
The Pathophysiology and Treatment of Pyoderma Gangrenosum—Current Options and New Perspectives
 , and
, and
Abstract
:1. Introduction
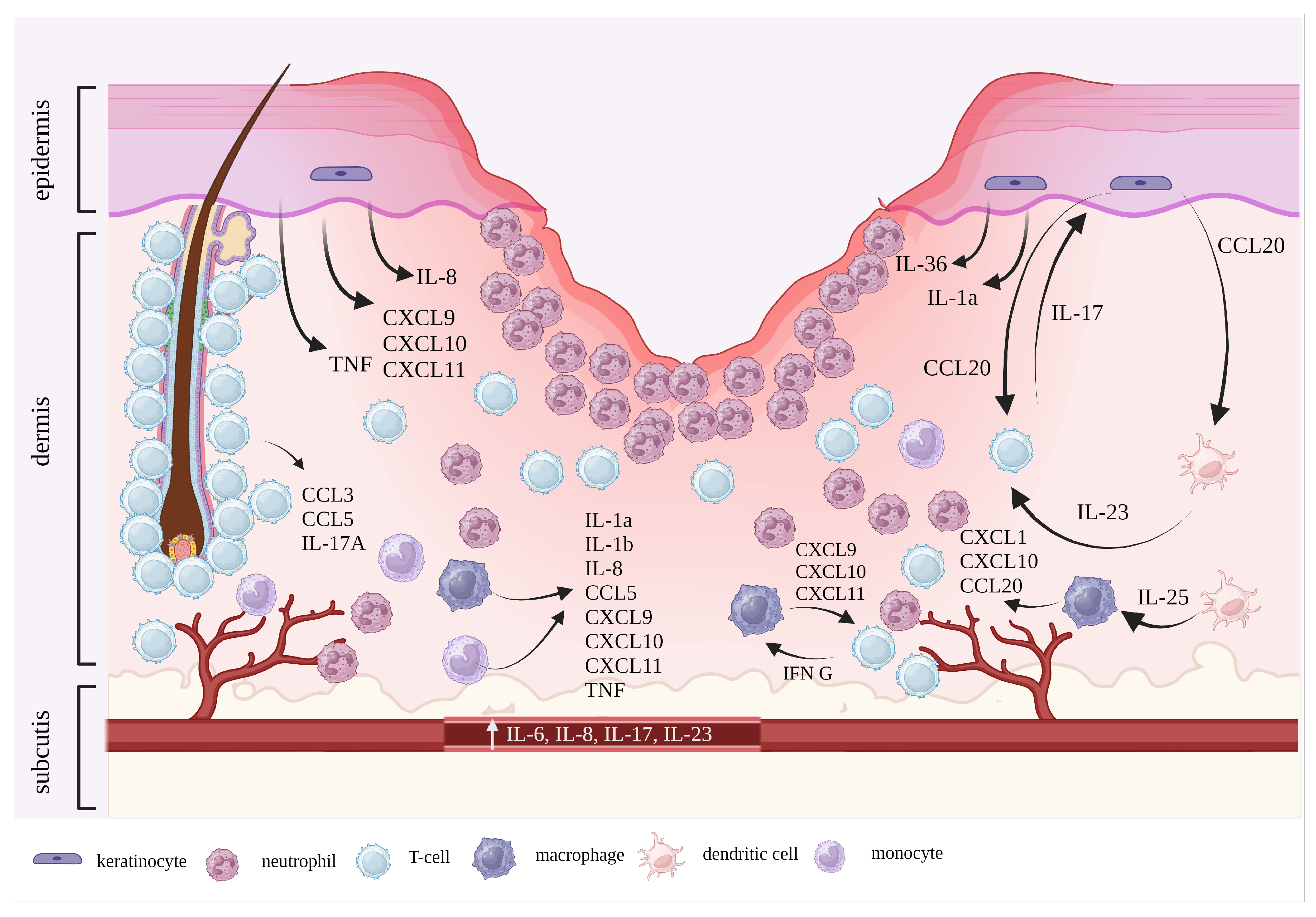
2. Pathophysiology
3. Treatment
3.1. Wound Management
3.2. Topical Therapies
3.3. Intralesional Therapies
3.4. Systemic Therapies
3.4.1. Corticosteroids
3.4.2. Cyclosporine A (CsA)
3.4.3. Other Immunosuppressants
Mycophenolate Mofetil (MMF)
Methotrexate (MTX)
Azathioprine
3.4.4. Immunosuppressive Antibiotics
Dapsone
Minocycline
3.4.5. Intravenous Immunoglobulin (IVIG)
3.4.6. Tumor Necrosis Factor-α (TNF-α) Inhibitors
Infliximab

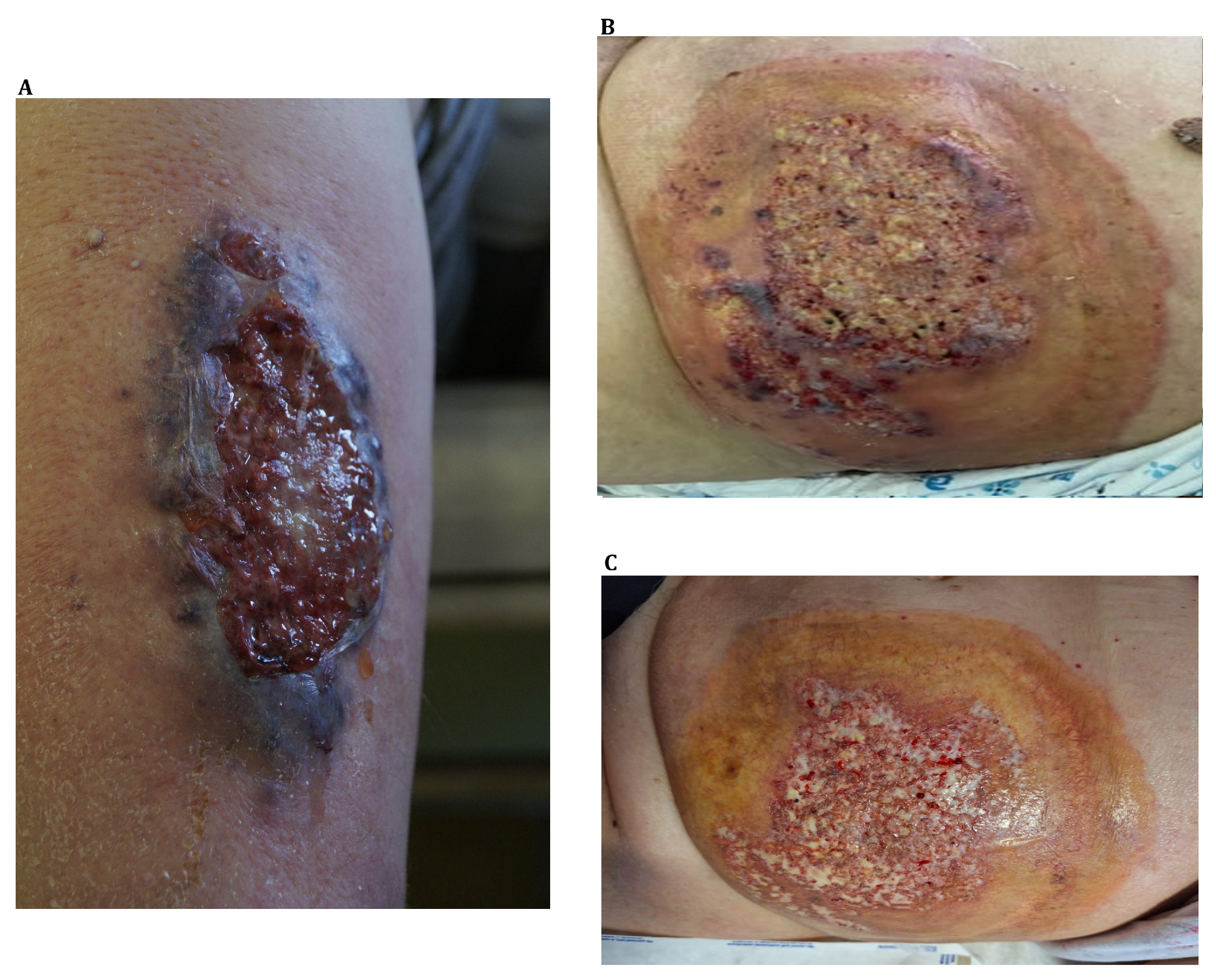{kind=link}
{kind=link}
| Authors (Year) | Biologic Drug | Dosage Regimen | Study Type | Comorbidities | Efficacy |
|---|---|---|---|---|---|
| T.N. Brooklyn et al. (2006) [64] | Infliximab | 5 mg/kg i.v. at weeks 0, 2, and 6, followed by infusions every 6 to 8 weeks or placebo at week 0 with possible switch at week 2 | Randomized placebo- controlled trial | CD (n = 12) UC (n = 6) no IBD (n = 11) | Out of 29 patients in infliximab group, 20 (67%) demonstrated adequate response |
| M. Regueiro et al. (2003) [106] | Infliximab | 5 mg/kg i.v. | Multicenter retrospective study | IBD in all cases | Complete healing in all 13 cases |
| F. Argüelles-Arias et al. (2013) [107] | Infliximab | 5 mg/kg i.v. | Retrospective observational study | IBD in all cases | Out of 24 patients, 22 (92%) demonstrated complete healing |
| T. Ljung et al. (2002) [109] | Infliximab | 5 mg/kg i.v. | Case series (n = 8) | CD | Complete healing in 3 (37%) cases, partial healing in 3 (37%) |
| F. Salehzadeh et al. (2019) [110] | Infliximab | 100 mg i.v. | Case report | none known | Full recovery in 2-year period |
| M. R. Kaur et al. (2005) [111] | Infliximab | 3 mg/kg i.v. | Case report | none known | Full recovery in 4-month period |
| L. Đ. Betetto et al. (2022) [112] | Infliximab | 10 mg/kg i.v. | Case report | UC | Satisfactory result |
Adalimumab
Etanercept
3.4.7. Ustekinumab
3.4.8. IL-1 Antagonists
3.4.9. IL-17 Inhibitors
IL-23 Inhibitors
4. Future Directions
4.1. Janus Kinase Inhibitors (JAKi)
| Authors (Year) | JAKi | Dosage Regimen | Age and Gender | Comorbidities | Efficacy |
|---|---|---|---|---|---|
| B. Kochar et al. (2019) [173] | Tofacitinib |
|
| all patients with Crohn’s disease and concomitant arthritis previously resistant to various biologics |
|
| P.S. Olavarria et al. (2021) [174] | Tofacitinib | 10 mg p.o. twice daily | 69-year old female | ulcerative colitis and arthralgias | Complete healing after 4 weeks |
| L. G. M. Castro (2023) [168] | Baricitinib Tofacitinib | 2 mg p.o. twice daily for 39 days 5 mg p.o. twice daily for 120 days | 73-year old male 79-year old female | familial Mediterranean fever none known | Complete healing with no relapse |
| M. R. dos Santos et al. (2023) [169] | Upadacitinib | 15 mg p.o. daily | 45-year old female | rheumatoid arthritis | Complete regression after 6 weeks |
| M. Scheinberg et al. (2021) [170] | Baricitinib | 4 mg p.o. daily | 71-year old female | IgA multiple myeloma in remission | Complete regression after 5 weeks |
| S. Nasifoglu et al. (2018) [171] | Ruxolitinib | NA | 64-year old female | polycythemia vera | Complete healing |
4.2. Spesolimab
4.3. Vilobelimab
| Study Number | Medication | Study Phase/Type | Estimated Enrollment | Estimated Study Completion |
|---|---|---|---|---|
| NCT05964413 [179] | Vilobelimab | 3 | 90 | 13 February 2026 |
| NCT04750213 [120] | Adalimumab | observational | 60 | 31 August 2025 |
| NCT06092216 [177] | Spesolimab | 4 | 20 | September 2025 |
| NCT04901325 [172] | Baricitinib | 2 | 10 | 5 December 2024 |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maverakis, E.; Ma, C.; Shinkai, K.; Fiorentino, D.; Callen, J.P.; Wollina, U.; Marzano, A.V.; Wallach, D.; Kim, K.; Schadt, C.; et al. Diagnostic Criteria of Ulcerative Pyoderma Gangrenosum: A Delphi Consensus of International Experts. JAMA Dermatol. 2018, 154, 461–466. [Google Scholar] [CrossRef]
- Jockenhöfer, F.; Wollina, U.; Salva, K.A.; Benson, S.; Dissemond, J. The PARACELSUS score: A novel diagnostic tool for pyoderma gangrenosum. Br. J. Dermatol. 2019, 180, 615–620. [Google Scholar] [CrossRef]
- Haag, C.; Hansen, T.; Hajar, T.; Latour, E.; Keller, J.; Shinkai, K.; Ortega-Loayza, A.G. Comparison of Three Diagnostic Frameworks for Pyoderma Gangrenosum. J. Investig. Dermatol. 2021, 141, 59–63. [Google Scholar] [CrossRef]
- Hughes, A.P.; Jackson, J.M.; Callen, J.P. Clinical features and treatment of peristomal pyoderma gangrenosum. JAMA 2000, 284, 1546–1548. [Google Scholar] [CrossRef] [PubMed]
- Tolkachjov, S.N.; Fahy, A.S.; Cerci, F.B.; Wetter, D.A.; Cha, S.S.; Camilleri, M.J. Postoperative Pyoderma Gangrenosum: A Clinical Review of Published Cases. Mayo Clin. Proc. 2016, 91, 1267–1279. [Google Scholar] [CrossRef]
- Borda, L.J.; Wong, L.L.; Marzano, A.V.; Ortega-Loayza, A.G. Extracutaneous involvement of pyoderma gangrenosum. Arch. Dermatol. Res. 2019, 311, 425–434. [Google Scholar] [CrossRef] [PubMed]
- George, C.; Deroide, F.; Rustin, M. Pyoderma gangrenosum—A guide to diagnosis and management. Clin. Med. 2019, 19, 224–228. [Google Scholar] [CrossRef]
- Honma, M.; Sugawara, M.; Ueno, N.; Honma, M.; Hinooka, R.; Tani, C. Clinical Characteristics of Peristomal Pyoderma Gangrenosum: A Single Center Retrospective Observational Study. J. Dermatol. 2022, 49, 1178–1182. [Google Scholar] [CrossRef]
- Xu, A.; Balgobind, A.; Strunk, A.; Garg, A.; Alloo, A. Prevalence estimates for pyoderma gangrenosum in the United States: An age- and sex-adjusted population analysis. J. Am. Acad. Dermatol. 2020, 83, 425–429. [Google Scholar] [CrossRef]
- Langan, S.M.; Groves, R.W.; Card, T.R.; Gulliford, M.C. Incidence, mortality, and disease associations of pyoderma gangrenosum in the United Kingdom: A retrospective cohort study. J. Investig. Dermatol. 2012, 132, 2166–2170. [Google Scholar] [CrossRef] [PubMed]
- Monari, P.; Moro, R.; Motolese, A.; Misciali, C.; Baraldi, C.; Fanti, P.A.; Caccavale, S.; Puviani, M.; Olezzi, D.; Zampieri, P.; et al. Epidemiology of pyoderma gangrenosum: Results from an Italian prospective multicentre study. Int. Wound J. 2018, 15, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Kolios, A.G.; Gübeli, A.; Meier, B.; Maul, J.-T.; Kündig, T.; Nilsson, J.; Hafner, J.; Guenova, E.; Kerl, K.; Anliker, M.; et al. Clinical Disease Patterns in a Regional Swiss Cohort of 34 Pyoderma Gangrenosum Patients. Dermatology 2017, 233, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Ighani, A.; Al-Mutairi, D.; Rahmani, A.; Weizman, A.V.; Piguet, V.; Alavi, A. Pyoderma gangrenosum and its impact on quality of life: A multicentre, prospective study. Br. J. Dermatol. 2019, 180, 672–673. [Google Scholar] [CrossRef]
