
Identification of Gene–Allele System Conferring Alkali-Tolerance at Seedling Stage in Northeast China Soybean Germplasm
 ,
,
Abstract
:1. Introduction
2. Results
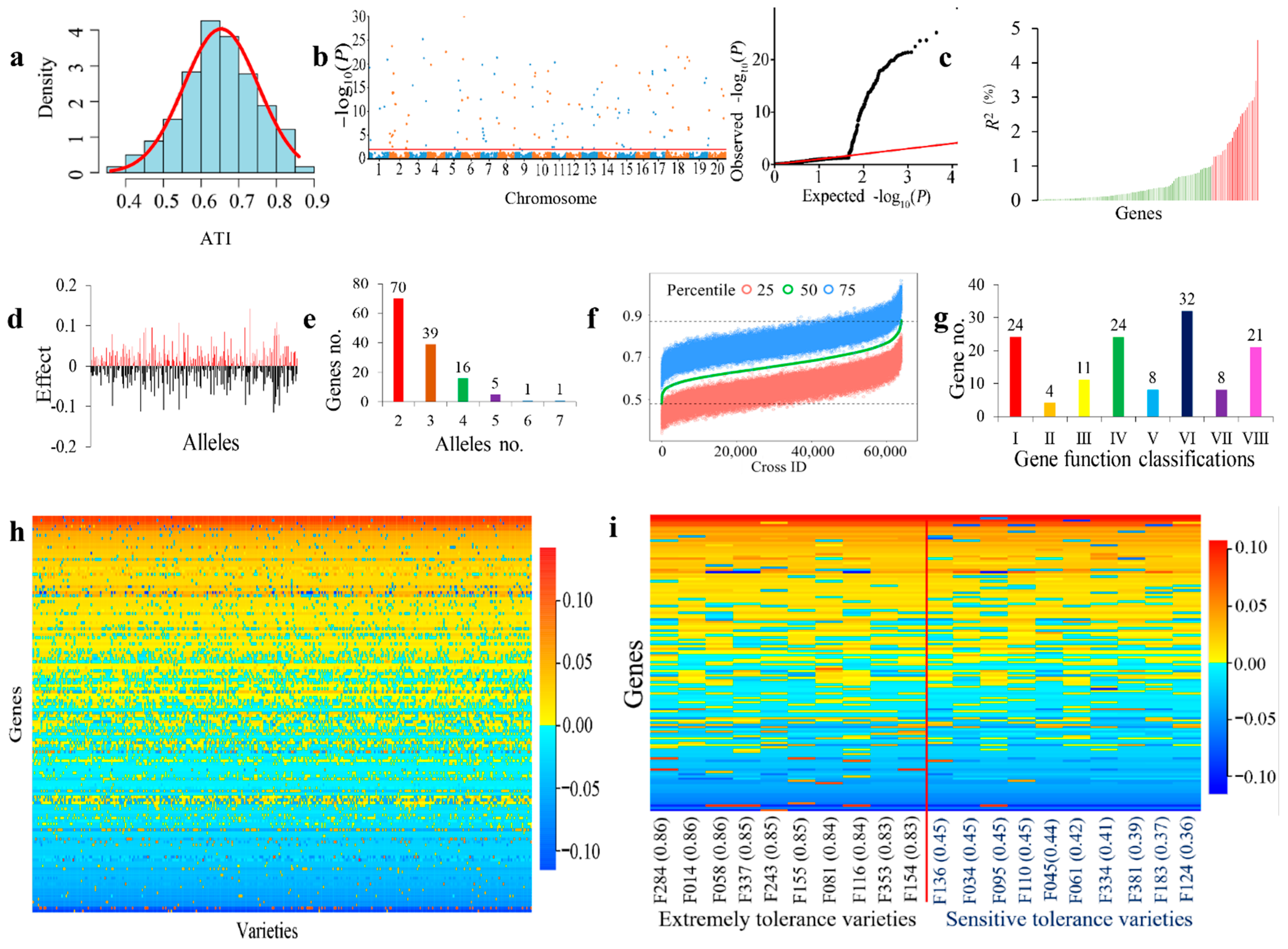
2.1. Wide Variation of Alkali-Tolerance in the NECSGP
2.2. Gene–Allele System of Alkali-Tolerance in the NECSGP
2.3. ATI Gene–Allele Matrix as a Compact Genetic Structure of the NECSGP
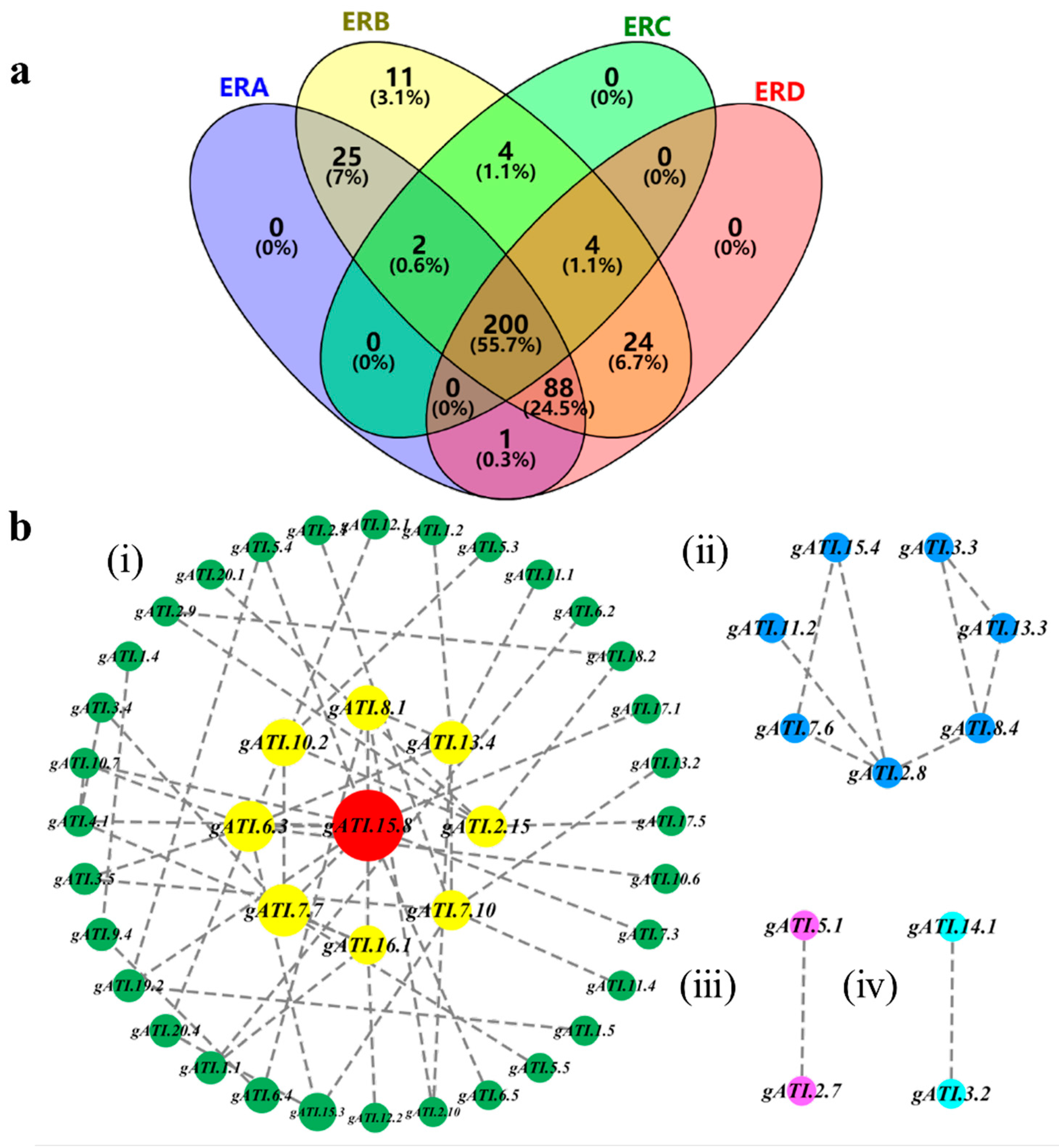
2.4. Population Genetic Differentiation among the Four Subregions
2.5. Recombination Potential and Prediction of Optimal Crosses for Alkali-Tolerance Improvement in the NECSGP
2.6. The Candidate Gene System of Alkali-Tolerance Inferred from Identified GASMs
3. Discussion
3.1. Advances in Exploring the Genetic System of Alkali-Tolerance
3.2. The Main Genetic Motivator of Alkali-Tolerance in the NECSGP by Using the GASM-RTM-GWAS Procedure
3.3. Breeding Strategy for Alkali-Tolerance Improvement in the NECSGP
4. Materials and Methods
4.1. Plant Materials
4.2. Experimental Design and Evaluation of ATI at the Seedling Stage
4.3. Method of Statistical Analysis
4.4. Genotyping and Assembly of GASM Markers
4.5. Identification of the ATI Gene–Allele System Using GASM-RTM-GWAS
4.6. Recombination Potential Prediction and Optimal Cross Design for ATI in the NECSGP
4.7. Functional Annotation and Interaction of Candidate Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vavilov, N.I.; Vavylov, M.I.; Dorofeev, V.F. Origin and Geography of Cultivated Plants; Cambridge University Press: Cambridge, UK, 1992. [Google Scholar]
- Liu, X.; He, J.; Wang, Y.; Xing, G.; Li, Y.; Yang, S.; Zhao, T.; Gai, J. Geographic differentiation and phylogeographic relationships among world soybean populations. Crop J. 2020, 8, 260–272. [Google Scholar] [CrossRef]
- Wysmierski, P.T.; Vello, N.A. The genetic base of Brazilian soybean cultivars: Evolution over time and breeding implications. Genet. Mol. Biol. 2013, 36, 547–555. [Google Scholar] [CrossRef]
- Gizlice, Z.; Carter, T., Jr.; Burton, J. Genetic base for North American public soybean cultivars released between 1947 and 1988. Crop Sci. 1994, 34, 1143–1151. [Google Scholar] [CrossRef]
- Pitman, M.G.; Läuchli, A. Global impact of salinity and agricultural ecosystems. In Salinity: Environment-Plants-Molecules; Springer: Dordrecht, The Netherlands, 2002; pp. 3–20. [Google Scholar]
- Tuyen, D.D.; Lal, S.K.; Xu, D.H. Identification of a major QTL allele from wild soybean (Glycine soja Sieb. & Zucc.) for increasing alkaline salt tolerance in soybean. Theor. Appl. Genet. 2010, 121, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.; Yang, J.; Liu, G. Northeast China; Saline-alkalized land; Agro-biological treatments. Soil 2006, 38, 256–262. [Google Scholar]
- Zhu, J. Plant salt tolerance. Trends Plant Sci. 2001, 6, 66–71. [Google Scholar] [CrossRef]
