
Aryl Hydrocarbon Receptor Regulates Muc2 Production Independently of IL-22 during Colitis

 , ,
, ,  , and
, and
Abstract
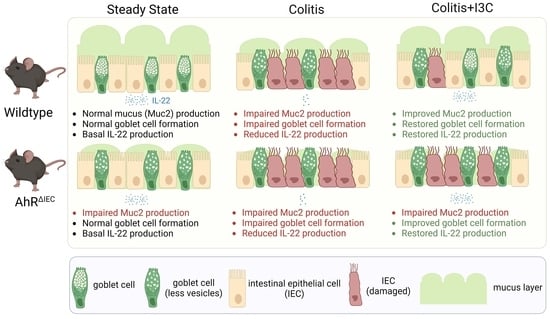
:
1. Introduction
2. Results
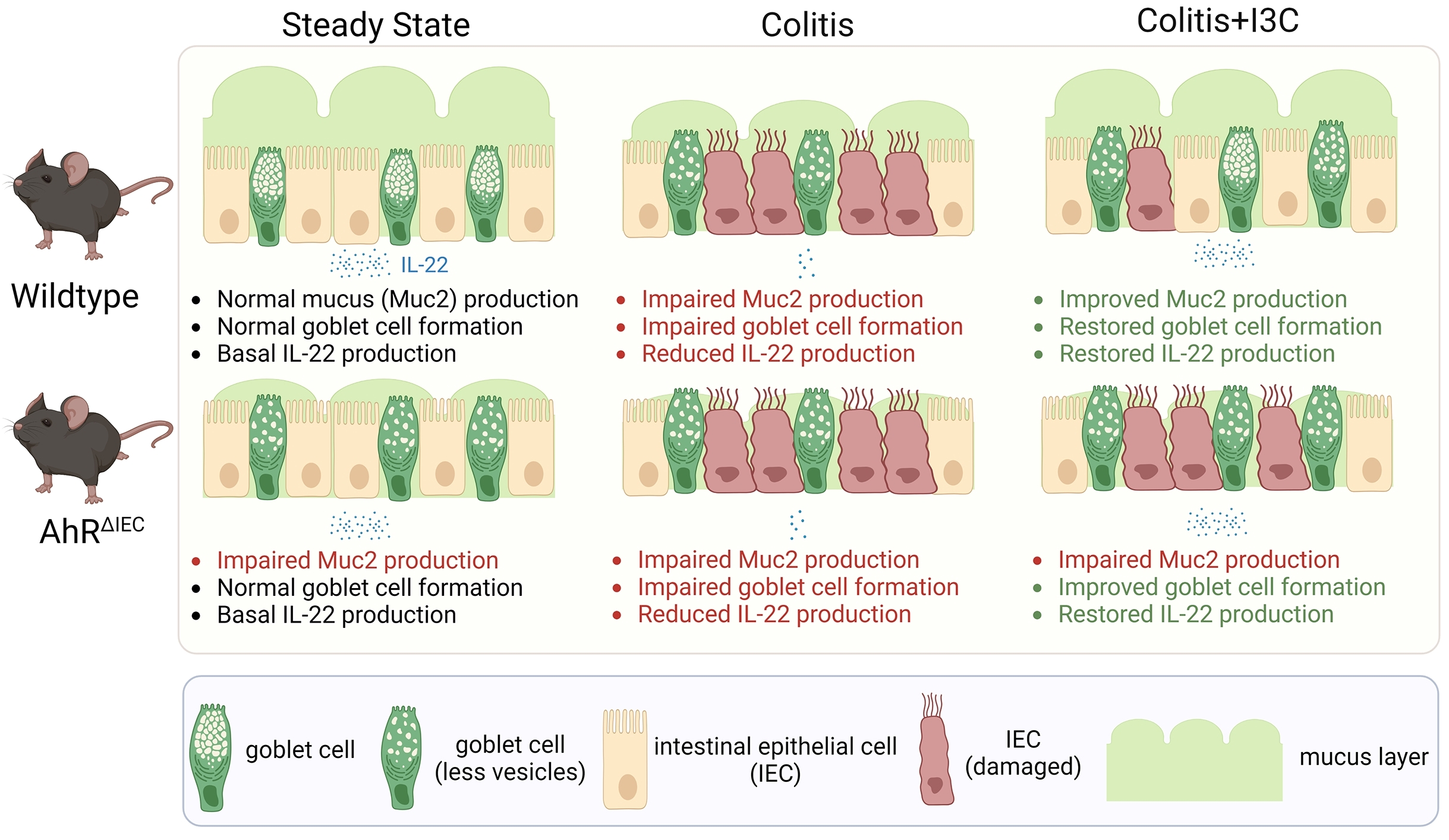
2.1. Gene Expression Analysis of Colonic Epithelial Cells (CECs) during I3C-Mediated Protection in the 2,4,6-Trinitrobenzene Sulfonic Acid (TNBS) Colitis Model
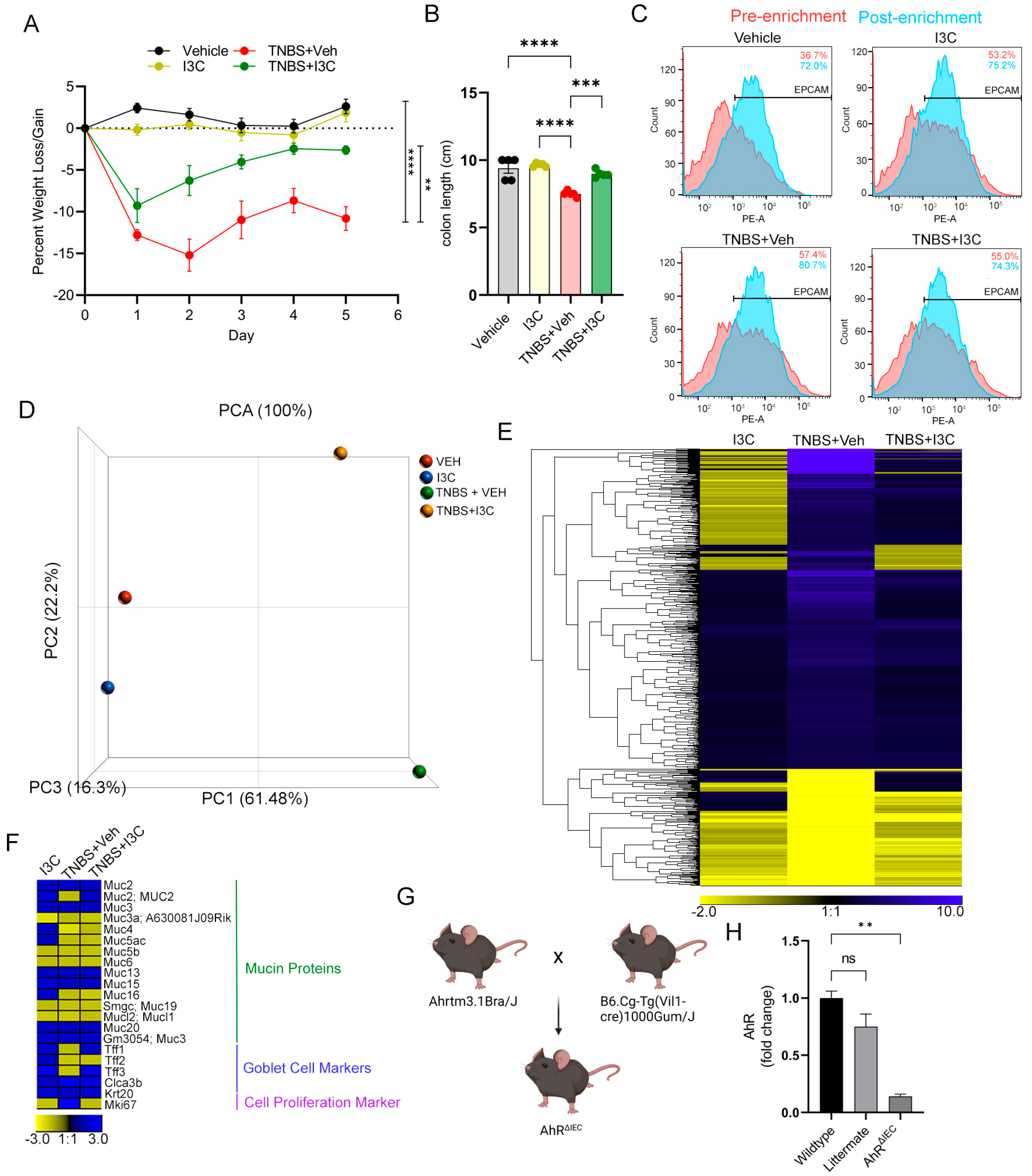
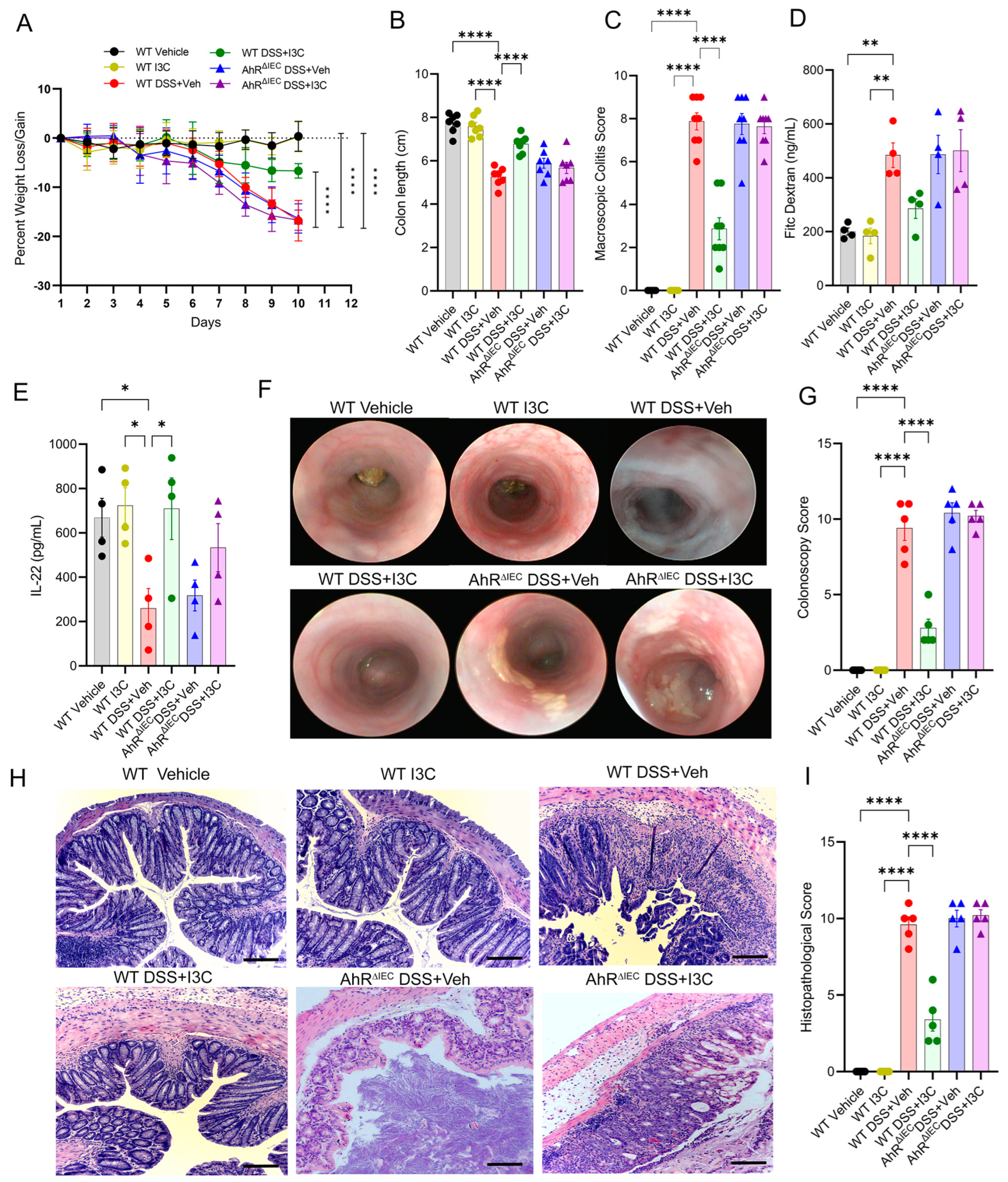
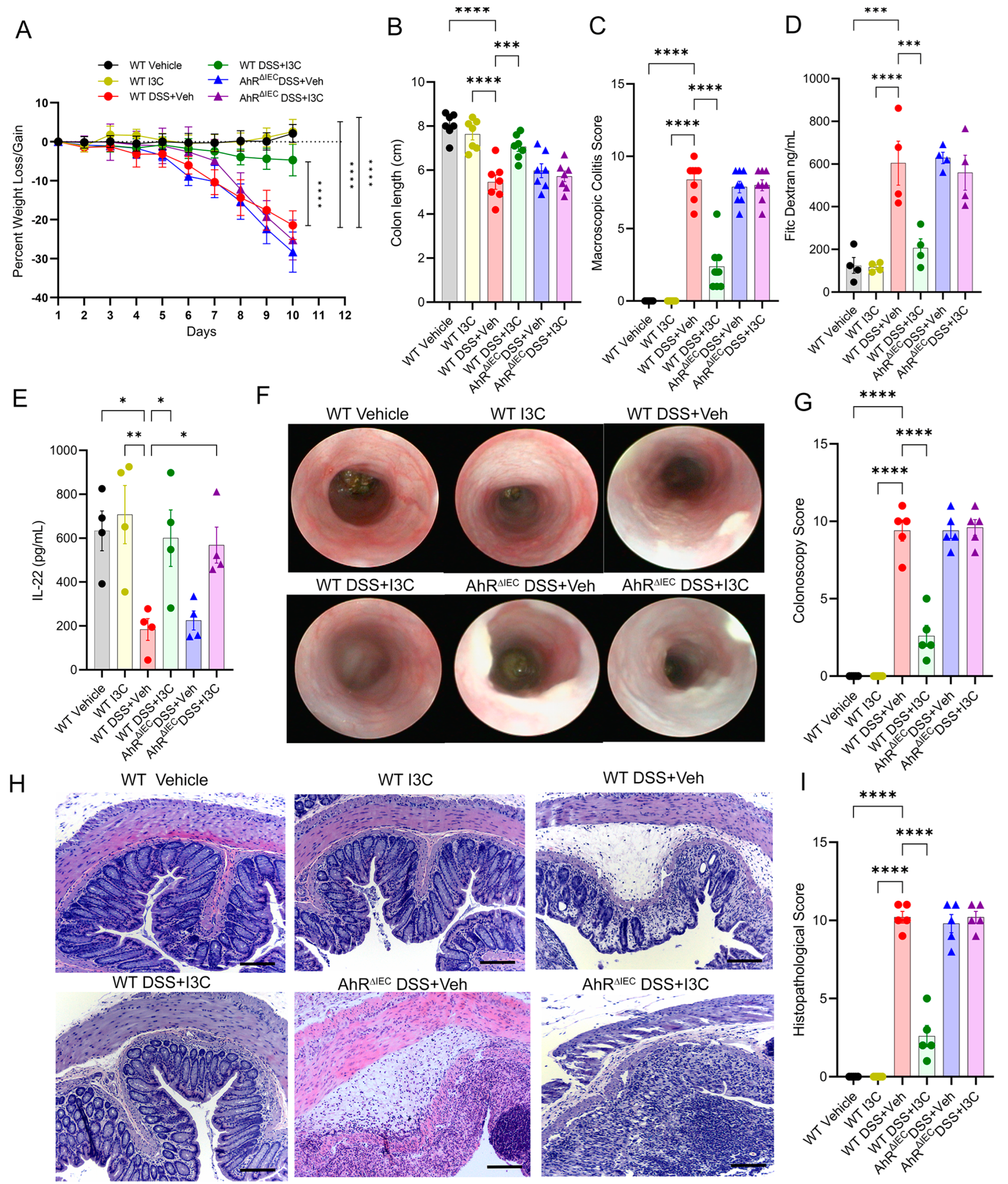
2.2. AhR Deficiency in IECs Leads to Loss of Protective Effects of I3C in the DSS-Induced Colitis Model despite Restoring IL-22 Production in the Colon
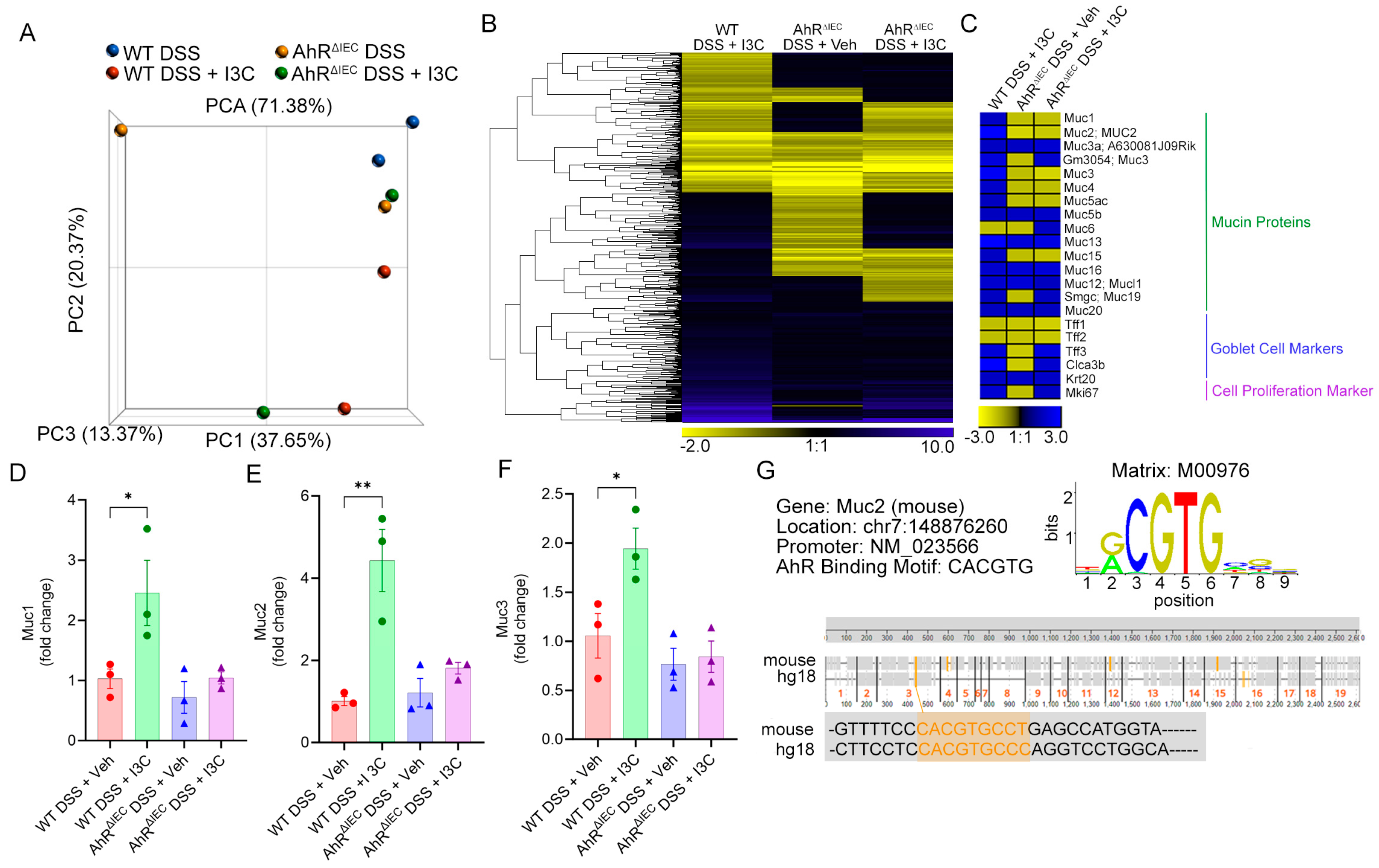
2.3. Deficiency of AhR in IECs In Vivo Impairs the Ability of I3C to Increase Mucus Production but Not Goblet Cell Development during Colitis
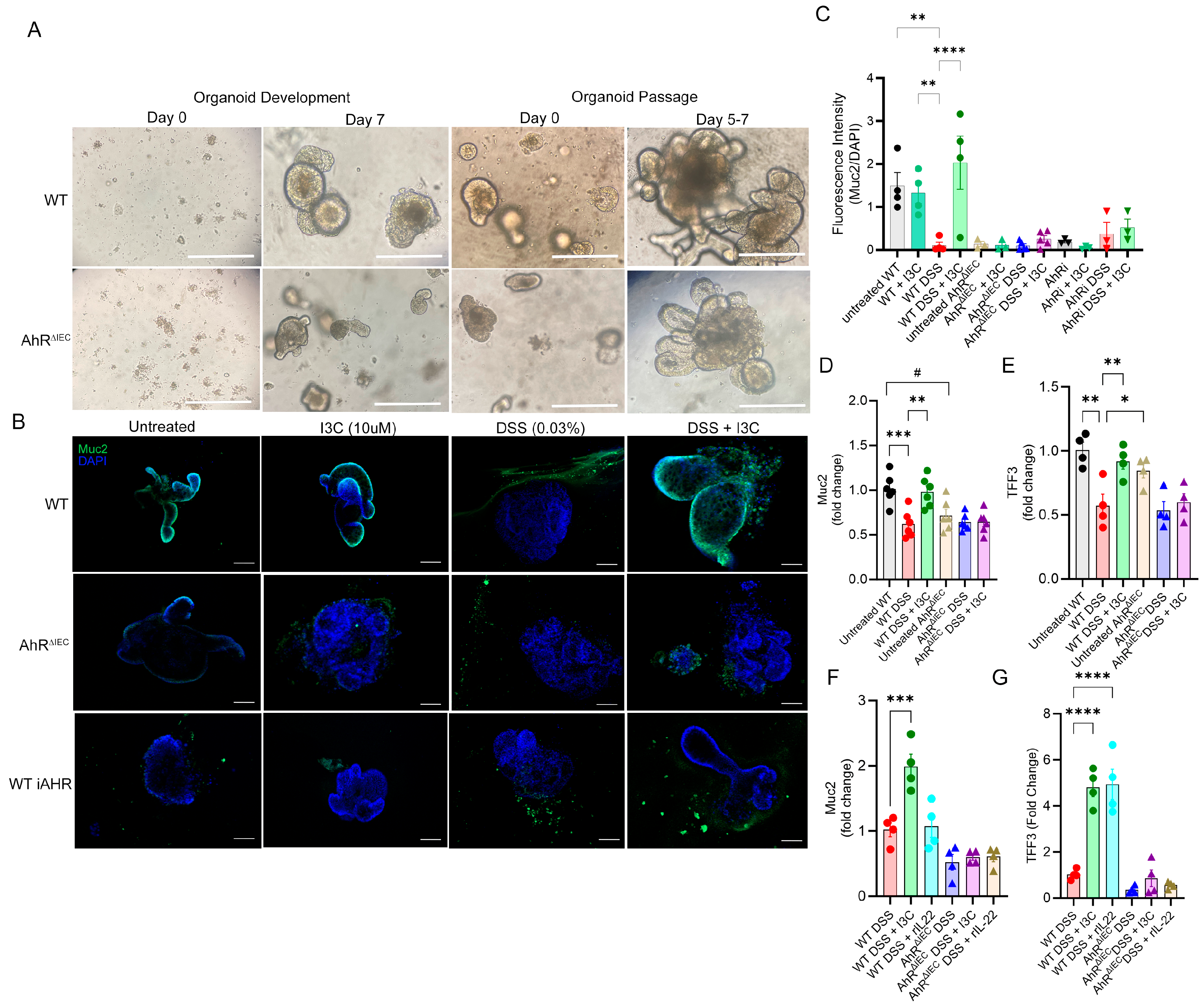
2.4. AhR Deficiency in Colon-Derived Organoids Results in Impaired Muc2 Production Independently of Goblet Cell Development
3. Discussion
4. Materials and Methods
4.1. Experimental Mice
4.2. Induction of Colitis and I3C Administration
4.3. Assessment of Colitis Disease Severity
4.4. Colon Histological Analysis
4.5. Evaluation of IL-22 Secretion
4.6. Transcriptome Microarray Analysis of Enriched CECs
4.7. Crypt Isolation and Colon Organoid Development
4.8. Immunofluorescence (IF) Staining and Imaging
4.9. In Silico Identification of DREs in Muc2 Promoter Region
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van der Sloot, K.W.J.; Weersma, R.K.; Alizadeh, B.Z.; Dijkstra, G. Identification of Environmental Risk Factors Associated with the Development of Inflammatory Bowel Disease. J. Crohns Colitis 2020, 14, 1662–1671. [Google Scholar] [CrossRef]
- Lautenschlager, S.A.; Barry, M.P.; Rogler, G.; Biedermann, L.; Schreiner, P.; Siebenhuner, A.R.; Swiss, I.B.D.C.S.G. Lifestyle factors associated with inflammatory bowel disease: Data from the Swiss IBD cohort study. BMC Gastroenterol. 2023, 23, 71. [Google Scholar] [CrossRef]
- GBD 2017 Inflammatory Bowel Disease Collaborators. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30. [Google Scholar]
- Yamazaki, M.; Chung, H.; Xu, Y.; Qiu, H. Trends in the prevalence and incidence of ulcerative colitis in Japan and the US. Int. J. Colorectal. Dis. 2023, 38, 135. [Google Scholar] [CrossRef] [PubMed]
