
Hydrophilic Interaction Liquid Chromatography–Hydrogen/Deuterium Exchange–Mass Spectrometry (HILIC-HDX-MS) for Untargeted Metabolomics
 , , and
, , and
Abstract
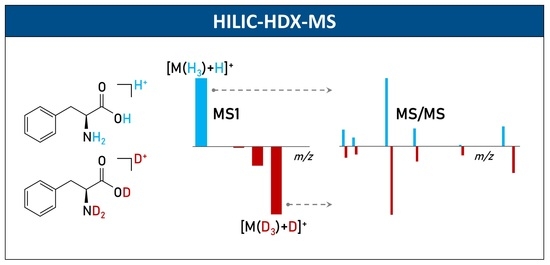
:
1. Introduction
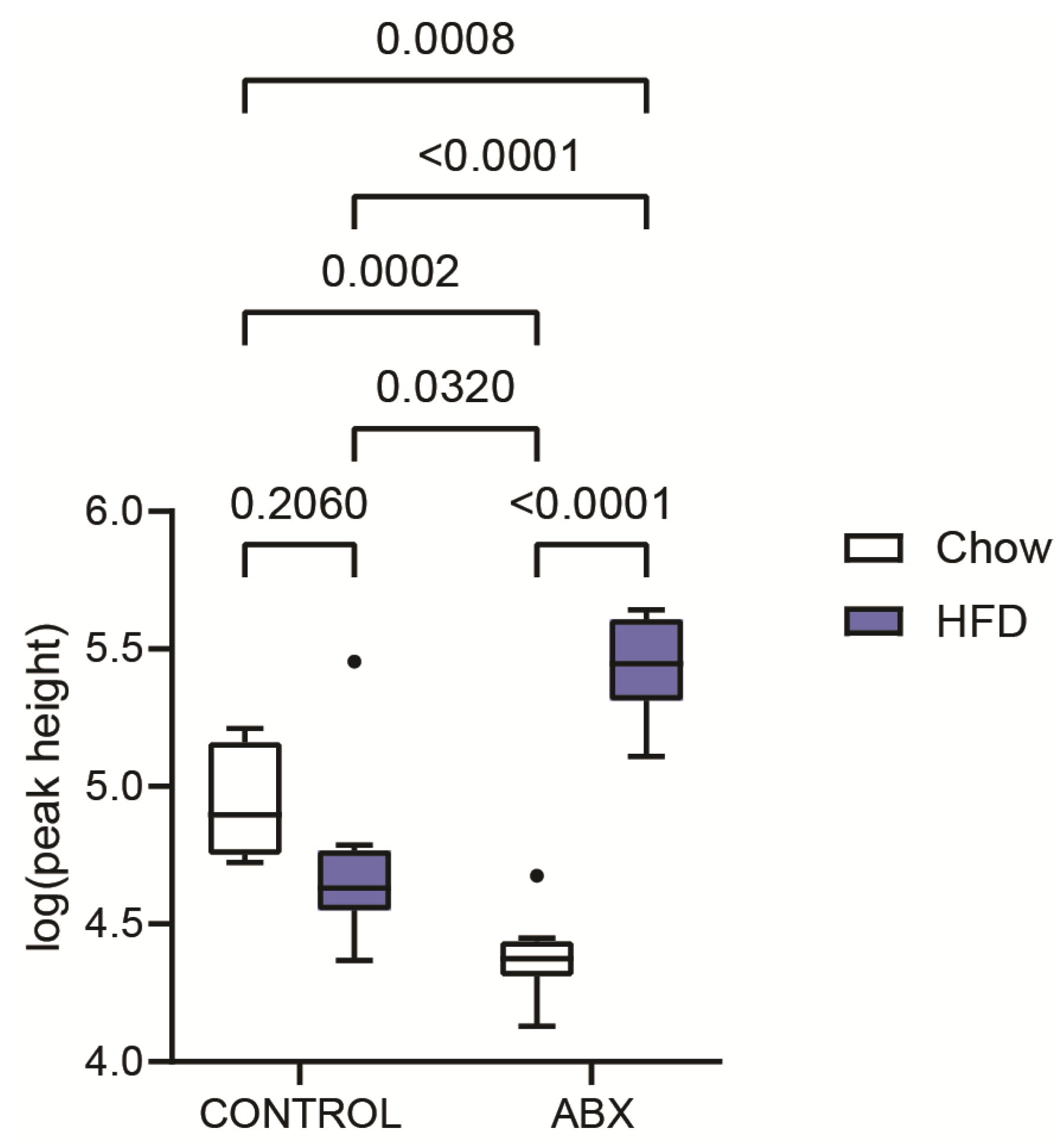
2. Results and Discussion
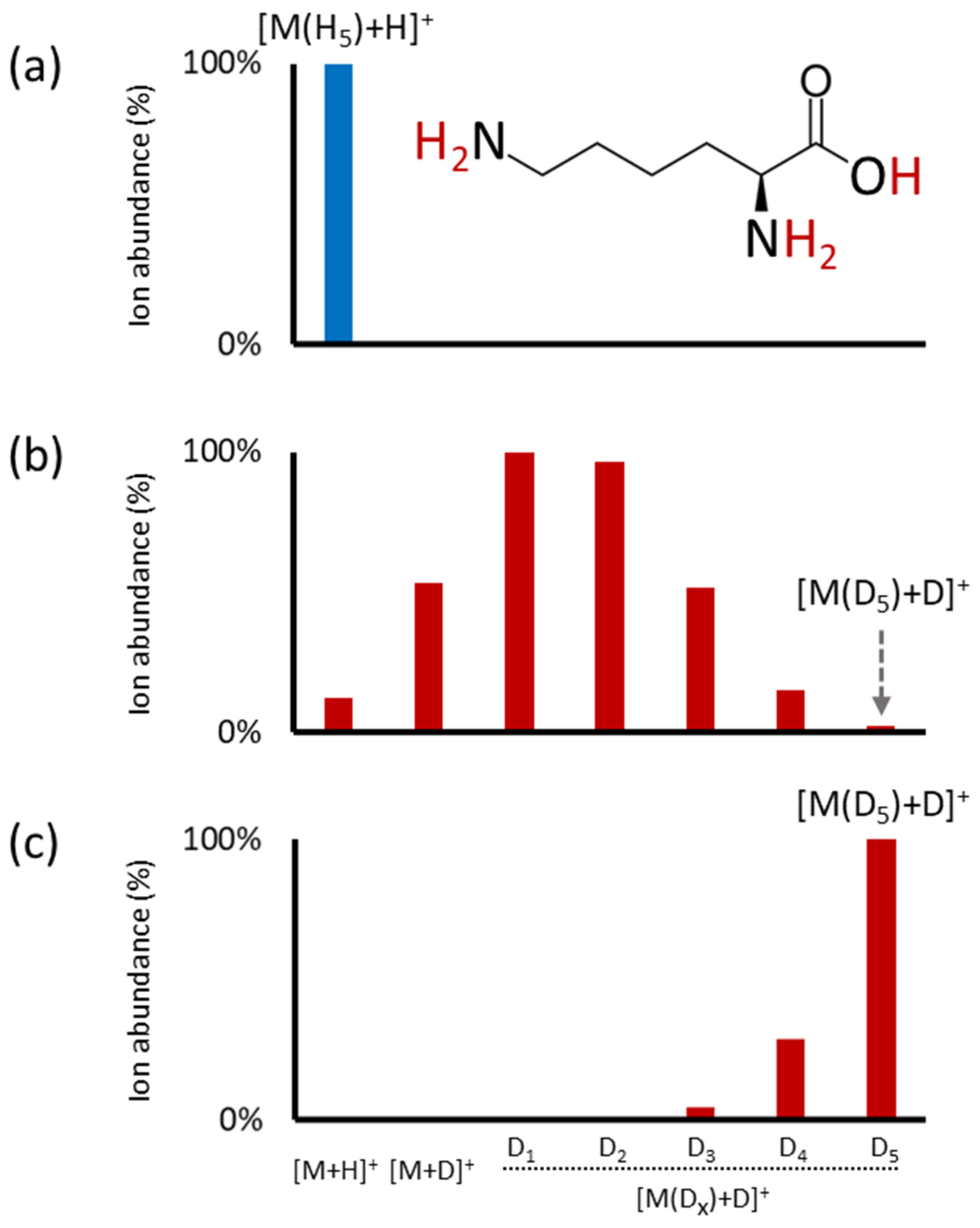
2.1. HILIC-MS with the Post-Column Addition of D2O
2.2. Partial and Full HILIC-HDX-MS
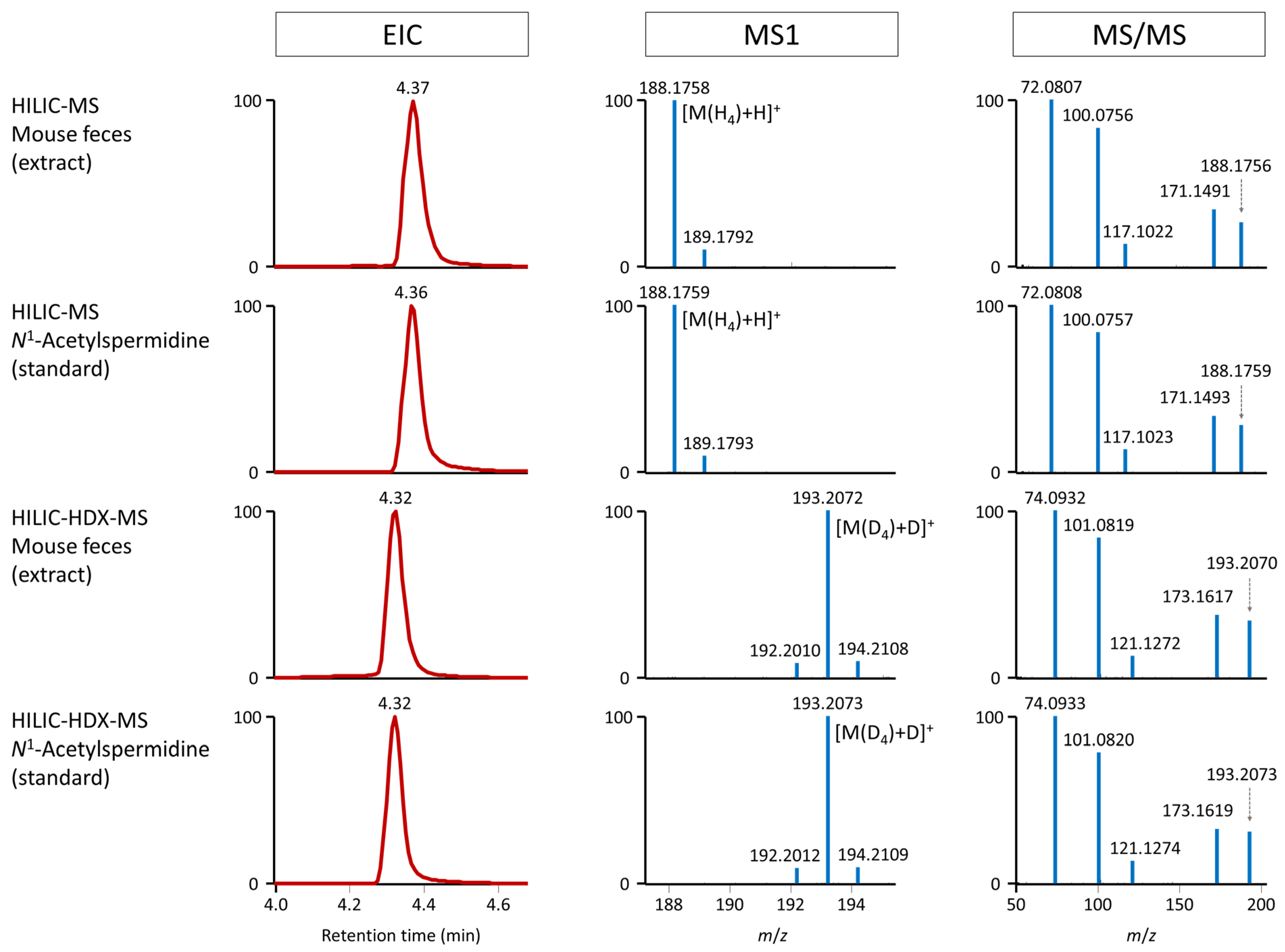
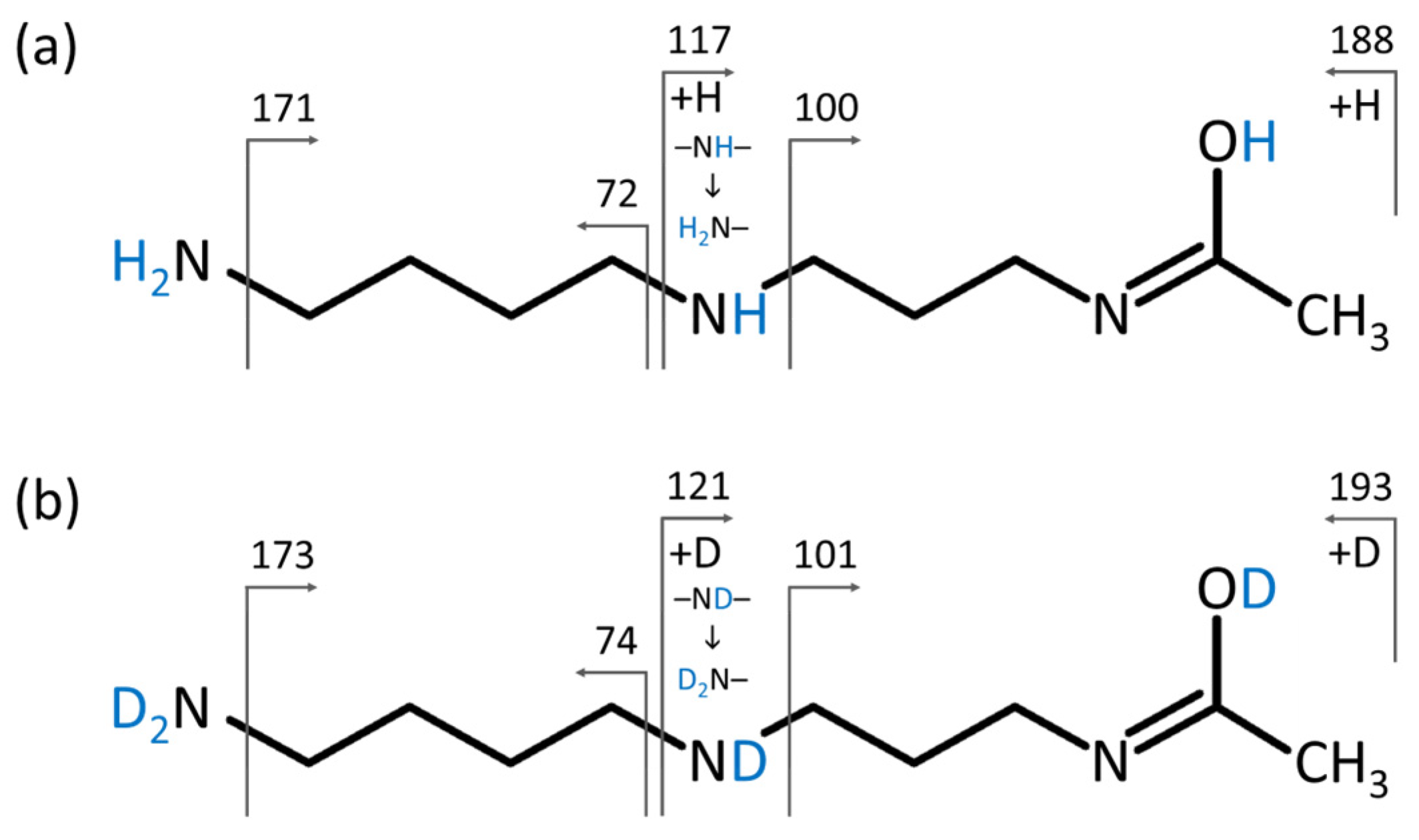
2.3. Structure Elucidation of N1-Acetylspermidine Using HILIC-HDX-MS
3. Materials and Methods
3.1. Materials and Reagents
3.2. Experiments with Animals
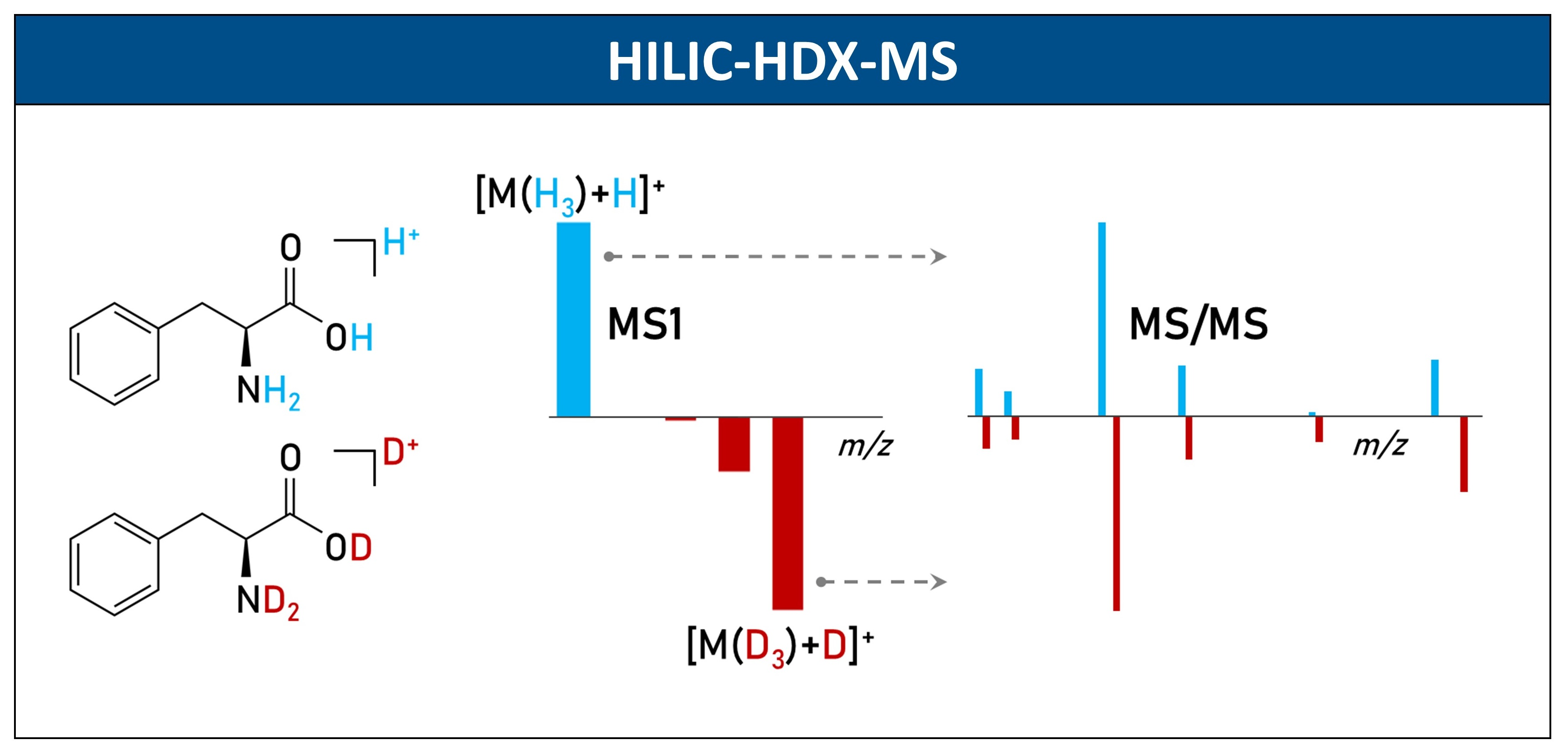
3.3. Sample Preparation
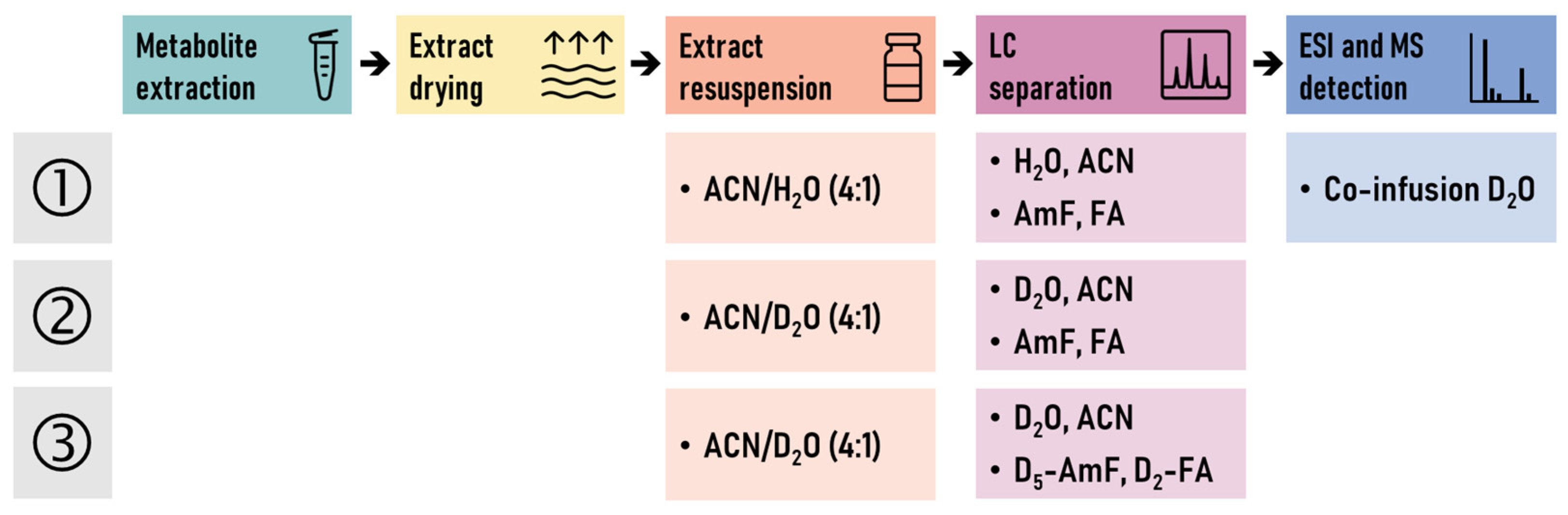
3.4. LC-MS Conditions
3.5. Data Processing
3.6. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dettmer, K.; Aronov, P.A.; Hammock, B.D. Mass spectrometry-based metabolomics. Mass Spectrom. Rev. 2007, 26, 51–78. [Google Scholar] [CrossRef] [PubMed]
- Lioupi, A.; Marinaki, M.; Virgiliou, C.; Begou, O.; Gika, H.; Wilson, I.; Theodoridis, G. Probing the polar metabolome by UHPLC-MS. TrAC-Trend Anal. Chem. 2023, 161, 117014. [Google Scholar] [CrossRef]
- Feizi, N.; Hashemi-Nasab, F.S.; Golpelichi, F.; Saburouh, N.; Parastar, H. Recent trends in application of chemometric methods for GC-MS and GCxGC-MS-based metabolomic studies. TrAC-Trend Anal. Chem. 2021, 138, 116239. [Google Scholar] [CrossRef]
- Rampler, E.; El Abiead, Y.; Schoeny, H.; Rusz, M.; Hildebrand, F.; Fitz, V.; Koellensperger, G. Recurrent topics in mass spectrometry-based metabolomics and lipidomics-standardization, coverage, and throughput. Anal. Chem. 2021, 93, 519–545. [Google Scholar] [CrossRef] [PubMed]
- Soga, T. Advances in capillary electrophoresis mass spectrometry for metabolomics. TrAC-Trend Anal. Chem. 2023, 158, 116883. [Google Scholar] [CrossRef]
- Rhee, E.P.; Waikar, S.S.; Rebholz, C.M.; Zheng, Z.H.; Perichon, R.; Clish, C.B.; Evans, A.M.; Avila, J.; Denburg, M.R.; Anderson, A.H.; et al. Variability of two metabolomic platforms in CKD. Clin. J. Am. Soc. Nephrol. 2019, 14, 40–48. [Google Scholar] [CrossRef]
- Dyar, K.A.; Lutter, D.; Artati, A.; Ceglia, N.J.; Liu, Y.; Armenta, D.; Jastroch, M.; Schneider, S.; de Mateo, S.; Cervantes, M.; et al. Atlas of circadian metabolism reveals system-wide coordination and communication between clocks. Cell 2018, 174, 1571–1585. [Google Scholar] [CrossRef]