- Luo, Q.; Yu, B.; Liu, Y. Differential sensitivity to chloride and sodium ions in seedlings of Glycine max and G. soja under NaCl stress. J. Plant Physiol. 2005, 162, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Zong, C. Gene-Allele Systems and Breeding Potential Prediction of Saline and Alkaline Tolerance at Seedling Stage in the Northeast China Soybean Germplasm Population. Ph.D. Thesis, Nanjing Agricultural University, Nanjing, China, 2022. [Google Scholar]
- Wang, H.; Wu, Z.; Han, J.; Zheng, W.; Yang, C. Comparison of ion balance and nitrogen metabolism in old and young leaves of alkali-stressed rice plants. PLoS ONE 2012, 7, e37817. [Google Scholar] [CrossRef] [PubMed]
- Parihar, P.; Singh, S.; Singh, R.; Singh, V.P.; Prasad, S.M. Effect of salinity stress on plants and its tolerance strategies: A review. Environ. Sci. Pollut. Res. 2015, 22, 4056–4075. [Google Scholar] [CrossRef]
- Rogovska, N.P.; Blackmer, A.M.; Mallarino, A.P. Relationships between soybean yield, soil pH, and soil carbonate concentration. Soil Sci. Soc. Am. J. 2007, 71, 1251–1256. [Google Scholar] [CrossRef]
- Yi, S.; Sun, Z.; Han, L.; Ju, G.; Qian, Y. Research Advance on Alkali-resistant Mechanism of Plants and Related Genes. World For. Res. 2011, 24, 28–32. [Google Scholar] [CrossRef]
- Hyten, D.L.; Song, Q.; Choi, I.-Y.; Yoon, M.-S.; Specht, J.E.; Matukumalli, L.K.; Nelson, R.L.; Shoemaker, R.C.; Young, N.D.; Cregan, P.B. High-throughput genotyping with the GoldenGate assay in the complex genome of soybean. Theor. Appl. Genet. 2008, 116, 945–952. [Google Scholar] [CrossRef]
- Sonah, H.; O’Donoughue, L.; Cober, E.; Rajcan, I.; Belzile, F. Identification of loci governing eight agronomic traits using a GBS-GWAS approach and validation by QTL mapping in soya bean. Plant Biotechnol. J. 2015, 13, 211–221. [Google Scholar] [CrossRef]
- Zhou, Z.; Jiang, Y.; Wang, Z.; Gou, Z.; Lyu, J.; Li, W.; Yu, Y.; Shu, L.; Zhao, Y.; Ma, Y. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 2015, 33, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Kan, G.; Hu, Z.; Cheng, H.; Zhang, Y.; Wang, Q.; Wang, H.; Yang, Y.; Li, H.; Hao, D. Use of single nucleotide polymorphisms and haplotypes to identify genomic regions associated with protein content and water-soluble protein content in soybean. Theor. Appl. Genet. 2014, 127, 1905–1915. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Tan, R.; Yuan, J.; Bales, C.; Du, W.; Zhang, S.; Chilvers, M.I.; Schmidt, C.; Song, Q.; Cregan, P.B. Genome-wide association mapping of quantitative resistance to sudden death syndrome in soybean. BMC Genom. 2014, 15, 809. [Google Scholar] [CrossRef] [PubMed]
- Dhanapal, A.P.; Ray, J.D.; Singh, S.K.; Hoyos-Villegas, V.; Smith, J.R.; Purcell, L.C.; Andy King, C.; Cregan, P.B.; Song, Q.; Fritschi, F.B. Genome-wide association study (GWAS) of carbon isotope ratio (δ 13 C) in diverse soybean [Glycine max (L.) Merr.] genotypes. Theor. Appl. Genet. 2015, 128, 73–91. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Meng, S.; Zhao, T.; Xing, G.; Yang, S.; Li, Y.; Guan, R.; Lu, J.; Wang, Y.; Xia, Q.; et al. An innovative procedure of genome-wide association analysis fits studies on germplasm population and plant breeding. Theor. Appl. Genet. 