baeksgaard, L.; Crosby, T.; Cunningham, M.; Cuffe, S.; Griffiths, G.O.; Parker, I.; et al. Trimodality Therapy versus Perioperative Chemotherapy in the Management of Locally Advanced Adenocarcinoma of the Oesophagus and Oesophagogastric Junction (Neo-AEGIS): An Open-Label, Randomised, Phase 3 Trial. Lancet Gastroenterol. Hepatol. 2023, 8, 1015–1027. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author/Year | Type of Cancer | Population | Concentration/Duration of Metformin Treatment | Results | Ref. |
|---|---|---|---|---|---|
| Lee et al. (2011) | EC, GC, CRC, HCC, PC | 480,984 adult Taiwanese participants with T2DM vs. 417,844 non-DM controls | Mean metformin dosage was expressed in daily 500 mg units | ↓ CRC and HCC incidences, depending on gender and cancer type (CRC in women, HCC in men), metformin HRs (95% CI): total 0.12 (0.08–0.19), CRC 0.36 (0.13–0.98), HCC 0.06 (0.02–0.16), PC 0.15 (0.03–0.79)], metformin dosage for a significant decrease in cancer incidence was ≤500 mg/day. | [33] |
| Tseng et al. (2017) | EC | 288,013 metformin-treated T2DM Taiwanese adults vs. 16,216 other antidiabetic-drug-treated T2DM Taiwanese adults | Duration of metformin ≥ 2 years | ↓ EC [HR (95% CI) 0.487 (0.347–0.684)] | [34] |
| Becker et al. (2013) | EC | All EC-T2DM patients in the GPRD (40–89 years of age, from1994–2010) vs. EC-free T2DM controls (up to 10 controls for each case) | Long-term (≥30 prescriptions) use | Not associated with a materially altered risk of esophageal cancer (adj. OR 1.23, 95% CI 0.92–1.65) | [35] |
| Wang et al. (2020) | ESCC Swedish | 411,603 T2DM adults vs. 4,116,030 non-T2DM controls | Long-term or 1-year use | ↓ ESCC [HR 0.68, 95% CI 0.54–0.85], especially in new-metformin users | [36] |
| Loomans-Kropp et al. (2021) | EAC | 1943 EAC cases vs. 19,430 controls | ≥2 prescriptions in the same drug category on different days and drug use must have occurred prior to study selection | Metformin use alone showed significant ↓ EAC risk among all participants [ OR 0.65; 95% CI 0.50, 0.82)] and those without BE [OR 0.99; 95% CI 0.28, 3.46] | [37] |
| Chen et al. (2020) | EC | Meta-analysis of 7 studies with 5,426,343 subjects | NA | ↓ EC [HR = 0.69, 95% CI 0.54 to 0.87, p < 0.001] | [38] |
| Wu et al. (2020) | EC | Meta-analysis of 5 studies | NA | Metformin did not reduce EC risk in T2DM patients (HR 0.88, 95% CI 0.60–1.28, p > 0.05). Subgroup analyses by geographic location showed that metformin ↓ EC in Asian patients withT2DM (HR 0.59, 95% CI 0.39–0.91, p = 0.02), without heterogeneity between studies (p = 0.80 and I2 = 0%). | [39] |
| Author/Year | Type of Cancer | Population | Concentration/Duration of Metformin Treatment | Results | Ref. |
|---|---|---|---|---|---|
| Wang et al. (2023) | EC | T2DM + no metformin (n = 379), no T2DM + no metformin (n = 3999), T2DM + metformin (n = 473) | Any dose | ↓ all-cause mortality in non-T2DM patients and metformin-T2DM patients, ↓ HRs of all-cause mortality with a higher daily dose of metformin (Ptrend = 0.04) | [40] |