- Abbondante, S.; Eckel-Mahan, K.L.; Ceglia, N.J.; Baldi, P.; Sassone-Corsi, P. Comparative circadian metabolomics reveal differential effects of nutritional challenge in the serum and liver. J. Biol. Chem. 2016, 291, 2812–2828. [Google Scholar] [CrossRef]
- Rakusanova, S.; Fiehn, O.; Cajka, T. Toward building mass spectrometry-based metabolomics and lipidomics atlases for biological and clinical research. TrAC-Trend Anal. Chem. 2023, 158, 116825. [Google Scholar] [CrossRef]
- da Silva, R.R.; Dorrestein, P.C.; Quinn, R.A. Illuminating the dark matter in metabolomics. Proc. Natl. Acad. Sci. USA 2015, 112, 12549–12550. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, N.F.; Mildau, K.; Meijer, D.; Louwen, J.J.R.; Bueschl, C.; Huber, F.; van der Hooft, J.J.J. Good practices and recommendations for using and benchmarking computational metabolomics metabolite annotation tools. Metabolomics 2022, 18, 103. [Google Scholar] [CrossRef]
- Minami, Y.; Kasukawa, T.; Kakazu, Y.; Iigo, M.; Sugimoto, M.; Ikeda, S.; Yasui, A.; van der Horst, G.T.J.; Soga, T.; Ueda, H.R. Measurement of internal body time by blood metabolomics. Proc. Natl. Acad. Sci. USA 2009, 106, 9890–9895. [Google Scholar] [CrossRef]
- Mahieu, N.G.; Patti, G.J. Systems-level annotation of a metabolomics data set reduces 25 000 features to fewer than 1000 unique metabolites. Anal. Chem. 2017, 89, 10397–10406. [Google Scholar] [CrossRef]
- Pinto, R.C.; Karaman, I.; Lewis, M.R.; Hällqvist, J.; Kaluarachchi, M.; Graça, G.; Chekmeneva, E.; Durainayagam, B.; Ghanbari, M.; Ikram, M.A.; et al. Finding correspondence between metabolomic features in untargeted liquid chromatography-mass spectrometry metabolomics datasets. Anal. Chem. 2022, 94, 5493–5503. [Google Scholar] [CrossRef]
- Kasukawa, T.; Sugimoto, M.; Hida, A.; Minami, Y.; Mori, M.; Honma, S.; Honma, K.; Mishima, K.; Soga, T.; Ueda, H.R. Human blood metabolite timetable indicates internal body time. Proc. Natl. Acad. Sci. USA 2012, 109, 15036–15041. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef]