2017, 130, 2327. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Zhang, Z.; He, J.; Zeng, W.; Cai, Z.; Lai, Z.; Pan, Y.; Hao, X.; Xing, G.; Wang, W.; et al. Gene–allele system of shade tolerance in southern China soybean germplasm revealed by genome-wide association study using gene–allele sequence as markers. Theor. Appl. Genet. 2023, 136, 152. [Google Scholar] [CrossRef] [PubMed]
- Bolon, Y.; Joseph, B.; Cannon, S.B.; Graham, M.A.; Diers, B.W.; Farmer, A.D.; May, G.D.; Muehlbauer, G.J.; Specht, J.E.; Tu, Z.J. Complementary genetic and genomic approaches help characterize the linkage group I seed protein QTL in soybean. BMC Plant Biol. 2010, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Simulation modeling in plant breeding: Principles and applications. Agric. Sci. China 2007, 6, 908–921. [Google Scholar] [CrossRef]
- Peleman, J.D.; Jeroen Rouppe, V.d.V. Breeding by design. Trends Plant Sci. 2003, 8, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Wan, J. Perspectives of Molecular Design Breeding in Crops. Acta Agron. Sin. 2006, 32, 455–462. [Google Scholar]
- Wang, J.; Wan, X.; Crossa, J.; Crouch, J.; Weng, J.; Zhai, H.; Wan, J. QTL mapping of grain length in rice (Oryza sativa L.) using chromosome segment substitution lines. Genet. Res. 2006, 88, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Gai, J.; Chen, L.; Zhang, Y.; Zhao, T.; Xing, G.; Xing, H. Genome-wide genetic dissection of germplasm resources and implications for breeding by design in soybean. Breed. Sci. 2012, 61, 495–510. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. Genetic Dissection of Seed Traits of the Chinese Soybean Landrace Population and Its Utilization in Breeding by Design. Ph.D. Thesis, Nanjing Agricultural University, Nanjing, China, 2014. [Google Scholar]
- Abel, G.H. Inheritance of the Capacity for Chloride Inclusion and Chloride Exclusion by Soybeans. Crop Sci. 1969, 9, 697–698. [Google Scholar] [CrossRef]
- Lam, H.; Chang, R.; Shao, G.; Liu, Z.T. Research on Tolerance to Stresses in Chinese Soybeans; China Agricultue Press: Beijing, China, 2009. [Google Scholar]
- Tuyen, D.; Zhang, H.; Xu, D. Validation and high-resolution mapping of a major quantitative trait locus for alkaline salt tolerance in soybean using residual heterozygous line. Mol. Breed. 2013, 31, 79–86. [Google Scholar] [CrossRef]
- Zhang, J. Transcriptomic Analysis of Response to NaHCO3 and QTL Mapping of NaHCO3 Tolerance Related Traits in Soybean. Ph.D. Thesis, Nanjing Agricultural University, Nanjing, China, 2016. [Google Scholar]
- Zhang, W.J.; Niu, Y.; Bu, S.H.; Li, M.; Feng, J.Y.; Zhang, J.; Yang, S.X.; Odinga, M.M.; Wei, S.P.; Liu, X.F.; et al. Epistatic association mapping for alkaline and salinity tolerance traits in the soybean germination stage. PLoS ONE 2014, 9, e84750. [Google Scholar] [CrossRef]