| Skinner et al. (2013) | EAC | 286 EAC patients treated with concurrent CRT followed by esophagectomy (29 T2DM + metformin patients, 21 T2DM + no metformin patients, 235 non-T2DM) | Any dose | ↑ CR rate in T2DM + metformin patients vs. T2DM + no metformin patients (34.5% vs. 4.8%, p = 0.01) and vs. non-T2DM (19.6%, p = 0.05), ↑ CR rate with ≥ 1500 mg/d metfromin, ↓ in field loco-regional failure following radiation (p = 0.05) | [41] |
| Spierings et al. (2015) | EC | 461 EC patients treated with concurrent CRT followed by esophagectomy (32 T2DM + metformin patients) | Any dose | No differences in pathological response rates or overall survival or disease-free survival between meformin to non-metformin users | [42] |
| Voorde et al. (2015) | EC | 196 EC adult patients (19 T2DM + metformin patients, 5 T2DM + no metformin patients, 172 non-T2DM) | Any dose | ↑ distant metastasis-free survival rate (p = 0.040), ↑ overall survival rate (p = 0.012), ↑ survival (p = 0.043) | [43] |
| Sakamoto et al. (2022) | EC, rectal cancer | meta-analysis of 5 studies with 2041 patients | NA | ↑ pCR rate (OR= 0.51 [0.34–0.76], p < 0.01), a positive correlation of metformin with EAC (coefficient = 0.13 [0.02–0.25], p = 0.03) and fluoropyrimidine anticancer drug use (coefficient = 0.01 [0.001–0.02], p = 0.03). | [44] |
| Author/Year | Type of Cancer | Population | Concentration/Duration of Metformin Treatment | Results | Ref. |
|---|---|---|---|---|---|
| Arai et al. (2022) | EC | 308,793 patients (1911 ESCC, 195 EAC) and 306,687 non-EC patients | Any dose | ↓ risk of ESCC (aOR 0.42, p < 0.0001) | [45] |
| Chak et al. (2015) | BE | 74 subjects with BE | Randomly assigned to groups given metformin daily (increasing to 2000 mg/day by week 4, n = 38) or placebo (n = 36) for 12 weeks. | No differences in esophageal levels of pS6K1or epithelial proliferation or apoptosis in esophageal tissues. | [46] |
| Agrawal et al. (2014) | BE, EC | 583 patients (115 EAC, 468 BE) | Any dose | No protective effect of metformin | [47] |
| Antonowicz et al. (2021) | EAC | Cell lines: FLO-1, OACM5.1, ESO26, KYAE-1, OE33, CPA, CPB, CPD | NA | ↑ short-chain alkanals and medium-chain alkanals -> ↓ genotoxicity | [48] |
| Author/Year | Type of EC/Cell Line/Animal Model | In Vitro/In Vivo | Outcomes | Ref. |
|---|---|---|---|---|
| Xu et al. (2013) | ESCC/Eca-109, TE-1 cells | In Vitro | ↑ cells in the G1/G0 phase ↓ cells in the S phase ↑ p21/p27 ↓ cyclin D1 expression activation of AMP/inhibition of mTOR signaling ↑ USP7 mRNA | [49] |
| Kobayashi et al. (2013) | ESCC/T.T, KYSE30,KYSE70 | In Vitro | ↑ cells in the G1/G0 phase ↓ cyclin D1/Cdk4/Cdk6/Rb expression | [50] |
| Damelin et al. (2014) | ESCC/WHCO1, WHCO5, SNO | In Vitro | ↑ cells in the G1/G0 phase | [51] |
| Cai et al. (2015) | ESCC/EC109,EC9706/8-week-old male athymic nude mice | In Vitro/In Vivo | ↑ cells in the G1/G0 phase, ↑ p53/p21/p27 ↓ cyclin D1 expression | [52] |
| Fujihara et al. (2015) | EAC/OE19, OE33, SK-GT4, OACM 5.1C/6-week-old male athymic nude mice | In Vitro/In Vivo | ↑ cells in the G1/G0 phase ↓ cyclin D1/Cdk4/Cdk6/Rb expression ↓ p-EGFR/p-IGF-1R/ErbB2/ErbB3/insulin-R/PYK ↓ VEGF/TIMP-1/TIMP-2 | [53] |
| Author/Year | Type of EC/Cell Line/Animal Model | In Vitro/In Vivo | Outcomes | Ref. |