- Chen, Z.Z.; Gerszten, R.E. Metabolomics and proteomics in type 2 diabetes. Circ. Res. 2020, 126, 1613–1627. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Hanash, S.; DeFelice, B.; Miyamoto, S.; Barnett, M.; Zhao, Y.; Goodman, G.; Feng, Z.; Gandara, D.; Fiehn, O.; et al. Diacetylspermine is a novel prediagnostic serum biomarker for non-small-cell lung cancer and has additive performance with pro-surfactant protein B. J. Clin. Oncol. 2015, 33, 3880–3886. [Google Scholar] [CrossRef] [PubMed]
- Low, D.Y.; Lefvre-Arbogast, S.; Gonzlez-Domnguez, R.; Urpi-Sarda, M.; Micheau, P.; Petera, M.; Centeno, D.; Durand, S.; Pujos-Guillot, E.; Korosi, A.; et al. Diet-related metabolites associated with cognitive decline revealed by untargeted metabolomics in a prospective cohort. Mol. Nutr. Food Res. 2019, 63, 1900177. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Cai, Y.; Yao, H.; Lin, C.S.; Xie, Y.Q.; Tang, S.Q.; Zhang, A.H. Small molecule metabolites: Discovery of biomarkers and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 132. [Google Scholar] [CrossRef]
- Cai, Y.P.; Zhou, Z.W.; Zhu, Z.J. Advanced analytical and informatic strategies for metabolite annotation in untargeted metabolomics. TrAC-Trend Anal. Chem. 2023, 158, 116903. [Google Scholar] [CrossRef]
- Zhu, Z.J.; Schultz, A.W.; Wang, J.H.; Johnson, C.H.; Yannone, S.M.; Patti, G.J.; Siuzdak, G. Liquid chromatography quadrupole time-of-flight mass spectrometry characterization of metabolites guided by the METLIN database. Nat. Protoc. 2013, 8, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Bittremieux, W.; Wang, M.X.; Dorrestein, P.C. The critical role that spectral libraries play in capturing the metabolomics community knowledge. Metabolomics 2022, 18, 94. [Google Scholar] [CrossRef] [PubMed]
- Chaleckis, R.; Meister, I.; Zhang, P.; Wheelock, C.E. Challenges, progress and promises of metabolite annotation for LC-MS-based metabolomics. Curr. Opin. Biotechnol. 2019, 55, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Alseekh, S.; Aharoni, A.; Brotman, Y.; Contrepois, K.; D’Auria, J.; Ewald, J.; Ewald, J.C.; Fraser, P.D.; Giavalisco, P.; Hall, R.D.; et al. Mass spectrometry-based metabolomics: A guide for annotation, quantification and best reporting practices. Nat. Methods 2021, 18, 747–756. [Google Scholar] [CrossRef]
- Purwaha, P.; Silva, L.P.; Hawke, D.H.; Weinstein, J.N.; Lorenzi, P.L. An artifact in LC-MS/MS measurement of glutamine and glutamic acid: In-source cyclization to pyroglutamic acid. Anal. Chem. 2014, 86, 5633–5637. [Google Scholar] [CrossRef]