- Fu, M.; Wang, Y.; Ren, H.; Du, W.; Wang, D.; Bao, R.; Yang, X.; Tian, Z.; Fu, L.; Cheng, Y. Genetic dynamics of earlier maturity group emergence in south-to-north extension of Northeast China soybeans. Theor. Appl. Genet. 2020, 133, 1839–1857. [Google Scholar] [CrossRef]
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef]
- Hanson, C.; Robinson, H.; Comstock, R. Biometrical studies of yield in segregating populations of Korean lespedeza. Agron. J. 1956, 48, 268–272. [Google Scholar] [CrossRef]
- Miller, M.R.; Dunham, J.P.; Amores, A.; Cresko, W.A.; Johnson, E.A. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 2007, 17, 240–248. [Google Scholar] [CrossRef]
- Murray, M.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4326. [Google Scholar] [CrossRef]
- Andolfatto, P.; Davison, D.; Erezyilmaz, D.; Hu, T.T.; Mast, J.; Sunayama-Morita, T.; Stern, D.L. Multiplexed shotgun genotyping for rapid and efficient genetic mapping. Genome Res. 2011, 21, 610–617. [Google Scholar] [CrossRef]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef]
- Li, R.; Yu, C.; Li, Y.; Lam, T.-W.; Yiu, S.-M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Liang, Y.; Huerta-Sanchez, E.; Jin, X.; Cuo, Z.X.P.; Pool, J.E.; Xu, X.; Jiang, H.; Vinckenbosch, N.; Korneliussen, T.S. Sequencing of 50 human exomes reveals adaptation to high altitude. Science 2010, 329, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Scheet, P.; Stephens, M. A fast and flexible statistical model for large-scale population genotype data: Applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 2006, 78, 629–644. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| ENV. † | Mid-Point Classic | SUM | Mean | Range | GCV ‡ (%) | h2 § (%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.38 | 0.43 | 0.48 | 0.53 | 0.58 | 0.63 | 0.68 | 0.73 | 0.78 | 0.83 | 0.88 | ||||||
| Total | 3 | 9 | 16 | 27 | 48 | 50 | 85 | 48 | 48 | 19 | 7 | 361 | 0.65 | 0.36~0.86 | 19.96 | 98.43 |
| ERA | 3 | 3 | 9 | 10 | 16 | 7 | 10 | 2 | 1 | 61 | 0.64 a * | 0.46~0.86 | 12.97 | 94.62 | ||
| ERB | 1 | 6 | 6 | 12 | 30 | 24 | 58 | 30 | 33 | 16 | 14 | 230 | 0.66 a | 0.46~0.86 | 14.83 | 96.57 |
| ERC | 1 | 2 | 1 | 1 | 1 | 2 | 8 | 0.69 a | 0.37~0.83 | 21.33 | 93.71 | |||||
| ERD | 1 | 3 | 2 | 4 | 11 | 5 | 14 | 7 | 8 | 3 | 4 | 62 | 0.64 | 0.39~0.85 | 16.95 | 95.36 |
| Gene Code | Gene Name | AN | R2 (%) | Gene Code | Gene Name | AN | R2 (%) | ||
|---|---|---|---|---|---|---|---|---|---|
| Main Effect | G × E | Main Effect | G × E | ||||||
| gATI.1.1 | Glyma01g02580 | 2 | 1.32 | gATI.10.8 | Glyma10g37420 | 2 | 0.56 | 0.04 | |
| gATI.1.2 | Glyma01g04515 | 3 | 0.09 | gATI.11.1 | Glyma11g01253 | 3 | 0.05 | ||
| gATI.1.3 | Glyma01g22861 | 2 | 0.19 | gATI.11.2 | Glyma11g03580 | 2 | 0.11 | ||
| gATI.1.4 | Glyma01g30320 | 2 | 0.28 | gATI.11.3 | Glyma11g07830 | 4 | 0.71 | ||
| gATI.1.5 | Glyma01g36070 | 2 | 1.69 | gATI.11.4 | Glyma11g15140 | 2 | 0.01 | 0.05 | |
| gATI.2.1 | Glyma02g00280 | 3 | 0.05 | 0.04 | gATI.11.5 | Glyma11g27510 | 2 | 0.1 | |
| gATI.2.2 | Glyma02g05640 | 3 | 0.08 | gATI.12.1 | Glyma12g03180 | 3 | 0.10 | ||