- Marzano, A.V.; Borghi, A.; Wallach, D.; Cugno, M. A Comprehensive Review of Neutrophilic Diseases. Clin. Rev. Allergy Immunol. 2018, 54, 114–130. [Google Scholar] [CrossRef]
- Wang, E.A.; Steel, A.; Luxardi, G.; Mitra, A.; Patel, F.; Cheng, M.Y.; Wilken, R.; Kao, J.; de Ga, K.; Sultani, H.; et al. Classic Ulcerative Pyoderma Gangrenosum Is a T Cell-Mediated Disease Targeting Follicular Adnexal Structures: A Hypothesis Based on Molecular and Clinicopathologic Studies. Front. Immunol. 2018, 8, 1980. [Google Scholar] [CrossRef]
- Weiss, D.I.; Ma, F.; Merleev, A.A.; Maverakis, E.; Gilliet, M.; Balin, S.J.; Bryson, B.D.; Ochoa, M.T.; Pellegrini, M.; Bloom, B.R.; et al. IL-1β Induces the Rapid Secretion of the Antimicrobial Protein IL-26 from Th17 Cells. J. Immunol. 2019, 203, 911–921. [Google Scholar] [CrossRef]
- Satoh, T.K.; Mellett, M.; Contassot, E.; French, L.E. Are neutrophilic dermatoses autoinflammatory disorders? Br. J. Dermatol. 2018, 178, 603–613. [Google Scholar] [CrossRef]
- Senra, L.; Mylonas, A.; Kavanagh, R.D.; Fallon, P.G.; Conrad, C.; Borowczyk-Michalowska, J.; Wrobel, L.J.; Kaya, G.; Yawalkar, N.; Boehncke, W.-H.; et al. IL-17E (IL-25) Enhances Innate Immune Responses during Skin Inflammation. J. Investig. Dermatol. 2019, 139, 1732–1742. [Google Scholar] [CrossRef]
- Smith, S.; Wu, P.W.; Seo, J.J.; Fernando, T.; Jin, M.; Contreras, J.; Montano, E.N.; Gabhann, J.N.; Cunningham, K.; Widaa, A.; et al. IL-16/miR-125a axis controls neutrophil recruitment in pristane-induced lung inflammation. JCI Insight 2018, 3, e120798. [Google Scholar] [CrossRef]
- Ergun, T. Pathergy Phenomenon. Front. Med. 2021, 8, 639404. [Google Scholar] [CrossRef]
- Le, S.T.; Wang, J.Z.; Alexanian, C.C.; Johng, S.Y.; Patel, F.B.; Wang, E.A.; Ma, C.; Wilken, R.; Cheng, M.Y.; Maverakis, E. End stage scurvy in the developed world: A diagnostic conundrum but not to be mistaken for pyoderma gangrenosum. Int. Wound J. 2019, 16, 1024. [Google Scholar] [CrossRef]
- Haag, C.K.; Nutan, F.; Cyrus, J.W.; Satpathy, J.; Shinkai, K.; Loayza, A.G.O. Pyoderma gangrenosum misdiagnosis resulting in amputation: A review. J. Trauma Acute Care Surg. 2019, 86, 307–313. [Google Scholar] [CrossRef]
- Bradsher, R.W. The Endemic Mimic: Blastomycosis An Illness Often Misdiagnosed. Trans. Am. Clin. Climatol. Assoc. 2014, 125, 188. [Google Scholar]
- Wallach, D.; Vignon-Pennamen, M.D. From acute febrile neutrophilic dermatosis to neutrophilic disease: Forty years of clinical research. J. Am. Acad. Dermatol. 2006, 55, 1066–1071. [Google Scholar] [CrossRef]
- Lorenz, U. SHP-1 and SHP-2 in T cells: Two phosphatases functioning at many levels. Immunol. Rev. 2009, 228, 342–359. [Google Scholar] [CrossRef]
- Zhang, J.; Somani, A.K.; Siminovitch, K.A. Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signalling. Semin. Immunol. 2000, 12, 361–378. [Google Scholar] [CrossRef] [PubMed]
- Nesterovitch, A.B.; Szanto, S.; Gonda, A.; Bardos, T.; Kis-Toth, K.; Adarichev, V.A.; Olasz, K.; Ghassemi-Najad, S.; Hoffman, M.D.; Tharp, M.D.; et al. Spontaneous insertion of a b2 element in the ptpn6 gene drives a systemic autoinflammatory disease in mice resembling neutrophilic dermatosis in humans. Am. J. Pathol. 2011, 178, 1701–1714. [Google Scholar] [CrossRef] [PubMed]
- Nesterovitch, A.B.; Gyorfy, Z.; Hoffman, M.D.; Moore, E.C.; Elbuluk, N.; Tryniszewska, B.; Rauch, T.A.; Simon, M.; Kang, S.; Fisher, G.J.; et al. Alteration in the gene encoding protein tyrosine phosphatase nonreceptor type 6 (PTPN6/SHP1) may contribute to neutrophilic dermatoses. Am. J. Pathol. 2011, 178, 1434–1441. [Google Scholar] [CrossRef] [PubMed]
- Nesterovitch, A.B.; Hoffman, M.D.; Simon, M.; Petukhov, P.A.; Tharp, M.D.; Glant, T.T. Mutations in the PSTPIP1 gene and aberrant splicing variants in patients with pyoderma gangrenosum. Clin. Exp. Dermatol. 2011, 36, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.J.; Allantaz, F.; Bennett, L.; Zhang, D.; Gao, X.; Wood, G.; Kastner, D.L.; Punaro, M.; Aksentijevich, I.; Pascual, V.; et al. Clinical, Molecular, and Genetic Characteristics of PAPA Syndrome: A Review. Curr. Genom. 2010, 11, 519. [Google Scholar] [CrossRef] [PubMed]
- Kastner, D.L.; Aksentijevich, I.; Goldbach-Mansky, R. Autoinflammatory disease reloaded: A clinical perspective. Cell 2010, 140, 784–790. [Google Scholar] [CrossRef]
- Marzano, A.V.; Borghi, A.; Meroni, P.L.; Cugno, M. Pyoderma gangrenosum and its syndromic forms: Evidence for a link with autoinflammation. Br. J. Dermatol. 2016, 175, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. A clinical perspective of IL-1β as the gatekeeper of inflammation. Eur. J. Immunol. 2011, 41, 1203–1217. [Google Scholar] [CrossRef]
- Wise, C.A.; Gillum, J.D.; Seidman, C.E.; Lindor, N.M.; Veile, R.; Bashiardes, S.; Lovett, M. Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum. Mol. Genet. 2002, 11, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Moltrasio, C.; Garcovich, S.; Marzano, A.V. PAPA spectrum disorders. G. Ital. Dermatol. Venereol. 2020, 155, 542–550. [Google Scholar] [CrossRef]
- Moura, R.R.; Brandão, L.; Moltrasio, C.; Agrelli, A.; Tricarico, P.M.; Maronese, C.A.; Crovella, S.; Marzano, A.V. Different molecular pathways are disrupted in Pyoderma gangrenosum patients and are associated with the severity of the disease. Sci. Rep. 2023, 13, 4919. [Google Scholar] [CrossRef]
- Marzano, A.V.; Genovese, G.; Moltrasio, C.; Tricarico, P.M.; Gratton, R.; Piaserico, S.; Garcovich, S.; Boniotto, M.; Brandão, L.; Moura, R.; et al. Whole-Exome Sequencing in 10 Unrelated Patients with Syndromic Hidradenitis Suppurativa: A Preliminary Step for a Genotype-Phenotype Correlation. Dermatology 2022, 238, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.M.; Sullivan, G.P.; Clancy, D.M.; Afonina, I.S.; Kulms, D.; Martin, S.J. Neutrophil-Derived Proteases Escalate Inflammation through Activation of IL-36 Family Cytokines. Cell Rep. 2016, 14, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, F.; Streilein, R.D.; Hall, R.P. Increased E-selectin, IL-8 and IL-10 gene expression in human skin after minimal trauma. Exp. Dermatol. 2003, 12, 777–783. [Google Scholar] [CrossRef]
- Maverakis, E.; Van Den Elzen, P.; Sercarz, E.E. Self-reactive T cells and degeneracy of T cell recognition: Evolving concepts-from sequence homology to shape mimicry and TCR flexibility. J. Autoimmun. 2001, 16, 201–209. [Google Scholar] [CrossRef]