- Tsugawa, H.; Kind, T.; Nakabayashi, R.; Yukihira, D.; Tanaka, W.; Cajka, T.; Saito, K.; Fiehn, O.; Arita, M. Hydrogen rearrangement rules: Computational MS/MS fragmentation and structure elucidation using MS-FINDER software. Anal. Chem. 2016, 88, 7946–7958. [Google Scholar] [CrossRef]
- Krettler, C.A.; Thallinger, G.G. A map of mass spectrometry-based fragmentation prediction and compound identification in metabolomics. Brief. Bioinform. 2021, 22, bbab073. [Google Scholar] [CrossRef]
- Duhrkop, K.; Shen, H.B.; Meusel, M.; Rousu, J.; Bocker, S. Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc. Natl. Acad. Sci. USA 2015, 112, 12580–12585. [Google Scholar] [CrossRef]
- Allen, F.; Pon, A.; Wilson, M.; Greiner, R.; Wishart, D. CFM-ID: A web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res. 2014, 42, W94–W99. [Google Scholar] [CrossRef]
- Wang, Y.F.; Kora, G.; Bowen, B.P.; Pan, C.L. MIDAS: A database-searching algorithm for metabolite identification in metabolomics. Anal. Chem. 2014, 86, 9496–9503. [Google Scholar] [CrossRef]
- Ruttkies, C.; Schymanski, E.L.; Wolf, S.; Hollender, J.; Neumann, S. MetFrag relaunched: Incorporating strategies beyond fragmentation. J. Cheminform. 2016, 8, 3. [Google Scholar] [CrossRef]
- Liu, D.Q.; Wu, L.M.; Sun, M.J.; MacGregor, P.A. On-line H/D exchange LC-MS strategy for structural elucidation of pharmaceutical impurities. J. Pharmaceut. Biomed. 2007, 44, 320–329. [Google Scholar] [CrossRef]
- Fischer, C.R.; Wilmes, P.; Bowen, B.P.; Northen, T.R.; Banfield, J.F. Deuterium-exchange metabolomics identifies N-methyl lyso phosphatidylethanolamines as abundant lipids in acidophilic mixed microbial communities. Metabolomics 2012, 8, 566–578. [Google Scholar] [CrossRef]
- Lam, W.; Ramanathan, R. In electrospray ionization source hydrogen/deuterium exchange LC-MS and LC-MS/MS for characterization of metabolites. J. Am. Soc. Mass Spectr. 2002, 13, 345–353. [Google Scholar] [CrossRef]
- Liu, D.Q.; Hop, C.E.C.A. Strategies for characterization of drug metabolites using liquid chromatography-tandem mass spectrometry in conjunction with chemical derivatization and on-line H/D exchange approaches. J. Pharm. Biomed. Anal. 2005, 37, 1–18. [Google Scholar] [CrossRef]