| gATI.2.3 | Glyma02g06730 | 5 | 3.48 | 0.07 | gATI.12.2 | Glyma12g08010 | 2 | 0.26 | |
| gATI.2.4 | Glyma02g08790 | 2 | 0.03 | gATI.12.3 | Glyma12g30080 | 3 | 0.24 | ||
| gATI.2.5 | Glyma02g09130 | 3 | 0.70 | gATI.13.1 | Glyma13g00490 | 3 | 0.38 | 0.04 | |
| gATI.2.6 | Glyma02g09240 | 3 | 0.41 | gATI.13.2 | Glyma13g01900 | 2 | 0.04 | ||
| gATI.2.7 | Glyma02g11151 | 4 | 0.78 | 0.08 | gATI.13.3 | Glyma13g23910 | 3 | 0.64 | |
| gATI.2.8 | Glyma02g11335 | 4 | 1.28 | gATI.13.4 | Glyma13g29225 | 3 | 0.12 | ||
| gATI.2.9 | Glyma02g14175 | 2 | 0.12 | gATI.13.5 | Glyma13g29360 | 2 | 0.45 | 0.02 | |
| gATI.2.10 | Glyma02g15470 | 3 | 0.28 | gATI.13.6 | Glyma13g29520 | 2 | 0.04 | ||
| gATI.2.11 | Glyma02g16850 | 3 | 0.39 | gATI.13.7 | Glyma13g40690 | 2 | 1.03 | ||
| gATI.2.12 | Glyma02g37010 | 2 | 0.05 | gATI.13.8 | Glyma13g42650 | 3 | 0.24 | 0.04 | |
| gATI.2.13 | Glyma02g38673 | 3 | 0.19 | gATI.14.1 | Glyma14g04260 | 3 | 0.14 | ||
| gATI.2.14 | Glyma02g40220 | 3 | 0.06 | gATI.14.2 | Glyma14g10780 | 7 | 0.98 | ||
| gATI.2.15 | Glyma02g44350 | 2 | 0.24 | gATI.14.3 | Glyma14g20110 | 2 | 0.31 | ||
| gATI.3.1 | Glyma03g07890 | 2 | 0.74 | gATI.14.4 | Glyma14g36130 | 2 | 0.25 | ||
| gATI.3.2 | Glyma03g27770 | 2 | 2.23 | gATI.14.5 | Glyma14g37280 | 3 | 2.84 | 0.05 | |
| gATI.3.3 | Glyma03g30270 | 4 | 2.14 | 0.04 | gATI.14.6 | Glyma14g38720 | 2 | 0.05 | |
| gATI.3.4 | Glyma03g36720 | 2 | 0.04 | gATI.15.1 | Glyma15g02310 | 2 | 0.35 | ||
| gATI.3.5 | Glyma03g37221 | 2 | 0.33 | 0.08 | gATI.15.2 | Glyma15g04006 | 3 | 0.32 | |
| gATI.4.1 | Glyma04g10720 | 2 | 2.00 | gATI.15.3 | Glyma15g07590 | 3 | 0.33 | ||
| gATI.4.2 | Glyma04g43300 | 2 | 2.88 | gATI.15.4 | Glyma15g16830 | 2 | 0.03 | ||
| gATI.5.1 | Glyma05g19630 | 2 | 0.02 | 0.02 | gATI.15.5 | Glyma15g19900 | 2 | 0.03 | |
| gATI.5.2 | Glyma05g24760 | 3 | 0.09 | gATI.15.6 | Glyma15g27480 | 5 | 0.36 | ||
| gATI.5.3 | Glyma05g27300 | 2 | 0.94 | gATI.15.7 | Glyma15g27750 | 2 | 0.37 | ||
| gATI.5.4 | Glyma05g27690 | 5 | 0.20 | 0.08 | gATI.15.8 | Glyma15g32540 | 2 | 0.03 | |
| gATI.5.5 | Glyma05g32890 | 4 | 0.81 | gATI.16.1 | Glyma16g08960 | 2 | 1.43 | ||
| gATI.6.1 | Glyma06g01490 | 2 | 0.49 | 0.03 | gATI.16.2 | Glyma16g28270 | 3 | 0.91 | |
| gATI.6.2 | Glyma06g05300 | 3 | 1.82 | gATI.16.3 | Glyma16g32650 | 5 | 0.89 | ||
| gATI.6.3 | Glyma06g07980 | 3 | 4.66 | gATI.16.4 | Glyma16g33831 | 4 | 0.10 | ||
| gATI.6.4 | Glyma06g17410 | 2 | 0.08 | gATI.17.1 | Glyma17g07120 | 3 | 1.66 | ||
| gATI.6.5 | Glyma06g19756 | 2 | 0.20 | gATI.17.2 | Glyma17g08230 | 2 | 0.95 | ||
| gATI.6.6 | Glyma06g47010 | 4 | 0.14 | gATI.17.3 | Glyma17g10650 | 4 | 0.39 | ||
| gATI.7.1 | Glyma07g00400 | 4 | 0.73 | gATI.17.4 | Glyma17g15720 | 2 | 0.03 | ||
| gATI.7.2 | Glyma07g05620 | 4 | 2.45 | 0.03 | gATI.17.5 | Glyma17g16831 | 3 | 0.11 | 0.11 |
| gATI.7.3 | Glyma07g06640 | 2 | 0.10 | gATI.17.6 | Glyma17g33020 | 2 | 0.09 | ||
| gATI.7.4 | Glyma07g07360 | 6 | 0.12 | 0.05 | gATI.17.7 | Glyma17g33930 | 2 | 2.56 | |