|---|---|---|---|---|
| Feng et al. (2014) | ESCC/EC109, EC9706/5–6-week-old female nude mice | In Vivo and In Vitro | Inhibition of ESCC cell growth, depolarization of the mitochondrial membrane, ↑ caspase-cleaved PARP/the ratio of Bax to Bcl-2 proteins, ↓ cellular proliferation represented by pH3 IHC ↑ AVOs, ↑ Beclin-1 ↓ p62 inhibition of Stat3/Bcl-2 pathway in an AMPK-dependent and -independent manner ↓ the growth of cultured ESCC cells -> ↓ tumor size and weight, inactivation of Stat3/Bcl-2 activity in vivo | [54] |
| Tang et al. (2017) | ESCC/Eca109, EC9706/6-week- old nude mice | In Vivo and In Vitro | ↓ PI3K/AKT/mTOR pathway | [57] |
| Wang et al. (2017) | ESCC/KYSE450/4–6-week-old male nude mice | In Vivo and In Vitro | ↓ 4EBP1/S6K1/p-4EBP1/p-S6K1 | [58] |
| Li et al. (2017) | ESCC/KYSE520, KYSE140, KYSE410, KYSE30, KYSE150, KYSE510/4–5-week-old female nude mice | In Vivo and In Vitro | ↑ cells in the G1/G0 phase, ↑ p21 ↓ cyclin D1 expression induction of mitochondrion-dependent apoptosis in ESCC cells, ↑ intracellular glutathione level/ROS | [59] |
| Shafaee et al. (2019) | ESCC/TE1, TE8, TE11 | In Vitro | ↑ p53 ↓ Bcl-2 expression | [56] |
| Peng et al. (2020) | ESCC/EC109 | In Vitro | ↓ of the growth of EC109 cells induction of apoptosis in ESCC cells ↓ phosphorylation of Stat3/ Bcl-2 pathway | [55] |
| Hong et al. (2022) | EAC/SK-GT4, FLO 1/4-week-old female mice | In Vivo and In Vitro | ↓ AXL expression -> ↓ p-AMPK/p-ULK1/LC3B-II ↓ of tumor growth | [60] |
| Author/Year | Type of EC/Cell Line/Animal Model | In Vitro/In Vivo | Outcomes | Ref. |
|---|---|---|---|---|
| Kobayashi et al. (2013) | ESCC/KYSE30 | In Vitro | ↑ 17 miRNAs ↓ 45 miRNAs | [50] |
| Isozaki et al. (2014) | ESCC/TE2, T.Tn | In Vitro | Arrest of the G0/G1 and G2/M phases of the cell cycle, regulation of [67] the miR-375-targeted genes | [67] |
| Wang et al. (2019) | ESCC/KYSE510, KYSE140 | In Vitro | Induction of human ESCC pyroptosis by targeting the miR-497/PELP1 axis | [69] |
| Liu et al. (2019) | ESCC/CE81T, TE1/6–8-week-old male NOD/SCID mice | In Vitro and In Vivo | ↑ glycolysis and oxidative phosphorylation via the Hsp27–AKT–HK2 pathway | [70] |
| Chen et al. (2021) | ESCC/Te13, Te1, Eca109/BALB/c nude mice | In Vitro and In Vivo | ↓ miR-340-5p in hypoxic exosomes -> ↑ the expression of KLF10 -> ↑ radiosensitivity | [68] |
| Fujihara et al. (2015) | EAC/OE19, OE33, SK-GT4, OACM 5.1C/6-week-old male athymic nude mice | In Vitro and In Vivo | ↑ 3 miRNAs ↓ 10 miRNAs | [53] |
| Author/Year | Drug Combination | Type of EC/Cell Line/Animal Model | In Vitro/In Vivo | Outcomes | Ref. |
|---|---|---|---|---|---|
| Damelin et al. (2014) | cisplatin | ESCC/WHCO1, WHCO5, SNO | In Vitro | ↑ glycolysis | [51] |
| intracellular NAD(P)H levels + ↓ intracellular thiols -> ↓ cisplatin-DNA adduct formation | |||||
| Yu et al. (2016) | cisplatin | ESCC/ECA109 | In Vitro | glucose-deprivation conditions -> ↑ metformin-associated cytotoxicity | [76] |
| ↓ cellular ATP levels | |||||
| ↓ AKT and AMPK signaling pathways -> ↓ DNA repair function -> ↑ cisplatin cytotoxicity | |||||