- Burisch, J.; Zhao, M.; Odes, S.; De Cruz, P.; Vermeire, S.; Bernstein, C.N.; Kaplan, G.G.; Duricova, D.; Greenberg, D.; Melberg, H.O.; et al. The cost of inflammatory bowel disease in high-income settings: A Lancet Gastroenterology & Hepatology Commission. Lancet Gastroenterol. Hepatol. 2023, 8, 458–492. [Google Scholar] [PubMed]
- Singh, S.; Qian, A.S.; Nguyen, N.H.; Ho, S.K.M.; Luo, J.; Jairath, V.; Sandborn, W.J.; Ma, C. Trends in U.S. Health Care Spending on Inflammatory Bowel Diseases, 1996–2016. Inflamm. Bowel Dis. 2022, 28, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Kaddoura, R.; Ghelani, H.; Alqutami, F.; Altaher, H.; Hachim, M.; Jan, R.K. Identification of Specific Biomarkers and Pathways in the Treatment Response of Infliximab for Inflammatory Bowel Disease: In-Silico Analysis. Life 2023, 13, 680. [Google Scholar] [CrossRef]
- Degli Esposti, L.; Daperno, M.; Dovizio, M.; Franchi, A.; Sangiorgi, D.; Savarino, E.V.; Scaldaferri, F.; Secchi, O.; Serra, A.; Perrone, V.; et al. A retrospective analysis of treatment patterns, drug discontinuation and healthcare costs in Crohn’s disease patients treated with biologics. Dig. Liver Dis. 2023, 55, 1214–1220. [Google Scholar] [CrossRef] [PubMed]
- Busbee, P.B.; Rouse, M.; Nagarkatti, M.; Nagarkatti, P.S. Use of natural AhR ligands as potential therapeutic modalities against inflammatory disorders. Nutr. Rev. 2013, 71, 353–369. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yang, K.; Han, B.; Sheng, B.; Yin, J.; Pu, A.; Li, L.; Sun, L.; Yu, M.; Qiu, Y.; et al. Aryl hydrocarbon receptor inhibits inflammation in DSS-induced colitis via the MK2/p-MK2/TTP pathway. Int. J. Mol. Med. 2018, 41, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Furumatsu, K.; Nishiumi, S.; Kawano, Y.; Ooi, M.; Yoshie, T.; Shiomi, Y.; Kutsumi, H.; Ashida, H.; Fujii-Kuriyama, Y.; Azuma, T.; et al. A role of the aryl hydrocarbon receptor in attenuation of colitis. Dig. Dis. Sci. 2011, 56, 2532–2544. [Google Scholar] [CrossRef] [PubMed]
- Arsenescu, R.; Arsenescu, V.; Zhong, J.; Nasser, M.; Melinte, R.; Dingle, R.W.; Swanson, H.; de Villiers, W.J. Role of the xenobiotic receptor in inflammatory bowel disease. Inflamm. Bowel Dis. 2011, 17, 1149–1162. [Google Scholar] [CrossRef] [PubMed]
- Busbee, P.B.; Menzel, L.; Alrafas, H.R.; Dopkins, N.; Becker, W.; Miranda, K.; Tang, C.; Chatterjee, S.; Singh, U.; Nagarkatti, M.; et al. Indole-3-carbinol prevents colitis and associated microbial dysbiosis in an IL-22-dependent manner. JCI Insight 2020, 5, e127551. [Google Scholar] [CrossRef] [PubMed]