- Marzano, A.V.; Damiani, G.; Ceccherini, I.; Berti, E.; Gattorno, M.; Cugno, M. Autoinflammation in pyoderma gangrenosum and its syndromic form (pyoderma gangrenosum, acne and suppurative hidradenitis). Br. J. Dermatol. 2017, 176, 1588–1598. [Google Scholar] [CrossRef]
- Braun-Falco, M.; Kovnerystyy, O.; Lohse, P.; Ruzicka, T. Pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH)--a new autoinflammatory syndrome distinct from PAPA syndrome. J. Am. Acad. Dermatol. 2012, 66, 409–415. [Google Scholar] [CrossRef]
- Marzano, A.V.; Ishak, R.S.; Colombo, A.; Caroli, F.; Crosti, C. Pyoderma gangrenosum, acne and suppurative hidradenitis syndrome following bowel bypass surgery. Dermatology 2012, 225, 215–219. [Google Scholar] [CrossRef]
- Marzano, A.V.; Ceccherini, I.; Gattorno, M.; Fanoni, D.; Caroli, F.; Rusmini, M.; Grossi, A.; De Simone, C.; Borghi, O.M.; Meroni, P.L.; et al. Association of pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH) shares genetic and cytokine profiles with other autoinflammatory diseases. Medicine 2014, 93, e187. [Google Scholar] [CrossRef]
- Lukens, J.R.; Kanneganti, T.-D. SHP-1 and IL-1α conspire to provoke neutrophilic dermatoses. Rare Dis. 2014, 2, e27742. [Google Scholar] [CrossRef]
- Lukens, J.R.; Vogel, P.; Johnson, G.R.; Kelliher, M.A.; Iwakura, Y.; Lamkanfi, M.; Kanneganti, T.-D. RIP1-driven autoinflammation targets IL-1α independently of inflammasomes and RIP3. Nature 2013, 498, 224. [Google Scholar] [CrossRef]
- Tartey, S.; Gurung, P.; Dasari, T.K.; Burton, A.; Kanneganti, T.D. ASK1/2 signaling promotes inflammation in a mouse model of neutrophilic dermatosis. J. Clin. Investig. 2018, 128, 2042–2047. [Google Scholar] [CrossRef]
- Tartey, S.; Gurung, P.; Samir, P.; Burton, A.; Kanneganti, T.-D. Cutting Edge: Dysregulated CARD9 Signaling in Neutrophils Drives Inflammation in a Mouse Model of Neutrophilic Dermatoses. J. Immunol. 2018, 201, 1639–1644. [Google Scholar] [CrossRef] [PubMed]
- Johnston, A.; Xing, X.; Wolterink, L.; Barnes, D.H.; Yin, Z.; Reingold, L.; Kahlenberg, J.M.; Harms, P.W.; Gudjonsson, J.E. IL-1 and IL-36 are dominant cytokines in generalized pustular psoriasis. J. Allergy Clin. Immunol. 2017, 140, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Carrier, Y.; Ma, H.-L.; Ramon, H.E.; Napierata, L.; Small, C.; O’Toole, M.; Young, D.A.; Fouser, L.A.; Nickerson-Nutter, C.; Collins, M.; et al. Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: Implications in psoriasis pathogenesis. J. Investig. Dermatol. 2011, 131, 2428–2437. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.; Horan, R.; Stefanska, A.; Carey, A.; Leon, G.; Aguilera, M.; Statovci, D.; Moran, T.; Fallon, P.; Shanahan, F.; et al. IL-36α expression is elevated in ulcerative colitis and promotes colonic inflammation. Mucosal Immunol. 2016, 9, 1193–1204. [Google Scholar] [CrossRef]
- Boutet, M.-A.; Bart, G.; Penhoat, M.; Amiaud, J.; Brulin, B.; Charrier, C.; Morel, F.; Lecron, J.-C.; Rolli-Derkinderen, M.; Bourreille, A.; et al. Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn’s disease. Clin. Exp. Immunol. 2016, 184, 159. [Google Scholar] [CrossRef]
- Hessam, S.; Sand, M.; Gambichler, T.; Skrygan, M.; Rüddel, I.; Bechara, F.G. Interleukin-36 in hidradenitis suppurativa: Evidence for a distinctive proinflammatory role and a key factor in the development of an inflammatory loop. Br. J. Dermatol. 2018, 178, 761–767. [Google Scholar] [CrossRef]
- Pappu, R.; Ramirez-Carrozzi, V.; Sambandam, A. The interleukin-17 cytokine family: Critical players in host defence and inflammatory diseases. Immunology 2011, 134, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Senra, L.; Stalder, R.; Martinez, D.A.; Chizzolini, C.; Boehncke, W.H.; Brembilla, N.C. Keratinocyte-Derived IL-17E Contributes to Inflammation in Psoriasis. J. Investig. Dermatol. 2016, 136, 1970–1980. [Google Scholar] [CrossRef] [PubMed]
- Marzano, A.V.; Fanoni, D.; Antiga, E.; Quaglino, P.; Caproni, M.; Crosti, C.; Meroni, P.L.; Cugno, M. Expression of cytokines, chemokines and other effector molecules in two prototypic autoinflammatory skin diseases, pyoderma gangrenosum and Sweet’s syndrome. Clin. Exp. Immunol. 2014, 178, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Yeon, H.B.; Lindor, N.M.; Seidman, J.G.; Seidman, C.E. Pyogenic arthritis, pyoderma gangrenosum, and acne syndrome maps to chromosome 15q. Am. J. Hum. Genet. 2000, 66, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Oka, M.; Berking, C.; Nesbit, M.; Satyamoorthy, K.; Schaider, H.; Murphy, G.; Ichihashi, M.; Sauter, E.; Herlyn, M. Interleukin-8 overexpression is present in pyoderma gangrenosum ulcers and leads to ulcer formation in human skin xenografts. Lab. Investig. 2000, 80, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Rubas, K.; Reich, A.; Nowicka-Suszko, D.; Maj, J. The role of interleukins 6, 8, 17 and 23 in the pathogenesis of pyoderma gangrenosum. J. Eur. Acad. Dermatol. Venereol. 2023, 37, e660–e662. [Google Scholar] [CrossRef] [PubMed]
- Su, W.P.D.; Sctiroeter, A.L.; Perry, H.O.; Powell, F.C. Histopathologic and immunopathologic study of pyoderma gangrenosum. J. Cutan. Pathol. 1986, 13, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Brooklyn, T.N.; Williams, A.M.; Dunnill, M.G.S.; Probert, C.S. T-cell receptor repertoire in pyoderma gangrenosum: Evidence for clonal expansions and trafficking. Br. J. Dermatol. 2007, 157, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23-IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef]
- Brooklyn, T.N.; Dunnill, M.G.S.; Shetty, A.; Bowden, J.J.; Williams, J.D.L.; Griffiths, C.E.M.; Forbes, A.; Greenwood, R.; Probert, C.S. Infliximab for the treatment of pyoderma gangrenosum: A randomised, double blind, placebo controlled trial. Gut 2006, 55, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Craig, F.F.; Thomas, K.S.; Mitchell, E.J.; Williams, H.C.; Norrie, J.; Mason, J.M.; Ormerod, A.D. UK Dermatology Clinical Trials Network’s STOP GAP trial (a multicentre trial of prednisolone versus ciclosporin for pyoderma gangrenosum): Protocol for a randomised controlled trial. Trials 2012, 13, 51. [Google Scholar] [CrossRef]
- Alavi, A.; French, L.E.; Davis, M.D.; Brassard, A.; Kirsner, R.S. Pyoderma Gangrenosum: An Update on Pathophysiology, Diagnosis and Treatment. Am. J. Clin. Dermatol. 2017, 18, 355–372. [Google Scholar] [CrossRef]
- Miller, J.; Yentzer, B.A.; Clark, A.; Jorizzo, J.L.; Feldman, S.R. Pyoderma gangrenosum: A review and update on new therapies. J. Am. Acad. Dermatol. 2010, 62, 646–654. [Google Scholar] [CrossRef]
- Croitoru, D.; Naderi-Azad, S.; Sachdeva, M.; Piguet, V.; Alavi, A. A Wound Care Specialist’s Approach to Pyoderma Gangrenosum. Adv. Wound Care 2020, 9, 686–694. [Google Scholar] [CrossRef]