| Wang et al. (2017) | cisplatin | ESCC/KYSE450/4–6-week-old male nude mice | In Vivo and In Vitro | ↑↑↑ anti-proliferation effects | [58] |
| Honjo et al. (2014) | 5-fluorouracil (5-FU) | EAC/FLO-1, BE3,SKGT-4, OE33,JHESO, OACP/nude mice | In Vivo and In Vitro | ↓ PI3K/mTOR signaling | [77] |
| ↓ CSCs -> inhibits EC cell proliferation | |||||
| Honjo et al. (2014) | 5-fluorouracil (5-FU) | ESCC/YES-6, KATO-TN/nude mice | In Vivo and In Vitro | ↓ PI3K/mTOR signaling | [77] |
| ↓ CSCs -> inhibits EC cell proliferation | |||||
| Feng et al. (2015) | IR | ESCC/ECa109 | In Vitro | ↑ cells undergoing G0/G1 cell cycle -> ↓ CDK4 and cyclin D1 | [78] |
| targeting the ATM and AMPK/mTOR/HIF-1a pathways -> ↓ HIF-1a -> ↑ radiosensitivity | |||||
| Nakayama et al. (2016) | IR | ESCC/TE-9 | In Vitro | ↓ TGF-β-Smad phosphorylation pathway, and a part of the non-Smad pathway -> suppression of IR-induced EMT | [79] |
| Chen et al. (2021) | IR | ESCC/Te13, Te1, Eca109/BALB/c nude mice | In Vivo and In Vitro | ↓ miR-340-5p in hypoxic exosomes -> ↑ the expression of KLF10 -> ↑ radiosensitivity | [68] |
| Jivan et al. (2015) | disulfiram (DSF), copper-DSF(Cu-DSF) | ESCC/WHCO1, WHCO5, SNO | In Vitro | ↑ copper transport -> ↑ DSF cytotoxicity | [80] |
| Qie et al. (2019) | CB-839 (glutaminase 1 inhibitor) | ESCC/TE1, TE7, TE8, TE10, TE15/4-week-old male athymic mice | In Vivo and In Vitro | direct mutation, or loss of regulatory E3 ubiquitin ligase Fbxo4 -> ↑↑ cyclin D1 -> Gln addiction -> induction of apoptosis and suppression of cell proliferation | [85] |
| Shafaee et al. (2019) | 2-deoxy-d-glucose (2DG) | ESCC/TE1, TE8, TE11 | In Vitro | ↑ p53 ↓ Bcl-2 expression | [56] |
| Wang et al. (2022) | 3-aminobenzamide (3-ABA) | ESCC/KYSE450, TE-10/5-week old BALB/c male nude mice | In Vivo and In Vitro | ↑ the suppressive effect of 3-ABA on ESCC cell growth | [86] |
| ↑ 51 proteins + ↓ 12proteins localised within the nucleus/cytoplasm/extracellular space -> ↓ growth of ESSC cells/↓ invasiveness |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papadakos, S.P.; Argyrou, A.; Lekakis, V.; Arvanitakis, K.; Kalisperati, P.; Stergiou, I.E.; Konstantinidis, I.; Schizas, D.; Koufakis, T.; Germanidis, G.; et al. Metformin in Esophageal Carcinoma: Exploring Molecular Mechanisms and Therapeutic Insights. Int. J. Mol. Sci. 2024, 25, 2978. https://doi.org/10.3390/ijms25052978
Papadakos SP, Argyrou A, Lekakis V, Arvanitakis K, Kalisperati P, Stergiou IE, Konstantinidis I, Schizas D, Koufakis T, Germanidis G, et al. Metformin in Esophageal Carcinoma: Exploring Molecular Mechanisms and Therapeutic Insights. International Journal of Molecular Sciences. 2024; 25(5):2978. https://doi.org/10.3390/ijms25052978
Chicago/Turabian StylePapadakos, Stavros P., Alexandra Argyrou, Vasileios Lekakis, Konstantinos Arvanitakis, Polyxeni Kalisperati, Ioanna E. Stergiou, Ippokratis Konstantinidis, Dimitrios Schizas, Theocharis Koufakis, Georgios Germanidis, and et al. 2024. "Metformin in Esophageal Carcinoma: Exploring Molecular Mechanisms and Therapeutic Insights" International Journal of Molecular Sciences 25, no. 5: 2978. https://doi.org/10.3390/ijms25052978