| gATI.7.5 | Glyma07g08290 | 2 | 0.05 | gATI.17.8 | Glyma17g36130 | 2 | 1.46 | 0.08 | |
| gATI.7.6 | Glyma07g10280 | 2 | 0.08 | gATI.18.1 | Glyma18g01490 | 2 | 2.71 | ||
| gATI.7.7 | Glyma07g14234 | 2 | 0.06 | gATI.18.2 | Glyma18g03930 | 2 | 0.02 | ||
| gATI.7.8 | Glyma07g31130 | 3 | 1.48 | 0.12 | gATI.18.3 | Glyma18g03975 | 2 | 0.68 | |
| gATI.7.9 | Glyma07g37810 | 2 | 0.16 | gATI.18.4 | Glyma18g04870 | 2 | 0.35 | ||
| gATI.7.10 | Glyma07g38180 | 3 | 2.90 | gATI.18.5 | Glyma18g11512 | 4 | 0.15 | ||
| gATI.8.1 | Glyma08g04620 | 4 | 0.18 | gATI.18.6 | Glyma18g15001 | 3 | 0.03 | 0.09 | |
| gATI.8.2 | Glyma08g10001 | 3 | 0.02 | gATI.18.7 | Glyma18g16761 | 3 | 0.27 | ||
| gATI.8.3 | Glyma08g18320 | 2 | 0.38 | gATI.18.8 | Glyma18g16780 | 2 | 0.71 | ||
| gATI.8.4 | Glyma08g42810 | 4 | 0.05 | gATI.18.9 | Glyma18g28130 | 2 | 3.00 | ||
| gATI.8.5 | Glyma08g45301 | 3 | 0.65 | gATI.18.10 | Glyma18g40780 | 3 | 1.71 | ||
| gATI.8.6 | Glyma08g45501 | 2 | 0.13 | gATI.18.11 | Glyma18g46101 | 3 | 2.62 | ||
| gATI.8.7 | Glyma08g45610 | 2 | 0.72 | 0.15 | gATI.18.12 | Glyma18g49450 | 2 | 0.12 | |
| gATI.9.1 | Glyma09g12180 | 3 | 0.03 | gATI.18.13 | Glyma18g50670 | 4 | 0.17 | 0.04 | |
| gATI.9.2 | Glyma09g15860 | 2 | 0.03 | gATI.18.14 | Glyma18g53285 | 3 | 2.06 | 0.07 | |
| gATI.9.3 | Glyma09g33220 | 4 | 2.52 | 0.05 | gATI.19.1 | Glyma19g31900 | 4 | 1.28 | |
| gATI.9.4 | Glyma09g40690 | 3 | 0.68 | gATI.19.2 | Glyma19g35820 | 2 | 0.78 | ||
| gATI.9.5 | Glyma09g41821 | 3 | 0.38 | 0.03 | gATI.19.3 | Glyma19g42710 | 2 | 0.04 | |
| gATI.10.1 | Glyma10g06480 | 2 | 0.08 | gATI.19.4 | Glyma19g43880 | 2 | 0.06 | ||
| gATI.10.2 | Glyma10g06600 | 2 | 0.80 | gATI.20.1 | Glyma20g01460 | 2 | 0.22 | 0.01 | |
| gATI.10.3 | Glyma10g07601 | 2 | 1.32 | gATI.20.2 | Glyma20g24740 | 5 | 0.95 | 0.06 | |
| gATI.10.4 | Glyma10g14620 | 2 | 0.06 | gATI.20.3 | Glyma20g30360 | 2 | 0.97 | ||
| gATI.10.5 | Glyma10g30100 | 3 | 0.75 | 0.04 | gATI.20.4 | Glyma20g30870 | 2 | 0.17 | |
| gATI.10.6 | Glyma10g30320 | 2 | 0.27 | ||||||
| gATI.10.7 | Glyma10g31560 | 2 | 1.32 | 0.02 | Total | 132 | a 359 (2.72) | 90.94 | 2.80 |
| Gene | Main Effect % | G × E % |
| Significant | 90.94 (126, 0.01~4.66) | 2.80 (35, 0.01~0.15) |
| LC major Gene | 59.86 (28, 1.03~4.66) | |
| SC major Gene | 31.08 (98, 0.01~0.98) | 2.80 (35, 0.01~0.15) |
| Unmapped minor Gene | 7.49 | |
| Total genetic contribution h2 | 98.43 | 2.80 |
| Allele | Main Effect | G × E |
| Positive allele | 131 (0.0004~0.1420) | 52 (0.001~0.107) |
| Negative allele | 125 (−0.1150~−0.0010) | 51 (−0.108~−0.001) |
| Total | 256 (−0.1150~0.1420) | 103 (−0.108~0.107) |
| Contrast | Total Alleles | Alleles Changed | |||
|---|---|---|---|---|---|
| Inherited | Emerged/Introduced | Excluded | Changed | ||
| ERB vs. ERD | 358 (176, 182) vs. 317 (150, 167) | 316 (153, 163) | 42 (22, 20) | 1 (1, 0) | 43 (23, 20) |
| ERC vs. ERD | 210 (109, 101) vs. 317 (150, 167) | 204 (100, 104) | 6 (1, 5) | 113 (54, 59) | 119 (55, 64) |