- Shah, R.P.; Garg, A.; Putlur, S.P.; Wagh, S.; Kumar, V.; Rao, V.; Singh, S.; Mandlekar, S.; Desikan, S. Practical and economical implementation of online H/D exchange in LC-MS. Anal. Chem. 2013, 85, 10904–10912. [Google Scholar] [CrossRef] [PubMed]
- Damont, A.; Legrand, A.; Cao, C.Q.; Fenaille, F.; Tabet, J.C. Hydrogen/deuterium exchange mass spectrometry in the world of small molecules. Mass Spectrom. Rev. 2023, 42, 1300–1331. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, T.; Tuerk, J.; Fuchs, R. Structural characterization of sulfadiazine metabolites using H/D exchange combined with various MS/MS experiments. J. Am. Soc. Mass Spectr. 2005, 16, 1687–1694. [Google Scholar] [CrossRef] [PubMed]
- Novak, T.J.; Helmy, R.; Santos, I. Liquid chromatography-mass spectrometry using the hydrogen/deuterium exchange reaction as a tool for impurity identification in pharmaceutical process development. J. Chromatogr. B 2005, 825, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Muz, M.; Krauss, M.; Kutsarova, S.; Schulze, T.; Brack, W. Mutagenicity in surface waters: Synergistic effects of carboline alkaloids and aromatic amines. Environ. Sci. Technol. 2017, 51, 1830–1839. [Google Scholar] [CrossRef]
- Acter, T.; Kim, D.; Ahmed, A.; Ha, J.H.; Kim, S. Application of atmospheric pressure photoionization H/D-exchange mass spectrometry for speciation of sulfur-containing compounds. J. Am. Soc. Mass Spectr. 2017, 28, 1687–1695. [Google Scholar] [CrossRef]
- Zherebker, A.; Kostyukevich, Y.; Kononikhin, A.; Roznyatovsky, V.A.; Popov, I.; Grishin, Y.K.; Perminova, I.V.; Nikolaev, E. High desolvation temperature facilitates the ESI-source H/D exchange at non-labile sites of hydroxybenzoic acids and aromatic amino acids. Analyst 2016, 141, 2426–2434. [Google Scholar] [CrossRef]
- Kostyukevich, Y.; Acter, T.; Zherebker, A.; Ahmed, A.; Kim, S.; Nikolaev, E. Hydrogen/deuterium exchange in mass spectrometry. Mass Spectrom. Rev. 2018, 37, 811–853. [Google Scholar] [CrossRef]
- De Vijlder, T.; Valkenborg, D.; Lemiere, F.; Romijn, E.P.; Laukens, K.; Cuyckens, F. A tutorial in small molecule identification via electrospray ionization-mass spectrometry: The practical art of structural elucidation. Mass Spectrom. Rev. 2018, 37, 607–629. [Google Scholar] [CrossRef] [PubMed]