- Van der Sluis, M.; De Koning, B.A.; De Bruijn, A.C.; Velcich, A.; Meijerink, J.P.; Van Goudoever, J.B.; Buller, H.A.; Dekker, J.; Van Seuningen, I.; Renes, I.B.; et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 2006, 131, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, S.; Wang, Y.; Hu, H.; Li, L.; Wu, Y.; Cao, D.; Cai, Y.; Zhang, J.; Zhang, X. Interleukin-22 regulates the homeostasis of the intestinal epithelium during inflammation. Int. J. Mol. Med. 2019, 43, 1657–1668. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Ogawa, A.; Mizoguchi, E.; Shimomura, Y.; Andoh, A.; Bhan, A.K.; Blumberg, R.S.; Xavier, R.J.; Mizoguchi, A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Investig. 2008, 118, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Kassouri, L.; Amiot, A.; Kirchgesner, J.; Treton, X.; Allez, M.; Bouhnik, Y.; Beaugerie, L.; Carbonnel, F.; Meyer, A. The outcome of Crohn’s disease patients refractory to anti-TNF and either vedolizumab or ustekinumab. Dig. Liver Dis. 2020, 52, 1148–1155. [Google Scholar] [CrossRef]
- Sabino, J.; Verstockt, B.; Vermeire, S.; Ferrante, M. New biologics and small molecules in inflammatory bowel disease: An update. Therap. Adv. Gastroenterol. 2019, 12, 1756284819853208. [Google Scholar] [CrossRef]
- Roda, G.; Jharap, B.; Neeraj, N.; Colombel, J.F. Loss of Response to Anti-TNFs: Definition, Epidemiology, and Management. Clin. Transl. Gastroenterol. 2016, 7, e135. [Google Scholar] [CrossRef]
- Ben-Horin, S.; Chowers, Y. Review article: Loss of response to anti-TNF treatments in Crohn’s disease. Aliment. Pharmacol. Ther. 2011, 33, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Kim, K.J.; Lee, S.O.; Yang, D.H.; Jung, K.W.; Duk Ye, B.; Byeon, J.S.; Myung, S.J.; Yang, S.K.; Kim, J.H.; et al. Ciprofloxacin usage and bacterial resistance patterns in Crohn’s disease patients with abscesses. J. Clin. Gastroenterol. 2014, 48, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Hashash, J.G.; Binion, D.G. Managing Clostridium difficile in inflammatory bowel disease (IBD). Curr. Gastroenterol. Rep. 2014, 16, 393. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, R.; Bernstein, C.N.; Gearry, R.; Hviid, A.; Kolho, K.L.; Kronman, M.P.; Shaw, S.; Van Kruiningen, H.; Colombel, J.F.; Atreja, A. Antibiotics associated with increased risk of new-onset Crohn’s disease but not ulcerative colitis: A meta-analysis. Am. J. Gastroenterol. 2014, 109, 1728–1738. [Google Scholar] [CrossRef]