- Almeida, I.R.; Coltro, P.S.; Gonçalves, H.O.C.; Westin, A.T.; Almeida, J.B.; Lima, R.V.K.S.; Silva, M.F.; Junior, J.A.F. The role of negative pressure wound therapy (NPWT) on the treatment of pyoderma gangrenosum: A systematic review and personal experience. Wound Repair Regen. 2021, 29, 486–494. [Google Scholar] [CrossRef]
- Bazaliński, D.; Krawiec, A.; Kucharzewski, M.; Więch, P. Negative Pressure Wound Therapy in Pyoderma Gangrenosum Treatment. Am. J. Case Rep. 2020, 21, 1–6. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Yanagi, T.; Sato, K.; Yoshimoto, N.; Hirata, Y.; Ujiie, I.; Nishimura, M.; Natsuga, K.; Shiiya, C.; Tsukinaga, I.; et al. Portable negative-pressure wound therapy for pyoderma gangrenosum: Report of two cases. J. Dermatol. 2018, 45, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, O.; Middleton, D.; Hunter, H. Negative-pressure wound therapy as an adjunct to treating pyoderma gangrenosum. J. R. Coll. Physicians Edinb. 2022, 52, 260–262. [Google Scholar] [CrossRef]
- Eisendle, K.; Thuile, T.; Deluca, J.; Pichler, M. Surgical Treatment of Pyoderma Gangrenosum with Negative Pressure Wound Therapy and Skin Grafting, Including Xenografts: Personal Experience and Comprehensive Review on 161 Cases. Adv. Wound Care 2020, 9, 405–425. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.S.; Ormerod, A.D.; Craig, F.E.; Greenlaw, N.; Norrie, J.; Mitchell, E.; Mason, J.M.; Johnston, G.A.; Wahie, S.; Williams, H.C.; et al. Clinical outcomes and response of patients applying topical therapy for pyoderma gangrenosum: A prospective cohort study. J. Am. Acad. Dermatol. 2016, 75, 940–949. [Google Scholar] [CrossRef]
- Lyon, C.C.; Stapleton, M.; Smith, A.J.; Mendelsohn, S.; Beck, M.H.; Griffiths, C.E.M. Topical tacrolimus in the management of peristomal pyoderma gangrenosum. J. Dermatol. Treat. 2001, 12, 13–17. [Google Scholar] [CrossRef]
- Chow, R.K.P.; Ho, V.C. Treatment of pyoderma gangrenosum. J. Am. Acad. Dermatol. 1996, 34, 1047–1060. [Google Scholar] [CrossRef]
- Mrowieiz, U.; Christophers, E. Clearing of pyoderma gangrenosum by intralesional cyclosporin A. Br. J. Dermatol. 1991, 125, 499. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, K.; Aflaki, E.; Akbarzade Jahromi, M.; Dastgheib, L. Successful Treatment of Classic Pyoderma Gangrenosum with Intralesional Infliximab Injection: A Case Report. Australas. J. Dermatol. 2023, 64, e252–e255. [Google Scholar] [CrossRef]
- Cozzani, E.; Gasparini, G.; Parodi, A. Pyoderma gangrenosum: A systematic review. G. Ital. Dermatol. Venereol. 2014, 149, 587–600. Available online: https://pubmed.ncbi.nlm.nih.gov/25213386/ (accessed on 20 April 2023).
- Pyoderma Gangrenosum: Treatment and Prognosis—UpToDate. Available online: https://www.uptodate.com/contents/pyoderma-gangrenosum-treatment-and-prognosis (accessed on 20 April 2023).
- Holt, P.J.; Davies, M.G.; Saunders, K.C.; Nuki, G. Pyoderma gangrenosum: Clinical and laboratory findings in 15 patients with special reference to polyarthritis. Medicine 1980, 59, 114–133. Available online: https://pubmed.ncbi.nlm.nih.gov/7360040/ (accessed on 20 April 2023). [CrossRef]
- Yamauchi, T.; Ishida, K.; Iwashima, Y.; Ikegaya, S.; Kawai, Y.; Ueda, T.; Wakahara, M.; Kumakiri, M. Successful treatment of pyoderma gangrenosum that developed in a patient with myelodysplastic syndrome. J. Infect. Chemother. 2003, 9, 268–271. [Google Scholar] [CrossRef]
- Ambooken, B.; Khader, A.; Muhammed, K.; Rajan, U.; Snigdha, O. Malignant pyoderma gangrenosum eroding the parotid gland successfully treated with dexamethasone pulse therapy. Int. J. Dermatol. 2014, 53, 1536–1538. [Google Scholar] [CrossRef]
- Ormerod, A.D.; Thomas, K.S.; Craig, F.E.; Mitchell, E.; Greenlaw, N.; Norrie, J.; Mason, J.M.; Walton, S.; Johnston, G.A.; Williams, H.C.; et al. Comparison of the two most commonly used treatments for pyoderma gangrenosum: Results of the STOP GAP randomised controlled trial. BMJ 2015, 350, h2958. [Google Scholar] [CrossRef] [PubMed]
- Perry, H.O. Pyoderma gangrenosum. South. Med. J. 1969, 62, 899–907. [Google Scholar] [CrossRef]
- Callen, J.P.; Jackson, J.M. Pyoderma gangrenosum: An update. Rheum. Dis. Clin. N. Am. 2007, 33, 787–802. [Google Scholar] [CrossRef]
- Graziano, F.; Macaluso, F.S.; Cassata, N.; Citrano, M.; Orlando, A. Pyoderma Gangrenosum in An Ulcerative Colitis Pediatric Patient During Vedolizumab Therapy Successfully Treated with Oral Cyclosporine. Inflamm. Bowel. Dis. 2021, 27, E110–E111. [Google Scholar] [CrossRef] [PubMed]
- Vidal, D.; Puig, L.; Gilaberte, M.; Alomar, A. Review of 26 cases of classical pyoderma gangrenosum: Clinical and therapeutic features. J. Dermatol. Treat. 2004, 15, 146–152. [Google Scholar] [CrossRef]
- Hasselmann, D.O.; Bens, G.; Tilgen, W.; Reichrath, J. Pyoderma gangrenosum: Clinical presentation and outcome in 18 cases and review of the literature. JDDG J. Der Dtsch. Dermatol. Ges. 2007, 5, 560–564. [Google Scholar] [CrossRef]
- Eaton, P.A.; Callen, J.P. Mycophenolate mofetil as therapy for pyoderma gangrenosum. Arch. Dermatol. 2009, 145, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kelly, R. Treatment of pyoderma gangrenosum with mycophenolate mofetil as a steroid-sparing agent. J. Am. Acad. Dermatol. 2013, 69, 565–569. [Google Scholar] [CrossRef]
- Hrin, M.L.; Bashyam, A.M.; Huang, W.W.; Feldman, S.R. Mycophenolate mofetil as adjunctive therapy to corticosteroids for the treatment of pyoderma gangrenosum: A case series and literature review. Int. J. Dermatol. 2021, 60, e486–e492. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Hrin, M.L.; Bowers, N.L.; Jorizzo, J.L.; Feldman, S.R.; Huang, W.W. Methotrexate for pyoderma gangrenosum: A retrospective case series of 33 patients. J. Am. Acad. Dermatol. 2023, 90, 642–644. [Google Scholar] [CrossRef]
- Sardar, P.; Guha, P.; Das, N.K.; Gharami, R.C.; Majumdar, S.; Banerjee, D.; Banerjee, R. Ulcerative pyoderma gangrenosum in mixed connective tissue disorder: A rare association and role of azathioprine in the management. Indian J. Dermatol. 2011, 56, 600–602. [Google Scholar] [CrossRef] [PubMed]
- Nazir, A.; Zafar, A. Management of Idiopathic Pyoderma Gangrenosum with Azathioprine As the Primary Adjunct in an Asian Man: A Case Report. Cureus 2022, 14, e25177. [Google Scholar] [CrossRef]
- Wozel, G.; Blasum, C. Dapsone in dermatology and beyond. Arch. Dermatol. Res. 2014, 306, 103–124. [Google Scholar] [CrossRef]
- Galun, E.; Flugelman, M.Y.; Rachmilewitz, D. Pyoderma gangrenosum complicating ulcerative colitis: Successful treatment with methylprednisolone pulse therapy and dapsone. Am. J. Gastroenterol. 1986, 81, 988–989. Available online: https://pubmed.ncbi.nlm.nih.gov/3766502/ (accessed on 20 April 2023).