- Ruttkies, C.; Schymanski, E.L.; Strehmel, N.; Hollender, J.; Neumann, S.; Williams, A.J.; Krauss, M. Supporting non-target identification by adding hydrogen deuterium exchange MS/MS capabilities to MetFrag. Anal. Bioanal. Chem. 2019, 411, 4683–4700. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.B.; Ford, L.A.; Evans, A.M.; Toal, D.R. Structure elucidation of metabolite x17299 by interpretation of mass spectrometric data. Metabolomics 2017, 13, 92. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.B.; Ford, L.A.; Evans, A.M.; Toal, D.R. Identification of an endogenous organosulfur metabolite by interpretation of mass spectrometric data. Org. Lett. 2018, 20, 2100–2103. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.Y.; Sun, Y.; Jia, S.L.; Yan, P.; Xiong, C.M.; Qi, M.L.; Wang, C.X.; Du, Z.F.; Jiang, H.L. Identification of endogenous carbonyl steroids in human serum by chemical derivatization, hydrogen/deuterium exchange mass spectrometry and the quantitative structure-retention relationship. J. Chromatogr. B 2023, 1226, 123776. [Google Scholar] [CrossRef] [PubMed]
- Kostyukeyich, Y.; Vladimirov, G.; Stekolschikova, E.; Ivanov, D.; Yablokov, A.; Zherebker, A.; Sosnin, S.; Orlov, A.; Fedoroy, M.; Khaitovich, P.; et al. Hydrogen/deuterium exchange aiding compound identification for LC-MS and MALDI imaging lipidomics. Anal. Chem. 2019, 91, 13465–13474. [Google Scholar] [CrossRef] [PubMed]
- Strehmel, N.; Böttcher, C.; Schmidt, S.; Scheel, D. Profiling of secondary metabolites in root exudates of Arabidopsis thaliana. Phytochemistry 2014, 108, 35–46. [Google Scholar] [CrossRef]
- Cajka, T.; Hricko, J.; Rudl Kulhava, L.; Paucova, M.; Novakova, M.; Kuda, O. Optimization of mobile phase modifiers for fast LC-MS-based untargeted metabolomics and lipidomics. Int. J. Mol. Sci. 2023, 24, 1987. [Google Scholar] [CrossRef] [PubMed]
- Hamuro, Y. Tutorial: Chemistry of hydrogen/deuterium exchange mass spectrometry. J. Am. Soc. Mass Spectrom. 2021, 32, 133–151. [Google Scholar] [CrossRef]
- Ball, D.; Nguyen, T.; Zhang, N.F.; D’Arcy, S. Using hydrogen-deuterium exchange mass spectrometry to characterize Mtr4 interactions with RNA. Method Enzymol. 2022, 673, 475–516. [Google Scholar]