- Shaw, S.Y.; Blanchard, J.F.; Bernstein, C.N. Association between the use of antibiotics and new diagnoses of Crohn’s disease and ulcerative colitis. Am. J. Gastroenterol. 2011, 106, 2133–2142. [Google Scholar] [CrossRef] [PubMed]
- Targownik, L.E.; Nugent, Z.; Singh, H.; Bernstein, C.N. Prevalence of and outcomes associated with corticosteroid prescription in inflammatory bowel disease. Inflamm. Bowel Dis. 2014, 20, 622–630. [Google Scholar] [CrossRef]
- Curtis, J.R.; Westfall, A.O.; Allison, J.; Bijlsma, J.W.; Freeman, A.; George, V.; Kovac, S.H.; Spettell, C.M.; Saag, K.G. Population-based assessment of adverse events associated with long-term glucocorticoid use. Arthritis Rheum. 2006, 55, 420–426. [Google Scholar] [CrossRef]
- Hoes, J.N.; Jacobs, J.W.; Verstappen, S.M.; Bijlsma, J.W.; Van der Heijden, G.J. Adverse events of low- to medium-dose oral glucocorticoids in inflammatory diseases: A meta-analysis. Ann. Rheum. Dis. 2009, 68, 1833–1838. [Google Scholar] [CrossRef]
- Khan, N.; Pernes, T.; Weiss, A.; Trivedi, C.; Patel, M.; Xie, D.; Yang, Y.X. Incidence of Infections and Malignancy Among Elderly Male Patients with IBD Exposed to Vedolizumab, Prednisone, and 5-ASA Medications: A Nationwide Retrospective Cohort Study. Adv. Ther. 2021, 38, 2586–2598. [Google Scholar] [CrossRef]
- Peng, C.; Wu, C.; Xu, X.; Pan, L.; Lou, Z.; Zhao, Y.; Jiang, H.; He, Z.; Ruan, B. Indole-3-carbinol ameliorates necroptosis and inflammation of intestinal epithelial cells in mice with ulcerative colitis by activating aryl hydrocarbon receptor. Exp. Cell Res. 2021, 404, 112638. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, J.; He, Q.; Yu, L.; Pham, Q.; Cheung, L.; Zhang, Z.; Kim, Y.S.; Smith, A.D.; Wang, T.T.Y. Dietary Indole-3-Carbinol Alleviated Spleen Enlargement, Enhanced IgG Response in C3H/HeN Mice Infected with Citrobacter rodentium. Nutrients 2020, 12, 3148. [Google Scholar] [CrossRef] [PubMed]
- Alkarkoushi, R.R.; Hui, Y.; Tavakoli, A.S.; Singh, U.; Nagarkatti, P.; Nagarkatti, M.; Chatzistamou, I.; Bam, M.; Testerman, T.L. Immune and microRNA responses to Helicobacter muridarum infection and indole-3-carbinol during colitis. World J. Gastroenterol. 2020, 26, 4763–4785. [Google Scholar] [CrossRef]