- Brown, R.E.; Lay, L.; Graham, D. Bilateral pyoderma gangrenosum of the hand: Treatment with dapsone. J. Hand Surg. Br. 1993, 18, 119–121. [Google Scholar] [CrossRef]
- Teasley, L.A.; Foster, C.S.; Baltatzis, S. Sclerokeratitis and facial skin lesions: A case report of pyoderma gangrenosum and its response to dapsone therapy. Cornea 2007, 26, 215–219. [Google Scholar] [CrossRef]
- Din, R.S.; Tsiaras, W.G.; Li, D.G.; Mostaghimi, A. Efficacy of Systemic Dapsone Treatment for Pyoderma Gangrenosum: A Retrospective Review. J. Drugs Dermatol. 2018, 17, 1058–1060. Available online: https://pubmed.ncbi.nlm.nih.gov/30365585/ (accessed on 20 April 2023). [PubMed]
- Shenefelt, P.D. Pyoderma gangrenosum associated with cystic acne and hidradenitis suppurativa controlled by adding minocycline and sulfasalazine to the treatment regimen. Cutis 1996, 57, 315–319. Available online: https://pubmed.ncbi.nlm.nih.gov/8726710/ (accessed on 20 April 2023).
- Reynolds, N.J.; Peachey, R.D. Response of atypical bullous pyoderma gangrenosum to oral minocycline hydrochloride and topical steroids. Acta. Derm. Venereol. 1990, 138, 538–539. Available online: https://pubmed.ncbi.nlm.nih.gov/1981437/ (accessed on 20 April 2023). [CrossRef]
- Miralles, E.S.; Nunez, M.; Perez, B.; Ledo, A. Minocycline hydrochloride hyperpigmentation complicating treatment of pyoderma gangrenosum. J. Dermatol. 1994, 21, 965–967. [Google Scholar] [CrossRef]
- Song, H.; Lahood, N.; Mostaghimi, A. Intravenous immunoglobulin as adjunct therapy for refractory pyoderma gangrenosum: Systematic review of cases and case series. Br. J. Dermatol. 2018, 178, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, U.; Romano, P.; Mulcahy, L.D.; Dooley, L.T.; Baker, D.G.; Gottlieb, A.B. Efficacy and safety of infliximab monotherapy for plaque-type psoriasis: A randomised trial. Lancet 2001, 357, 1842–1847. [Google Scholar] [CrossRef] [PubMed]
- Regueiro, M.; Valentine, J.; Plevy, S.; Fleisher, M.R.; Lichtenstein, G.R. Infliximab for treatment of pyoderma gangrenosum associated with inflammatory bowel disease. Am. J. Gastroenterol. 2003, 98, 1821–1826. [Google Scholar] [CrossRef] [PubMed]
- Argüelles-Arias, F.; Castro-Laria, L.; Lobatón, T.; Aguas-Peris, M.; Rojas-Feria, M.; Acosta, M.B.-D.; Soto-Escribano, P.; Calvo-Moya, M.; Ginard-Vicens, D.; Chaparro-Sánchez, M.; et al. Characteristics and treatment of pyoderma gangrenosum in inflammatory bowel disease. Dig. Dis. Sci. 2013, 58, 2949–2954. [Google Scholar] [CrossRef]
- Tumor Necrosis Factor-Alpha Inhibitors: An Overview of Adverse Effects—UpToDate. Available online: https://www.uptodate.com/contents/tumor-necrosis-factor-alpha-inhibitors-an-overview-of-adverse-effects (accessed on 20 April 2023).
- Ljung, T.; Staun, M.; Grove, O.; Fausa, O.; Vatn, M.H.; Hellström, P.M. Pyoderma gangrenosum associated with crohn disease: Effect of TNF-alpha blockade with infliximab. Scand. J. Gastroenterol. 2002, 37, 1108–1110. [Google Scholar] [CrossRef] [PubMed]
- Salehzadeh, F.; Mohammadikebar, Y.; Enteshary, A.; Ghanbarpour, O.; Mirzarahimi, M. Infliximab in treatment of idiopathic refractory childhood pyoderma gangrenosum (PG). Biologics 2019, 13, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.R.; Lewis, H.M. Severe recalcitrant pyoderma gangrenosum treated with infliximab. Br. J. Dermatol. 2005, 153, 689–691. [Google Scholar] [CrossRef] [PubMed]
- Betetto, L.Đ.; Točkova, O.; Suhodolčan, A.B. Mucocutaneous pyoderma gangrenosum: A case report and literature review. Acta Dermatovenerol. Alp. Pannonica Adriat. 2022, 31, S10–S13. [Google Scholar] [CrossRef]
- Yamasaki, K.; Yamanaka, K.; Zhao, Y.; Iwano, S.; Takei, K.; Suzuki, K.; Yamamoto, T. Adalimumab in Japanese patients with active ulcers of pyoderma gangrenosum: Final analysis of a 52-week phase 3 open-label study. J. Dermatol. 2022, 49, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Seishima, M.; Sasaki, M.; Sugie, S. Successful treatment of pyoderma gangrenosum using adalimumab in a patient undergoing hemodialysis. J. Dermatol. 2022, 49, e435–e436. [Google Scholar] [CrossRef] [PubMed]
- Ohmura, S.; Homma, Y.; Hanai, S.; Otsuki, Y.; Miyamoto, T. Successful switching treatment of adalimumab for refractory pyoderma gangrenosum in a patient with rheumatoid arthritis with prior use of tumour necrosis factor inhibitors: A case report and review of the literature. Mod. Rheumatol. Case Rep. 2023, 7, 9–13. [Google Scholar] [CrossRef]
- Campanati, A.; Brisigotti, V.; Ganzetti, G.; Molinelli, E.; Giuliodori, K.; Consales, V.; Racchini, S.; Bendia, E.; Offidani, A. Finally, recurrent pyoderma gangrenosum treated with Adalimumab: Case report and review of the literature. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 1245–1247. [Google Scholar] [CrossRef]
- Kikuchi, N.; Hiraiwa, T.; Ohashi, T.; Hanami, Y.; Satoh, M.; Takenoshita, H.; Yamamoto, T. Pyoderma gangrenosum possibly triggered by adalimumab. Eur. J. Dermatol. 2012, 22, 804–805. [Google Scholar] [CrossRef]
- Benzaquen, M.; Monnier, J.; Beaussault, Y.; Rouby, F.; Berbis, P. Pyoderma gangrenosum arising during treatment of psoriasis with adalimumab: Effectiveness of ustekinumab. Australas J. Dermatol. 2017, 58, e270–e271. [Google Scholar] [CrossRef]
- Tan, Y.; Kavaklieva, S.; Wood, F. Pyoderma gangrenosum induced by adalimumab in a seropositive rheumatoid arthritis patient: A paradoxical effect of adalimumab? Rheumatology 2021, 60, e288–e289. [Google Scholar] [CrossRef]
- A Study to Assess Adverse Events and Change in Disease State in Adult Participants Being Treated with Humira in Participants Diagnosed with Pyoderma Gangrenosum (PG). Available online: https://clinicaltrials.gov/study/NCT04750213?cond=Pyoderma%20Gangrenosum&rank=4&limit=10&aggFilters=status:rec%20not&page=1 (accessed on 2 January 2024).