- Hatvany, J.B.; Liyanage, O.T.; Gallagher, E.S. Effect of pH on in-electrospray hydrogen/deuterium exchange of carbohydrates and peptides. J. Am. Soc. Mass Spectrom. 2024; online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Castillo, P.; Kuda, O.; Kopecky, J.; Pomar, C.A.; Palou, A.; Palou, M.; Picó, C. Reverting to a healthy diet during lactation normalizes maternal milk lipid content of diet-induced obese rats and prevents early alterations in the plasma lipidome of the offspring. Mol. Nutr. Food Res. 2022, 66, 2200204. [Google Scholar] [CrossRef] [PubMed]
- Greiner, P.; Houdek, P.; Sládek, M.; Sumová, A. Early rhythmicity in the fetal suprachiasmatic nuclei in response to maternal signals detected by omics approach. PLoS Biol. 2022, 20, e3001637. [Google Scholar] [CrossRef]
- Benova, A.; Ferencakova, M.; Bardova, K.; Funda, J.; Prochazka, J.; Spoutil, F.; Cajka, T.; Dzubanova, M.; Balcaen, T.; Kerckhofs, G.; et al. Novel thiazolidinedione analog reduces a negative impact on bone and mesenchymal stem cell properties in obese mice compared to classical thiazolidinediones. Mol. Metab. 2022, 65, 101598. [Google Scholar] [CrossRef]
- Janovska, P.; Melenovsky, V.; Svobodova, M.; Havlenova, T.; Kratochvilova, H.; Haluzik, M.; Hoskova, E.; Pelikanova, T.; Kautzner, J.; Monzo, L.; et al. Dysregulation of epicardial adipose tissue in cachexia due to heart failure: The role of natriuretic peptides and cardiolipin. J. Cachexia Sarcopenia Muscle 2020, 11, 1614–1627. [Google Scholar] [CrossRef]
- Grazul, H.; Kanda, L.L.; Gondek, D. Impact of probiotic supplements on microbiome diversity following antibiotic treatment of mice. Gut Microbes 2016, 7, 101–114. [Google Scholar] [CrossRef]
- Hricko, J.; Kulhava, L.R.; Paucova, M.; Novakova, M.; Kuda, O.; Fiehn, O.; Cajka, T. Short-term stability of serum and liver extracts for untargeted metabolomics and lipidomics. Antioxidants 2023, 12, 986. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Ikeda, K.; Takahashi, M.; Satoh, A.; Mori, Y.; Uchino, H.; Okahashi, N.; Yamada, Y.; Tada, I.; Bonini, P.; et al. A lipidome atlas in MS-DIAL 4. Nat. Biotechnol. 2020, 38, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Arita, M.; Kanazawa, M.; Ogiwara, A.; Bamba, T.; Fukusaki, E. MRMPROBS: A data assessment and metabolite identification tool for large-scale multiple reaction monitoring based widely targeted metabolomics. Anal. Chem. 2013, 85, 5191–5199. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Specification | HILIC-MS | HILIC-MS with D2O Co-Infusion | Partial HILIC-HDX-MS | Full HILIC-HDX-MS |