- Schanz, O.; Chijiiwa, R.; Cengiz, S.C.; Majlesain, Y.; Weighardt, H.; Takeyama, H.; Forster, I. Dietary AhR Ligands Regulate AhRR Expression in Intestinal Immune Cells and Intestinal Microbiota Composition. Int. J. Mol. Sci. 2020, 21, 3189. [Google Scholar] [CrossRef]
- Benson, J.M.; Beamer, C.A.; Seaver, B.P.; Shepherd, D.M. Indole-3-Carbinol Exerts Sex-Specific Effects in Murine Colitis. Eur. J. Inflamm. 2012, 10, 335–346. [Google Scholar] [CrossRef]
- Li, Y.; Innocentin, S.; Withers, D.R.; Roberts, N.A.; Gallagher, A.R.; Grigorieva, E.F.; Wilhelm, C.; Veldhoen, M. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell 2011, 147, 629–640. [Google Scholar] [CrossRef]
- Nolan, L.S.; Mihi, B.; Agrawal, P.; Gong, Q.; Rimer, J.M.; Bidani, S.S.; Gale, S.E.; Goree, M.; Hu, E.; Lanik, W.E.; et al. Indole-3-Carbinol-Dependent Aryl Hydrocarbon Receptor Signaling Attenuates the Inflammatory Response in Experimental Necrotizing Enterocolitis. Immunohorizons 2021, 5, 193–209. [Google Scholar] [CrossRef]
- Yin, J.; Yang, K.; Zhou, C.; Xu, P.; Xiao, W.; Yang, H. Aryl hydrocarbon receptor activation alleviates dextran sodium sulfate-induced colitis through enhancing the differentiation of goblet cells. Biochem. Biophys. Res. Commun. 2019, 514, 180–186. [Google Scholar] [CrossRef]
- Shah, K.; Maradana, M.R.; Joaquina Delas, M.; Metidji, A.; Graelmann, F.; Llorian, M.; Chakravarty, P.; Li, Y.; Tolaini, M.; Shapiro, M.; et al. Cell-intrinsic Aryl Hydrocarbon Receptor signalling is required for the resolution of injury-induced colonic stem cells. Nat. Commun. 2022, 13, 1827. [Google Scholar] [CrossRef]
- Turner, J.E.; Stockinger, B.; Helmby, H. IL-22 mediates goblet cell hyperplasia and worm expulsion in intestinal helminth infection. PLoS Pathog. 2013, 9, e1003698. [Google Scholar] [CrossRef] [PubMed]
- He, G.W.; Lin, L.; DeMartino, J.; Zheng, X.; Staliarova, N.; Dayton, T.; Begthel, H.; van de Wetering, W.J.; Bodewes, E.; van Zon, J.; et al. Optimized human intestinal organoid model reveals interleukin-22-dependency of paneth cell formation. Cell Stem Cell 2022, 29, 1333–1345.e6. [Google Scholar] [CrossRef] [PubMed]
- Patnaude, L.; Mayo, M.; Mario, R.; Wu, X.; Knight, H.; Creamer, K.; Wilson, S.; Pivorunas, V.; Karman, J.; Phillips, L.; et al. Mechanisms and regulation of IL-22-mediated intestinal epithelial homeostasis and repair. Life Sci. 2021, 271, 119195. [Google Scholar] [CrossRef] [PubMed]