- Ariane, M.; Bouaziz, J.D.; de Masson, A.; Jachiet, M.; Bagot, M.; Lepelletier, C. Efficacy and safety of etanercept for postoperative pyoderma gangrenosum after infliximab serum sickness. Dermatol. Ther. 2019, 32, e12774. [Google Scholar] [CrossRef]
- Kim, F.S.; Pandya, A.G. The use of etanercept in the treatment of peristomal pyoderma gangrenosum. Clin. Exp. Dermatol. 2012, 37, 442–443. [Google Scholar] [CrossRef]
- Roy, D.B.; Conte, E.T.; Cohen, D.J. The treatment of pyoderma gangrenosum using etanercept. J. Am. Acad. Dermatol. 2006, 54, S128–S134. [Google Scholar] [CrossRef]
- Rogge, F.J.; Pacifico, M.; Kang, N. Treatment of pyoderma gangrenosum with the anti-TNFalpha drug—Etanercept. J. Plast. Reconstr. Aesthet. Surg. 2008, 61, 431–433. [Google Scholar] [CrossRef]
- Haridas, V.; Shetty, P.; Dsouza, L.C.; Dinesh, U.S.; Haridas, K.; Bargale, A. Pyoderma gangrenosum in Sjögren’s syndrome and its successful treatment with topical application of etanercept. Int. J. Rheum. Dis. 2017, 20, 657–659. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, G.; Jorizzo, J.L. Use of etanercept in treatment of pyoderma gangrenosum in a patient with autoimmune hepatitis. J. Dermatol. Treat. 2005, 16, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Pastor, N.; Betlloch, I.; Pascual, J.C.; Blanes, M.; Bañuls, J.; Silvestre, J.F. Pyoderma gangrenosum treated with anti-TNF alpha therapy (etanercept). Clin. Exp. Dermatol. 2006, 31, 152–153. [Google Scholar] [CrossRef] [PubMed]
- McGowan, J.W., 4th; Johnson, C.A.; Lynn, A. Treatment of pyoderma gangrenosum with etanercept. J. Drugs Dermatol. 2004, 3, 441–444. Available online: https://pubmed.ncbi.nlm.nih.gov/15303791/ (accessed on 20 April 2023). [PubMed]
- Guedes, R.; Moreira, A.; Menezes, N.; Baptista, A.; Varela, P. Treatment of thalidomide resistant pyoderma gangrenosum with etanercept. Acta Dermatovenerol. Croat. 2012, 20, 175–180. Available online: https://pubmed.ncbi.nlm.nih.gov/23069303/ (accessed on 20 April 2023). [PubMed]
- Abdallah, H.B.; Fogh, K.; Bech, R. Pyoderma gangrenosum and tumour necrosis factor alpha inhibitors: A semi-systematic review. Int. Wound J. 2019, 16, 511–521. [Google Scholar] [CrossRef]
- Kowalzick, L.; Bertolini, J.; Baumann, C.; Walther, B.; Truhm, B.; Eickenscheidt, L. Paradoxical Reaction to Etanercept: Development of Pyoderma Gangraenosum during Therapy of Psoriasis Arthritis. J. Dtsch. Dermatol. Ges. 2013, 11, 447–449. [Google Scholar] [CrossRef]
- Kleinpenning, M.M.; Langewouters, A.M.G.; Van De Kerkhof, P.C.M.; Greebe, R.J. Severe pyoderma gangrenosum unresponsive to etanercept and adalimumab. J. Dermatol. Treat. 2011, 22, 261–265. [Google Scholar] [CrossRef]
- Vallerand, I.A.; Hardin, J. Ustekinumab for the treatment of recalcitrant pyoderma gangrenosum: A case report. SAGE Open Med. Case Rep. 2019, 7, 2050313X1984520. [Google Scholar] [CrossRef]
- González, J.L.; Sáez, M.L.; Moraleda, I.M.; Martínez, Á.H. Pyoderma gangrenosum solved by ustekinumab therapy. Gastroenterol. Hepatol. 2021, 44, 299–300. [Google Scholar] [CrossRef]
- Fahmy, M.; Ramamoorthy, S.; Hata, T.; Sandborn, W.J. Ustekinumab for peristomal pyoderma gangrenosum. Am. J. Gastroenterol. 2012, 107, 794–795. [Google Scholar] [CrossRef] [PubMed]
- Low, Z.M.; Mar, A. Treatment of severe recalcitrant pyoderma gangrenosum with ustekinumab. Australas J. Dermatol. 2018, 59, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Cámara, P.G.; Ara, M.L.Z.; López, S.G. Ustekinumab in a patient with pyoderma gangrenosum and refractory Crohn’s disease. Med. Clin. 2019, 153, e35–e36. [Google Scholar] [CrossRef]
- Piqueras-García, J.; Sahuquillo-Torralba, A.J.; Torres-Navarro, I.; Botella-Estrada, R. Pyoderma Gangrenosum with Ulcerative Colitis Successfully Treated with Ustekinumab. Actas Dermosifiliogr. 2019, 110, 776–778. [Google Scholar] [CrossRef] [PubMed]
- Nieto, D.; Sendagorta, E.; Rueda, J.M.; Herranz, P. Successful treatment with ustekinumab and vacuum-assisted closure therapy in recalcitrant myelodysplastic syndrome-associated pyoderma gangrenosum: Case report and literature review. Clin. Exp. Dermatol. 2019, 44, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Goldminz, A.M.; Botto, N.C.; Gottlieb, A.B. Severely recalcitrant pyoderma gangrenosum successfully treated with ustekinumab. J. Am. Acad. Dermatol. 2012, 67, e237–e238. [Google Scholar] [CrossRef] [PubMed]
- Petty, A.J.; Whitley, M.J.; Balaban, A.; Ellington, K.; Marano, A.L. Pyoderma gangrenosum induced by secukinumab in a patient with psoriasis successfully treated with ustekinumab. JAAD Case Rep. 2020, 6, 731–733. [Google Scholar] [CrossRef] [PubMed]
- Cosgarea, I.; Lovric, Z.; Körber, A.; Dissemond, J. Successful treatment of refractory pyoderma gangrenosum with ustekinumab only after excision of renal cell carcinoma. Int. Wound J. 2016, 13, 1041–1042. [Google Scholar] [CrossRef]
- Guenova, E.; Teske, A.; Fehrenbacher, B.; Hoerber, S.; Adamczyk, A.; Schaller, M.; Hoetzenecker, W.; Biedermann, T. Interleukin 23 expression in pyoderma gangrenosum and targeted therapy with ustekinumab. Arch. Dermatol. 2011, 147, 1203–1205. [Google Scholar] [CrossRef]
- Nunes, G.; Patita, M.; Fernandes, V. Refractory Pyoderma Gangrenosum in a Patient with Crohn’s Disease: Complete Response to Ustekinumab. J. Crohns Colitis 2019, 13, 812–813. [Google Scholar] [CrossRef] [PubMed]
- Kolios, A.; Maul, J.; Meier, B.; Kerl, K.; Traidl-Hoffmann, C.; Hertl, M.; Zillikens, D.; Röcken, M.; Ring, J.; Facchiano, A.; et al. Canakinumab in adults with steroid-refractory pyoderma gangrenosum. Br. J. Dermatol. 2015, 173, 1216–1223. [Google Scholar] [CrossRef]
- Acierno, S.; Angrisani, F.; Marino, A.; Caporali, R.F.; Cimaz, R.; Giani, T. Canakinumab treatment in a young girl with refractory chronic recurrent multifocal osteomyelitis associated with pyoderma gangrenosum. Int. J. Rheum. Dis. 2022, 25, 1333–1338. [Google Scholar] [CrossRef]
- Jaeger, T.; Andres, C.; Grosber, M.; Zirbs, M.; Hein, R.; Ring, J.; Traidl-Hoffmann, C. Pyoderma gangrenosum and concomitant hidradenitis suppurativa--rapid response to canakinumab (anti-IL-1β). Eur. J. Dermatol. 2013, 23, 408–410. [Google Scholar] [CrossRef]
- O’Connor, C.; Gallagher, C.; Hollywood, A.; Paul, L.; O’Connell, M. Anakinra for recalcitrant pyoderma gangrenosum. Clin. Exp. Dermatol. 2021, 46, 1558–1560. [Google Scholar] [CrossRef] [PubMed]
- Coe, J.; Kudva, S.; Shams, K. Matching the dose to the disease: Successful treatment of recalcitrant pyoderma gangrenosum using high-dose secukinumab. Dermatol. Ther. 2022, 35, e15669. [Google Scholar] [CrossRef] [PubMed]
- Kao, A.S.; King, A.D.; Bardhi, R.; Daveluy, S. Targeted therapy with ixekizumab in pyoderma gangrenosum: A case series and a literature overview. JAAD Case Rep. 2023, 37, 49–53. [Google Scholar] [CrossRef]
- Tee, M.W.; Avarbock, A.B.; Ungar, J.; Frew, J.W. Rapid resolution of pyoderma gangrenosum with brodalumab therapy. JAAD Case Rep. 2020, 6, 1167–1169. [Google Scholar] [CrossRef]
- McPhie, M.L.; Kirchhof, M.G. Pyoderma gangrenosum treated with secukinumab: A case report. SAGE Open Med. Case Rep. 2020, 8, 2050313X2094043. [Google Scholar] [CrossRef]
- García, M.M.; González, M.M.; Lobato, J.M.P. Secukinumab for pyoderma gangrenosum: A case report. Med. Clin. 2019, 152, 246. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhou, S.; Xiong, H.; Yang, J.; Yang, Z.; Zhou, N.; Mao, J.; Li, M. A Case of Paradoxical Reactions to Biologic Therapy for Psoriasis. Clin. Cosmet. Investig. Dermatol. 2023, 16, 1493–1497. [Google Scholar] [CrossRef] [PubMed]
- Orita, A.; Hoshina, D.; Hirosaki, K. Pyoderma gangrenosum caused by secukinumab successfully treated with risankizumab: A case report and literature review. Clin. Exp. Dermatol. 2022, 47, 1372–1374. [Google Scholar] [CrossRef]
- Sadik, C.D.; Thieme, M.; Zillikens, D.; Terheyden, P. First emergence of pyoderma gangraenosum, palmoplantar pustulosis and sacroiliitis in a psoriasis patient associated with switching from secukinumab to brodalumab. J. Eur. Acad. Dermatol. Venereol. 2019, 33, e406–e407. [Google Scholar] [CrossRef]