|---|---|---|---|---|
| Mobile Phase A | H2O 10 mM Ammonium Formate 0.125% Formic Acid | H2O 10 mM Ammonium Formate 0.125% Formic Acid | D2O 10 mM Ammonium Formate 0.125% Formic Acid | D2O 10 mM D5-Ammonium Formate 0.125% D2-Formic Acid |
| Mobile Phase B | 95% Acetonitrile/5% H2O 10 mM Ammonium Formate 0.125% Formic Acid | 95% Acetonitrile/5% H2O 10 mM Ammonium Formate 0.125% Formic Acid | 95% Acetonitrile/5% D2O 7.5 mM Ammonium Formate 0.125% Formic Acid | 95% Acetonitrile/5% D2O 7.5 mM D5-Ammonium Formate 0.125% D2-Formic Acid |
| Resuspension Solvent | Acetonitrile/H2O (4:1, v/v) with CUDA, Val-Tyr-Val (IS) | Acetonitrile/H2O (4:1, v/v) with CUDA, Val-Tyr-Val (IS) | Acetonitrile/D2O (4:1, v/v) with CUDA, Val-Tyr-Val (IS) | Acetonitrile/D2O (4:1, v/v) with CUDA, Val-Tyr-Val (IS) |
| Column Flow Rate | 0.4 mL/min | 0.4 mL/min | 0.4 mL/min | 0.4 mL/min |
| Co-Infusion Solvent | — | D2O | — | — |
| Co-Infusion Flow Rate | — | 0.05, 0.1 mL/min | — | — |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cajka, T.; Hricko, J.; Rakusanova, S.; Brejchova, K.; Novakova, M.; Rudl Kulhava, L.; Hola, V.; Paucova, M.; Fiehn, O.; Kuda, O. Hydrophilic Interaction Liquid Chromatography–Hydrogen/Deuterium Exchange–Mass Spectrometry (HILIC-HDX-MS) for Untargeted Metabolomics. Int. J. Mol. Sci. 2024, 25, 2899. https://doi.org/10.3390/ijms25052899
Cajka T, Hricko J, Rakusanova S, Brejchova K, Novakova M, Rudl Kulhava L, Hola V, Paucova M, Fiehn O, Kuda O. Hydrophilic Interaction Liquid Chromatography–Hydrogen/Deuterium Exchange–Mass Spectrometry (HILIC-HDX-MS) for Untargeted Metabolomics. International Journal of Molecular Sciences. 2024; 25(5):2899. https://doi.org/10.3390/ijms25052899
Chicago/Turabian StyleCajka, Tomas, Jiri Hricko, Stanislava Rakusanova, Kristyna Brejchova, Michaela Novakova, Lucie Rudl Kulhava, Veronika Hola, Michaela Paucova, Oliver Fiehn, and Ondrej Kuda. 2024. "Hydrophilic Interaction Liquid Chromatography–Hydrogen/Deuterium Exchange–Mass Spectrometry (HILIC-HDX-MS) for Untargeted Metabolomics" International Journal of Molecular Sciences 25, no. 5: 2899. https://doi.org/10.3390/ijms25052899