- Madison, B.B.; Dunbar, L.; Qiao, X.T.; Braunstein, K.; Braunstein, E.; Gumucio, D.L. Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J. Biol. Chem. 2002, 277, 33275–33283. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, K.; Fu, J.; Johansson, M.E.; Liu, X.; Gao, N.; Wu, Q.; Song, J.; McDaniel, J.M.; McGee, S.; Chen, W.; et al. Core 1- and 3-derived O-glycans collectively maintain the colonic mucus barrier and protect against spontaneous colitis in mice. Mucosal Immunol. 2017, 10, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, K.S.; Kissoon-Singh, V.; Gibson, D.L.; Ma, C.; Montero, M.; Sham, H.P.; Ryz, N.; Huang, T.; Velcich, A.; Finlay, B.B.; et al. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog. 2010, 6, e1000902. [Google Scholar] [CrossRef] [PubMed]
- Grondin, J.A.; Kwon, Y.H.; Far, P.M.; Haq, S.; Khan, W.I. Mucins in Intestinal Mucosal Defense and Inflammation: Learning From Clinical and Experimental Studies. Front. Immunol. 2020, 11, 2054. [Google Scholar] [CrossRef] [PubMed]
- Babickova, J.; Tothova, L.; Lengyelova, E.; Bartonova, A.; Hodosy, J.; Gardlik, R.; Celec, P. Sex Differences in Experimentally Induced Colitis in Mice: A Role for Estrogens. Inflammation 2015, 38, 1996–2006. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Prokopec, S.D.; Watson, J.D.; Sun, R.X.; Pohjanvirta, R.; Boutros, P.C. Male and female mice show significant differences in hepatic transcriptomic response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. BMC Genom. 2015, 16, 625. [Google Scholar] [CrossRef] [PubMed]
- Maite, C.B.; Roy, M.; Emilie, V. The Effect of Sex-Specific Differences on IL-10(-/-) Mouse Colitis Phenotype and Microbiota. Int. J. Mol. Sci. 2023, 24, 10364. [Google Scholar]
- Walisser, J.A.; Glover, E.; Pande, K.; Liss, A.L.; Bradfield, C.A. Aryl hydrocarbon receptor-dependent liver development and hepatotoxicity are mediated by different cell types. Proc. Natl. Acad. Sci. USA 2005, 102, 17858–17863. [Google Scholar] [CrossRef]
- Kawada, M.; Arihiro, A.; Mizoguchi, E. Insights from advances in research of chemically induced experimental models of human inflammatory bowel disease. World J. Gastroenterol. 2007, 13, 5581–5593. [Google Scholar] [CrossRef]