- Kao, A.S.; King, A.D.; Daveluy, S. Successful treatment of cabozantinib-induced pyoderma gangrenosum with ixekizumab therapy: A case report. Dermatol. Ther. 2022, 35, e15716. [Google Scholar] [CrossRef]
- Pollack, I.R.; Wolner, Z.J.; Hammett, J.; Swerlick, R.A. Pyoderma gangrenosum in a patient on ixekizumab. JAAD Case Rep. 2021, 16, 152–154. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Oak, A.S.W.; Elewski, B.E. Use of IL-23 Inhibitors for the Treatment of Plaque Psoriasis and Psoriatic Arthritis: A Comprehensive Review. Am. J. Clin. Dermatol. 2021, 22, 173–192. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K. New treatment of pyoderma gangrenosum and hidradenitis suppurativa: A review. J. Dermatol. 2023, 51, 172–179. [Google Scholar] [CrossRef]
- Leow, L.J.; Zubrzycki, N. Recalcitrant Ulcerative Pyoderma Gangrenosum of the Leg Responsive to Tildrakizumab: A Case Report. Clin. Cosmet. Investig. Dermatol. 2022, 15, 1729–1736. [Google Scholar] [CrossRef]
- John, J.M.; Sinclair, R.D. Tildrakizumab for treatment of refractory pyoderma gangrenosum of the penis and polymyalgia rheumatica: Killing two birds with one stone. Australas. J. Dermatol. 2020, 61, 170–171. [Google Scholar] [CrossRef]
- Çalışkan, E.; Edek, Y.C.; Adışen, E.; İlter, N. Peristomal pyoderma gangrenosum treated with interleukin 23 inhibitor treatment: A case report. J. Eur. Acad. Dermatol. Venereol. 2023. [Google Scholar] [CrossRef]
- Baier, C.; Barak, O. Guselkumab as a treatment option for recalcitrant pyoderma gangrenosum. JAAD Case Rep. 2020, 8, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Reese, A.M.; Erickson, K.; Reed, K.B.; Ortega-Loayza, A.G. Modified dose of guselkumab for treatment of pyoderma gangrenosum. JAAD Case Rep. 2022, 21, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Burgdorf, B.; Schlott, S.; Ivanov, I.H.; Dissemond, J. Successful treatment of a refractory pyoderma gangrenosum with Risankizumab. Int. Wound J. 2020, 17, 1086–1088. [Google Scholar] [CrossRef] [PubMed]
- Piñeiro, L.V.; Martínez, M.L.; Pin, A.I.C. Off-label use of guselkumab for pyoderma gangrenosum. Med. Clin. 2023, 161, 226–227. [Google Scholar] [CrossRef]
- Castro, L.G.M. JAK inhibitors: A novel, safe, and efficacious therapy for pyoderma gangrenosum. Int. J. Dermatol. 2023, 62, 1088–1093. [Google Scholar] [CrossRef]
- Dos Santos, M.R.D.; Ianhez, M.; Ribeiro, B.N.; de Queiroz, B.B.; Miot, H.A. Refractory pyoderma gangrenosum associated with rheumatoid arthritis successfully treated with upadacitinib. Comments on: ‘JAK inhibitors: A novel, safe, and efficacious therapy for pyoderma gangrenosum. Int. J. Dermatol. 2023, 62, e595–e598. [Google Scholar] [CrossRef] [PubMed]
- Scheinberg, M.; Machado, L.A.; Castro, L.G.M.; Ferreira, S.B.; Michalany, N. Successful treatment of ulcerated pyoderma gangrenosum with baricitinib, a novel JAK inhibitor. J. Transl. Autoimmun. 2021, 4, 100099. [Google Scholar] [CrossRef]
- Nasifoglu, S.; Heinrich, B.; Welzel, J. Successful therapy for pyoderma gangrenosum with a Janus kinase 2 inhibitor. Br. J. Dermatol. 2018, 179, 504–505. [Google Scholar] [CrossRef]
- Baricitinib in the Treatment of Adults with Pyoderma Gangrenosum (PG). Available online: https://clinicaltrials.gov/study/NCT04901325?cond=Pyoderma%20Gangrenosum&rank=8&limit=10&aggFilters=status:rec%20not&page=1 (accessed on 2 January 2024).
- Kochar, B.; Herfarth, N.; Mamie, C.; Navarini, A.A.; Scharl, M.; Herfarth, H.H. Tofacitinib for the Treatment of Pyoderma Gangrenosum. Clin. Gastroenterol. Hepatol. 2019, 17, 991–993. [Google Scholar] [CrossRef]
- Olavarría, P.S.; Iturria, S.R.; Castillejo, Ó.N. Tofacitinib, a useful option for the treatment of pyoderma gangrenosum in an ulcerative colitis patient. Rev. Esp. De Enfermedades Dig. 2021, 113, 733–734. [Google Scholar] [CrossRef]
- Guénin, S.H.; Khattri, S.; Lebwohl, M.G. Spesolimab use in treatment of pyoderma gangrenosum. JAAD Case Rep. 2023, 34, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chen, X.; Guo, Q.; Qiao, Z.; Wang, N.; Pai, P.; Liu, X. Rapid Response to Spesolimab in a Patient with severe refractory Pyoderma Gangrenosum. Clin. Exp. Dermatol. 2023, 49, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Spesolimab in Pyoderma Gangrenosum. Available online: https://clinicaltrials.gov/study/NCT06092216?cond=Pyoderma%20Gangrenosum&rank=2&limit=10&aggFilters=status:rec%20not&page=1 (accessed on 2 January 2024).
- Exploratory Study of IFX-1 in Patients with Pyoderma Gangrenosum. Available online: https://clinicaltrials.gov/study/NCT03971643?cond=Pyoderma%20Gangrenosum&rank=12&limit=10&page=2 (accessed on 2 January 2024).
- Phase III Trial to Investigate Efficacy and Safety of Vilobelimab in Ulcerative Pyoderma Gangrenos. Available online: https://clinicaltrials.gov/study/NCT05964413?cond=Pyoderma%20Gangrenosum&rank=5&limit=10&aggFilters=status:rec%20not&page=1 (accessed on 2 January 2024).


| Authors (Year) | Systemic Drug | Dosage Regimen | Method of Administration | Others |
|---|---|---|---|---|
| T. Yamauchi et al. (2003) [82] | Methylprednisolone | 1 g for 3 days | i.v. | Dosage reduced within 2 weeks—therapy maintained with 30 mg prednisolone daily for 6 months |
| B. Ambooken et al. (2014) [83] | Dexamethasone | 100 mg in 500 mL 5% dextrose infused over 3–4 h on 3 consecutive days | i.v. | 9 pulses at 28 days intervals |
| A. D. Ormerod et al. (2015) [84] | Prednisolone | 0.75 mg/kg/day; maximum dose 75 mg/day | p.o. | - |
| A. D. Ormerod et al. (2015) [84] | Cyclosporine A | 4 mg/kg/day; maximum dose 400 mg/day | p.o. | - |
| P. A. Eaton et al. (2009). [90] | Mycophenolate mofetil | Initial dose 0.5/day or 1g/day; maximal dosages from 0.5 g 4 times daily to 2 g twice daily | p.o. | - |
| J. Li et al. (2013). [91] | Mycophenolate mofetil | 1 g or 2 g total daily | p.o. | The maintenance dose was 2 g or 3 g total daily; the average duration of treatment was 12.1 months |
| M. L. Hrin et al. (2021) [92] | Mycophenolate mofetil | 1g to 2.5 g daily | p.o. | - |
| J. A.Williams et al. (2023) [93] | Methotrexate | 5–25 mg | ND | 97% received concominant prednisone |
| P. Sardar et al. (2011) [94] | Azathioprine | 1 mg/kg daily | p.o. | Patient was unresponsive to systemic steroid and dapsone |
| E. Galun (1986) [97]; R.E. Brown (1993) [98] L. A. Teasley et al. (2007) [99]; R. S. Din et al. (2018) [100] | Dapsone | 50–200 mg daily | p.o. | Screening for glucose-6-phosphate dehydrogenase (G6PD) before and during treatment due to hematologic toxicity |
| P. D. Shenefelt et al. (1990) [101]; N. J. Reynolds et al. (1996) [102] | Minocycline | 100 mg twice daily | p.o. | Combination with other therapeutics |
| H. Song et al. (2018) [104] | Intravenous immunoglobulin | 2 g/kg | i.v. | The mean time to initial response of 3–5 weeks |
| Authors (Year) | Biologic Drug | Dosage Regimen | Study Type | Comorbidities | Efficacy |
|---|---|---|---|---|---|
| F. Argüelles-Arias et al. (2013) [107] | Adalimumab | 160/80 mg given s.c. at 0 and 2 weeks, and then every 2 weeks | Retrospective observational study | IBD in all cases | 7 patients, complete response |
| K. Yamasaki et al. (2022) [113] | Adalimumab | 160 mg s.c. at week 0, 80 mg at week 2, and then 40 mg every week from week 4 | Open-label study | UC; RA; hypertension; hyperlipidemia; hyperuricemia; osteoporosis | 12 (54.5%) of 22 patients reached a satisfactory outcome |
| M. Seishima et al. (2022) [114] | Adalimumab | 160 and 80 mg given s.c., biweekly, and then 40 mg weekly | Case report | History of systemic sarcoidosis; renal failure | Satisfactory result |
| S. Ohmura et al. (2023) [115] | Adalimumab | NA | Case report | RA | Satisfactory result |
| A. Campanati et al. (2015) [116] | Adalimumab | 160 mg s.c. at week 0, 80 mg at week 1, and then 40 mg every 2 weeks | Case report | CD | Complete healing after 12 weeks |
| Authors (Year) | Biologic Drug | Dosage Regimen | Study Type | Comorbidities | Efficacy |