| ERA vs. ERD | 316 (159, 157) vs. 317 (150, 167) | 289 (145, 144) | 27 (11, 16) | 28 (14, 14) | 55 (25, 30) |
| RA vs. ERB | 316 (159, 157) vs. 358 (176, 182) | 315 (156, 159) | 1 (1, 0) | 43 (19, 24) | 44 (20, 24) |
| ERC vs. ERB | 210 (109, 101) vs. 358 (176, 182) | 210 (101, 109) | 0 | 148 (74, 74) | 148 (74, 74) |
| Region/ Subregion | ATI Range of Accessions | No. of Predicted Crosses | Predicted via the Linkage Model | Predicted via the Independent Assortment Model | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean | Range | High Tolerance Cross No. (ATI > 0.86) | Percentage (%) | Mean | Range | High Tolerance Cross No. (ATI > 0.86) | Percentage (%) | |||
| Total | 0.36~0.86 | 64,980 | 0.75 | 0.47~1.03 | 4388 | 6.75 | 0.75 | 0.48~0.98 | 4580 | 7.05 |
| ERA | 0.46~0.86 | 2775 | 0.74 | 0.53~0.97 | 150 | 5.41 | 0.74 | 0.54~0.96 | 145 | 5.23 |
| ERB | 0.46~0.86 | 32,896 | 0.75 | 0.47~1.00 | 2096 | 6.37 | 0.76 | 0.48~0.98 | 2190 | 6.66 |
| ERC | 0.37~0.83 | 28 | 0.79 | 0.62~0.92 | 6 | 21.42 | 0.79 | 0.62~0.94 | 6 | 21.43 |
| ERD | 0.39~0.85 | 210 | 0.76 | 0.55~0.93 | 19 | 9.05 | 0.76 | 0.56~0.93 | 26 | 12.38 |
| P1 | P2 | Predicted Cross (75th Percentile) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Code | Name | MG | ATI | Code | Name | MG | ATI | Mean of Parents | SD (%) | Predicted ATI |
| F134 | Nenfeng 15 | I | 0.82 | F81 | Amsoy | I | 0.84 | 0.84 | 26.07 | 1.03 |
| F243 | Kenjian 35 | 0 | 0.85 | F81 | Amsoy | I | 0.84 | 0.85 | 21.48 | 1.00 |
| F243 | Kenjian 35 | 0 | 0.85 | F81 | Amsoy | I | 0.84 | 0.85 | 21.48 | 1.00 |
| F104 | Jiyu 93 | I | 0.81 | F284 | Mufeng No. 3 | 0 | 0.86 | 0.83 | 23.12 | 0.99 |
| F104 | Jiyu 93 | I | 0.81 | F134 | Nenfeng 15 | I | 0.82 | 0.82 | 25.49 | 0.99 |
| F104 | Jiyu 93 | I | 0.81 | F387 | Jiyu 86 | III | 0.83 | 0.82 | 23.11 | 0.99 |
| F134 | Nenfeng 15 | I | 0.82 | F284 | Mufeng No. 3 | 0 | 0.86 | 0.84 | 20.9 | 0.99 |
| F104 | Jiyu 93 | I | 0.81 | F243 | Kenjian 35 | 0 | 0.85 | 0.82 | 23.53 | 0.99 |
| F233 | Kennong 24 | I | 0.82 | F81 | Amsoy | I | 0.84 | 0.84 | 20.06 | 0.99 |
| F104 | Jiyu 93 | I | 0.81 | F135 | Nenfeng 17 | 0 | 0.81 | 0.82 | 24.08 | 0.99 |
| Gene Code | Gene Name | R2 | Superior Allele | Allele Effect | In Accessions and Parents | No. of Links | Gene Function and Classification |
|---|---|---|---|---|---|---|---|
| gATI.1.1 | Glyma01g02580 | 1.32 | gATI.1.1.a2 | 0.016 | √ | 2 | |
| gATI.1.5 | Glyma01g36070 | 1.69 | gATI.1.5.a1 | 0.024 | √ | 1 | |
| gATI.2.3 | Glyma02g06730 | 3.48 | gATI.2.3.a2 | 0.027 | √ | ||
| gATI.2.8 | Glyma02g11335 | 1.28 | gATI.2.8.a3 * | 0.079 | √ | 4 | Cell–cell signaling (I) |
| gATI.2.15 | Glyma02g44350 | 0.24 | gATI.2.15.a1 | 0.043 | √ | 5 | Acetyl-CoA metabolic process (IV) |
| gATI.3.2 | Glyma03g27770 | 2.23 | gATI.3.2.a1 | 0.023 | √ | 1 | |
| gATI.3.3 | Glyma03g30270 | 2.14 | gATI.3.3.a1 | 0.051 | √ | 2 | |
| gATI.4.1 | Glyma04g10720 | 2.00 | gATI.4.1.a1 * | 0.021 | √ | 4 | Biological process (VI) |
| gATI.4.2 | Glyma04g43300 | 2.88 | gATI.4.2.a2 | 0.048 | √ | ||