- Centofanti, F.; Alonzi, T.; Latini, A.; Spitalieri, P.; Murdocca, M.; Chen, X.; Cui, W.; Shang, Q.; Goletti, D.; Shi, Y.; et al. Indole-3-carbinol in vitro antiviral activity against SARS-Cov-2 virus and in vivo toxicity. Cell Death Discov. 2022, 8, 491. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Reed, G.A.; Arneson, D.W.; Putnam, W.C.; Smith, H.J.; Gray, J.C.; Sullivan, D.K.; Mayo, M.S.; Crowell, J.A.; Hurwitz, A. Single-dose and multiple-dose administration of indole-3-carbinol to women: Pharmacokinetics based on 3,3’-diindolylmethane. Cancer Epidemiol. Biomark. Prev. A Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2006, 15, 2477–2481. [Google Scholar] [CrossRef] [PubMed]
- Kodani, T.; Rodriguez-Palacios, A.; Corridoni, D.; Lopetuso, L.; Di Martino, L.; Marks, B.; Pizarro, J.; Pizarro, T.; Chak, A.; Cominelli, F. Flexible colonoscopy in mice to evaluate the severity of colitis and colorectal tumors using a validated endoscopic scoring system. J. Vis. Exp. 2013, 80, e50843. [Google Scholar]
- Saeed, A.I.; Rieder, S.A.; Price, R.L.; Barker, J.; Nagarkatti, P.; Nagarkatti, M. Acute lung injury induced by Staphylococcal enterotoxin B: Disruption of terminal vessels as a mechanism of induction of vascular leak. Microsc. Microanal. 2012, 18, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.A.; Kellermann, C.A.; Burnat, G.; Hahn, E.G.; Rau, T.; Konturek, P.C. Mice lacking cannabinoid CB1-, CB2-receptors or both receptors show increased susceptibility to trinitrobenzene sulfonic acid (TNBS)-induced colitis. J. Physiol. Pharmacol. 2010, 61, 89–97. [Google Scholar] [PubMed]
- Gunasekera, D.C.; Ma, J.; Vacharathit, V.; Shah, P.; Ramakrishnan, A.; Uprety, P.; Shen, Z.; Sheh, A.; Brayton, C.F.; Whary, M.T.; et al. The development of colitis in Il10(-/-) mice is dependent on IL-22. Mucosal Immunol. 2020, 13, 493–506. [Google Scholar] [CrossRef]
- Overcast, G.R.; Meibers, H.E.; Eshleman, E.M.; Saha, I.; Waggoner, L.; Patel, K.N.; Jain, V.G.; Haslam, D.B.; Alenghat, T.; VanDussen, K.L.; et al. IEC-intrinsic IL-1R signaling holds dual roles in regulating intestinal homeostasis and inflammation. J. Exp. Med. 2023, 220, e20212523. [Google Scholar] [CrossRef]
- Wen, Y.A.; Li, X.; Goretsky, T.; Weiss, H.L.; Barrett, T.A.; Gao, T. Loss of PHLPP protects against colitis by inhibiting intestinal epithelial cell apoptosis. Biochim. Biophys. Acta 2015, 1852 Pt A, 2013–2023. [Google Scholar] [CrossRef]