|---|---|---|---|---|---|
| M. Ariane et al. (2019) [121] | Etanercept | 50 mg per week s.c. | Case report | None known; breast plastic surgery | Complete remission |
| F. S. Kim et al. (2012) [122] | Etanercept | 50 mg twice weekly; at 9 months, 50 mg per week s.c. | Case report | CD | Satisfactory result |
| D. B. Roy et al. (2006) [123] | Etanercept | 25 mg twice weekly s.c. | Case reports (n = 3) |
| Complete healing after 2 months in patients 1 and 3; satisfactory result in patient 2 |
| F. J. Rogge et al. (2008) [124] | Etanercept | 50 mg per week s.c. | Case report | None known | Complete healing after 7 months |
| V. Haridas et al. (2017) [125] | Etanercept | 1 mg/2 × 2 cm area—topical | Case report | Sjogren’s syndrome | Satisfactory result |
| G. Goldenberg et al. (2005) [126] | Etanercept | 25 mg twice weekly s.c. | Case report | Autoimmune hepatitis | Complete healing after 5 months |
| N. Pastor et al. (2005) [127] | Etanercept | 25 mg twice weekly s.c. | Case report | NA | Complete healing after 8 weeks |
| JW 4th McGowan et al. (2004) [128] | Etanercept | NA | Case report | NA | Satisfactory result |
| R. Guedes et al. (2012) [129] | Etanercept | NA | Case report | NA | Satisfactory result |
| M. M. Kleinpenning et al. (2011) [132] | Etanercept | 50 mg twice weekly s.c. | Case report | Hypogammaglobulinemia | Insufficient clinical improvement |
| Authors (Year) | Biologic Drug | Dosage Regimen | Study Type | Comorbidities | Efficacy |
|---|---|---|---|---|---|
| M. Benzaquen et al. (2017) [118] | Ustekinumab | 45 mg s.c. | Case report | psoriasis | Satisfactory result |
| I. A. Vallerand et al. (2019) [133] | Ustekinumab | 520 mg iv. Infusion at week 0, 90 mg s.c. at week 8 and then every 8 weeks | Case report | MG, DM, hypertension, dyslipidemia, CKD, gout, and obstructive sleep apnea | Complete healing after 6 months |
| J. López González et al. (2021) [134] | Ustekinumab | 260 mg iv. Infusion, then 90 mg s.c. every 8 weeks | Case report | CD | Satisfactory result |
| M. Fahmy et al. (2012) [135] | Ustekinumab | 90 mg s.c. at weeks 0 and 2, then every 8 weeks beginning at week 10 | Case report | UC | Complete healing by week 10 |
| Z. M. Low et al. (2018) [136] | Ustekinumab | 90 mg s.c. at weeks 0 and 4, then every 6 weeks, and later 45 mg every 3 weeks | Case report | NA | Significant improvement at 3 months |
| P. García Cámara et al. (2019) [137] | Ustekinumab | 520 mg iv. Infusion at week 0, then 90 mg s.c. every 8 weeks | Case report | CD | Complete healing after 12 months |
| J. Piqueras-García et al. (2019) [138] | Ustekinumab | 90 mg s.c. at weeks 0, 4, 10, and every 8 weeks thereafter | Case report | UC | Satisfactory result |
| D. Nieto et al. (2019) [139] | Ustekinumab | 90 mg s.c. every 8 weeks | Case report | Myelodysplastic syndrome | Complete healing after 20 weeks |
| A. M. Goldminz et al. (2012) [140] | Ustekinumab | 90 mg s.c. at weeks 0 and 4, and then every 8 weeks | Case report | None known | Satisfactory results after 22 weeks |
| A. J. Petty et al. (2020) [141] | Ustekinumab | 90 mg s.c. at weeks 0 and 4, and then every 8 weeks | Case report | Psoriasis and palmoplantar pustulosis | Satisfactory results after 2 doses |
| I. Cosgarea et al. (2016) [142] | Ustekinumab | NA | Case report | Renal cell carcinoma, chronic venous insufficiency, diabetes, hypertension | Complete healing after 3 months |
| E. Guenova et al. (2011) [143] | Ustekinumab | 45 mg s.c. at week 0 and week 4 | Case report | None known | Complete healing after 14 weeks |
| G. Nunes et al. (2019) [144] | Ustekinumab | 520 mg iv. Infusion, then 90 mg s.c. every 8 weeks | Case report | CD | Satisfactory result |
| Authors (Year) | Biologic Drug | Dosage Regimen | Study Type | Comorbidities | Efficacy |
|---|---|---|---|---|---|
| A. G. A. Kolios et al. (2015) [145] | Canakinumab | 150 mg s.c. at weeks 0 and 2, then 150–300 mg at week 4 if needed | Prospective, open-label study | none known | Complete healing in 4 out of 5 patients |
| S. Acierno et al. (2022) [146] | Canakinumab | 4 mg/kg s.c. every 4 weeks, after a year, 4 mg/kg every 8 weeks, because of exacerbation of the disease after a year of remission, return to the dosage of 4 mg/kg every 4 weeks | Case report | refractory chronic recurrent multifocal osteomyelitis | Satisfactory response |
| T. Jaeger et al. (2013) [147] | Canakinumab | 150 mg s.c. every 3–6 weeks, a total of 8 injections | Case report | HS | Complete remission in 1 year |
| C. O’Connor et al. (2021) [148] | Anakinra | 2 mg/kg s.c. daily in 4 weeks, then 100 mg daily | Case report |
| Complete healing in 4 months |
| Authors (Year) | Biologic Drug | Dosage Regimen | Study Type | Comorbidities | Efficacy |
|---|---|---|---|---|---|
| J. Coe et al. (2022) [149] | Secukinumab | 300 mg s.c. four weekly; after 2 months, 300 mg two weekly | Case report | Depression, osteoarthritis, hiatus hernia, Gilbert’s syndrome, and previous hepatitis A infection | Complete healing after a year of high-dose therapy |
| A.S. Kao et al. (2023) [150] | Ixekizumab | 160 mg s.c. at week 0, then 80 mg every 2 weeks until week 12, then 80 mg every 4 weeks | Case series |
|
|
| M. W. Tee et al. [151] | Brodalumab | 210 s.c. every week | Case series |
| Complete healing in both cases |
| M.L. McPhie et al. (2020) [152] | Secukinumab | 300 mg s.c. at weeks 0, 1, 2, 3, and 4, followed by monthly maintenance dosing | Case report | NA | Complete healing |
| M.M. Garcia et al. (2018) [153] | Secukinumab | 300 mg s.c. at weeks 0, 1, 2, 3, and 4, then every 4 weeks; beginning week 16, 300 mg every other week | Case report | RA, post-surgery for Quervain’s tenosynovitis | Partial response after 20 months of treatment |
| Authors (Year) | Biologic Drug | Dosage Regimen | Study Type | Comorbidities | Efficacy |
|---|---|---|---|---|---|
| L. J. Leow et al. (2022) [161] | Tildrakizumab | 100 mg s.c. on week 0 and 4, then every 8 weeks | Case report | NA | Constant improvement after 82 weeks |
| E. Çalışkan et al. (2023) [163] | Risankizumab | NA | Case report | ankylosing spondylitis, ileostomy due to megacolon toxicum | Refractory to treatment; closed primary ostomy—regression of lesions; no new lesions at the side of new ostomy |
| C. Baier et al. (2020) [164] | Guselkumab | 100 mg s.c. monthly | Case report | monoclonal gammopathy of undetermined significance and type 2 diabetes | Complete healing within 3 months |
| A. M. Reese et al. (2022) [165] | Guselkumab | 200 mg s.c. at week 0, 100 mg at week 4, then every 6 weeks | Case report | type 2 diabetes mellitus | Complete healing after 4 doses |
| J. M. John et al. (2020) [162] | Tildrakizumab | 100 mg s.c. on week 0, 4, then every 12 weeks | Case report | gout, polymyalgia rheumatica, renal impairment | Almost complete healing |
| B. Burgdorf et al. (2020) [166] | Risankizumab | 150 mg s.c. on weeks 0, 4, then every 12 weeks | Case report | none | Significant improvement |
| L.V. Piñeiro et al. (2023) [167] | Guselkumab | 100 mg s.c. at week 0, 4, then every 8 weeks | Case report | NA | Complete healing with residual post-inflammatory lesions |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Łyko, M.; Ryguła, A.; Kowalski, M.; Karska, J.; Jankowska-Konsur, A. The Pathophysiology and Treatment of Pyoderma Gangrenosum—Current Options and New Perspectives. Int. J. Mol. Sci. 2024, 25, 2440. https://doi.org/10.3390/ijms25042440
Łyko M, Ryguła A, Kowalski M, Karska J, Jankowska-Konsur A. The Pathophysiology and Treatment of Pyoderma Gangrenosum—Current Options and New Perspectives. International Journal of Molecular Sciences. 2024; 25(4):2440. https://doi.org/10.3390/ijms25042440
Chicago/Turabian StyleŁyko, Magdalena, Anna Ryguła, Michał Kowalski, Julia Karska, and Alina Jankowska-Konsur. 2024. "The Pathophysiology and Treatment of Pyoderma Gangrenosum—Current Options and New Perspectives" International Journal of Molecular Sciences 25, no. 4: 2440. https://doi.org/10.3390/ijms25042440