| gATI.6.2 | Glyma06g05300 | 1.82 | gATI.6.2.a3 | 0.083 | √ | 1 | |
| gATI.6.3 | Glyma06g07980 | 4.66 | gATI.6.3.a3 | 0.095 | √ | 6 | Positive regulation of transcription (III) |
| gATI.7.2 | Glyma07g05620 | 2.45 | gATI.7.2.a4 | 0.053 | √ | ||
| gATI.7.7 | Glyma07g14234 | 0.06 | gATI.7.7.a2 * | 0.042 | √ | 5 | Metabolic process (IV) |
| gATI.7.8 | Glyma07g31130 | 1.48 | gATI.7.8.a2 | 0.077 | √ | ||
| gATI.7.10 | Glyma07g38180 | 2.90 | ATI.7.10.a3 | 0.028 | √ | 6 | Response to wounding (I) |
| gATI.8.1 | Glyma08g04620 | 0.18 | gATI.8.1.a2 | 0.039 | √ | 4 | Protein dephosphorylation (III) |
| gATI.8.4 | Glyma08g42810 | 0.05 | gATI.8.4.a1 | 0.017 | √ | 3 | |
| gATI.9.3 | Glyma09g33220 | 2.52 | gATI.9.3.a3 | 0.082 | √ | ||
| gATI.10.2 | Glyma10g06600 | 0.80 | gATI.10.2.a2 | 0.023 | √ | 3 | |
| gATI.10.3 | Glyma10g07601 | 1.32 | gATI.10.3.a1 | 0.015 | √ | ||
| gATI.10.7 | Glyma10g31560 | 1.32 | gATI.10.7.a1 | 0.018 | √ | 3 | Ubiquitin-dependent protein catabolic process (IV) |
| gATI.13.4 | Glyma13g29225 | 0.12 | gATI.13.4.a1 | 0.029 | √ | 6 | NA (VII) |
| gATI.13.7 | Glyma13g40690 | 1.03 | gATI.13.7.a1 | 0.012 | √ | ||
| gATI.14.5 | Glyma14g37280 | 2.84 | gATI.14.5.a3 | 0.068 | NA | ||
| gATI.15.8 | Glyma15g32540 | (0.03) | gATI.15.8.a1 | 0.005 | √ | 11 | Translation (III) |
| gATI.16.1 | Glyma16g08960 | 1.43 | gATI.16.1.a1 | 0.096 | √ | 5 | Fatty acid beta-oxidation (IV) |
| gATI.17.1 | Glyma17g07120 | 1.66 | gATI.17.1.a1 | 0.033 | √ | 1 | |
| gATI.17.7 | Glyma17g33930 | 2.56 | gATI.17.7.a2 | 0.015 | √ | ||
| gATI.17.8 | Glyma17g36130 | 1.46 | gATI.17.8.a1 | 0.061 | √ | ||
| gATI.18.1 | Glyma18g01490 | 2.71 | gATI.18.1.a1 | 0.045 | √ | ||
| gATI.18.9 | Glyma18g28130 | 3.00 | gATI.18.9.a2 | 0.076 | √ | ||
| gATI.18.10 | Glyma18g40780 | 1.71 | gATI.18.10.a1 | 0.108 | √ | ||
| gATI.18.11 | Glyma18g46101 | 2.62 | gATI.18.11.a1 | 0.078 | √ | ||
| gATI.18.14 | Glyma18g53285 | 2.06 | gATI.18.14.a3 | 0.048 | NA | ||
| gATI.19.1 | Glyma19g31900 | 1.28 | gATI.19.1.a2 | 0.05 | √ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zong, C.; Zhao, J.; Wang, Y.; Wang, L.; Chen, Z.; Qi, Y.; Bai, Y.; Li, W.; Wang, W.; Ren, H.; et al. Identification of Gene–Allele System Conferring Alkali-Tolerance at Seedling Stage in Northeast China Soybean Germplasm. Int. J. Mol. Sci. 2024, 25, 2963. https://doi.org/10.3390/ijms25052963
Zong C, Zhao J, Wang Y, Wang L, Chen Z, Qi Y, Bai Y, Li W, Wang W, Ren H, et al. Identification of Gene–Allele System Conferring Alkali-Tolerance at Seedling Stage in Northeast China Soybean Germplasm. International Journal of Molecular Sciences. 2024; 25(5):2963. https://doi.org/10.3390/ijms25052963
Chicago/Turabian StyleZong, Chunmei, Jinming Zhao, Yanping Wang, Lei Wang, Zaoye Chen, Yuxin Qi, Yanfeng Bai, Wen Li, Wubin Wang, Haixiang Ren, and et al. 2024. "Identification of Gene–Allele System Conferring Alkali-Tolerance at Seedling Stage in Northeast China Soybean Germplasm" International Journal of Molecular Sciences 25, no. 5: 2963. https://doi.org/10.3390/ijms25052963