- Girish, N.; Liu, C.Y.; Gadeock, S.; Gomez, M.L.; Huang, Y.; Sharifkhodaei, Z.; Washington, M.K.; Polk, D.B. Persistence of Lgr5+ colonic epithelial stem cells in mouse models of inflammatory bowel disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 321, G308–G324. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, J.M.; Lee, E.J.; Hwang, W.B.; Kim, D.J. Indole-3-Carbinol Promotes Goblet-Cell Differentiation Regulating Wnt and Notch Signaling Pathways AhR-Dependently. Mol. Cells 2018, 41, 290–300. [Google Scholar]
- O’Rourke, K.P.; Dow, L.E.; Lowe, S.W. Immunofluorescent Staining of Mouse Intestinal Stem Cells. Bio Protoc. 2016, 6, e1732. [Google Scholar] [CrossRef] [PubMed]
- Kreft, L.; Soete, A.; Hulpiau, P.; Botzki, A.; Saeys, Y.; De Bleser, P. ConTra v3: A tool to identify transcription factor binding sites across species, update 2017. Nucleic Acids Res. 2017, 45, W490–W494. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Forward | Reverse |
|---|---|---|
| AhR | CAGTGGGAATAAGGCAAGAGTGA | GGTACAAGTGCACATGCCTGC |
| Vil1 | GCCTTCTCCTCTAGGCTCGT | AGGCAAATTTTGGTGTACGG |
| Vil1 IP | - | TATAGGGCAGAGCTGGAGGA |
| GAPDH | AACAGCAACTCCCACTCTTC | CCTGTTGCTGTAGCCGTATT |
| Muc1 | TGGATTGTTTCTGCAGATTTT | CCTGACCTGAACTTGATGCT |
| Muc2 | CTACCATTACCACCACTAC | GTCTCTCGATCACCACCATTT |
| Muc3 | TGTTCAGCTTTACTGTGTTTCAA | TTGCATGTCTCCTCAGGATT |
| Tff3 | TAATGCTGTTGGTGGTCCTG | CAGCCACGGTTGTTACACTG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saxena, A.; Mitchell, C.; Bogdon, R.; Roark, K.; Wilson, K.; Staley, S.; Hailey, M.; Williams, M.C.; Rutkovsky, A.; Nagarkatti, P.; et al. Aryl Hydrocarbon Receptor Regulates Muc2 Production Independently of IL-22 during Colitis. Int. J. Mol. Sci. 2024, 25, 2404. https://doi.org/10.3390/ijms25042404
Saxena A, Mitchell C, Bogdon R, Roark K, Wilson K, Staley S, Hailey M, Williams MC, Rutkovsky A, Nagarkatti P, et al. Aryl Hydrocarbon Receptor Regulates Muc2 Production Independently of IL-22 during Colitis. International Journal of Molecular Sciences. 2024; 25(4):2404. https://doi.org/10.3390/ijms25042404
Chicago/Turabian StyleSaxena, Archana, Chandani Mitchell, Raymond Bogdon, Kasie Roark, Kiesha Wilson, Shanieka Staley, Michelle Hailey, Michal Claire Williams, Alex Rutkovsky, Prakash Nagarkatti, and et al. 2024. "Aryl Hydrocarbon Receptor Regulates Muc2 Production Independently of IL-22 during Colitis" International Journal of Molecular Sciences 25, no. 4: 2404. https://doi.org/10.3390/ijms25042404