

Role of Leptin in Obesity, Cardiovascular Disease, and Type 2 Diabetes
 , , , , ,
, , , , , 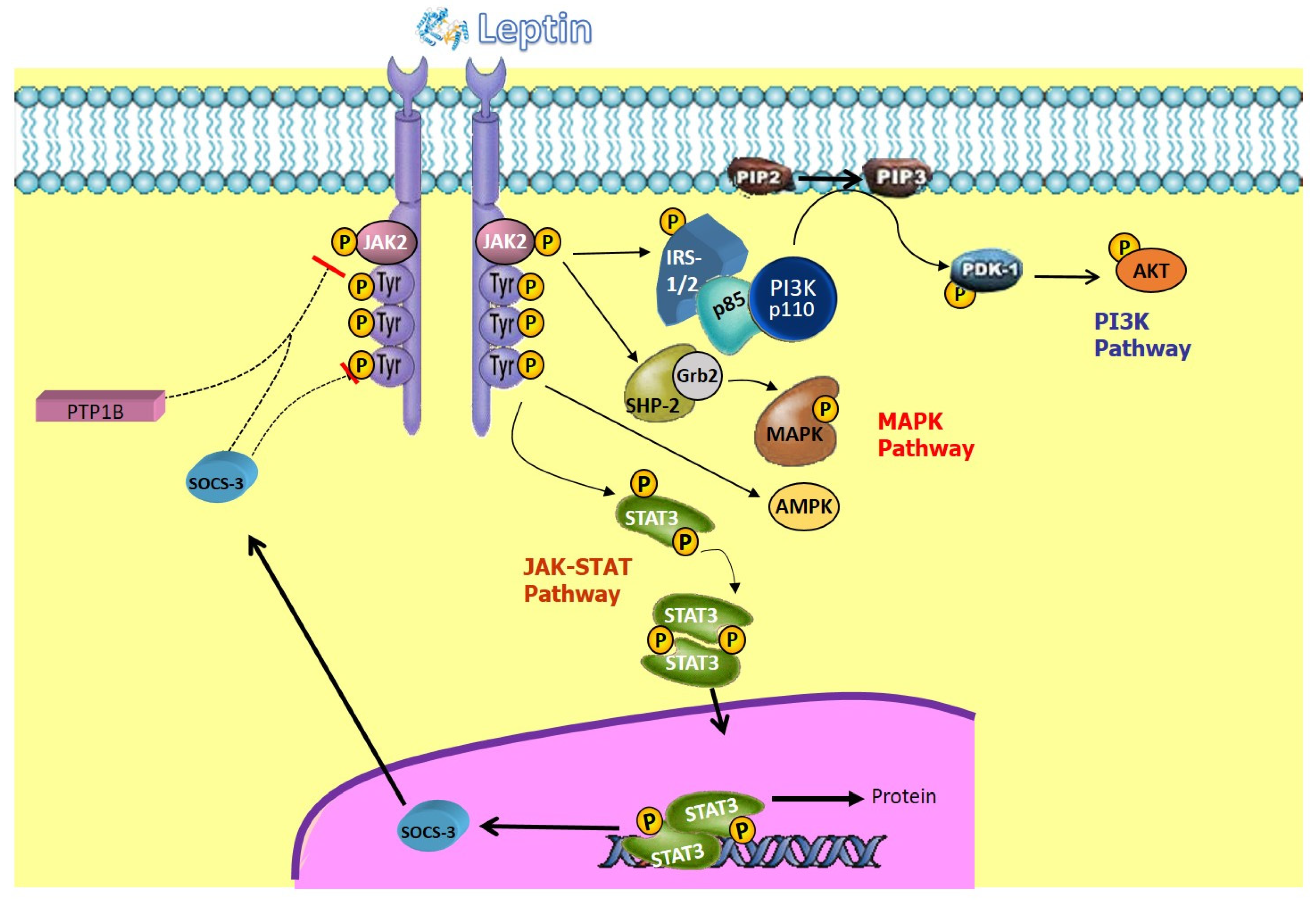{kind=link}
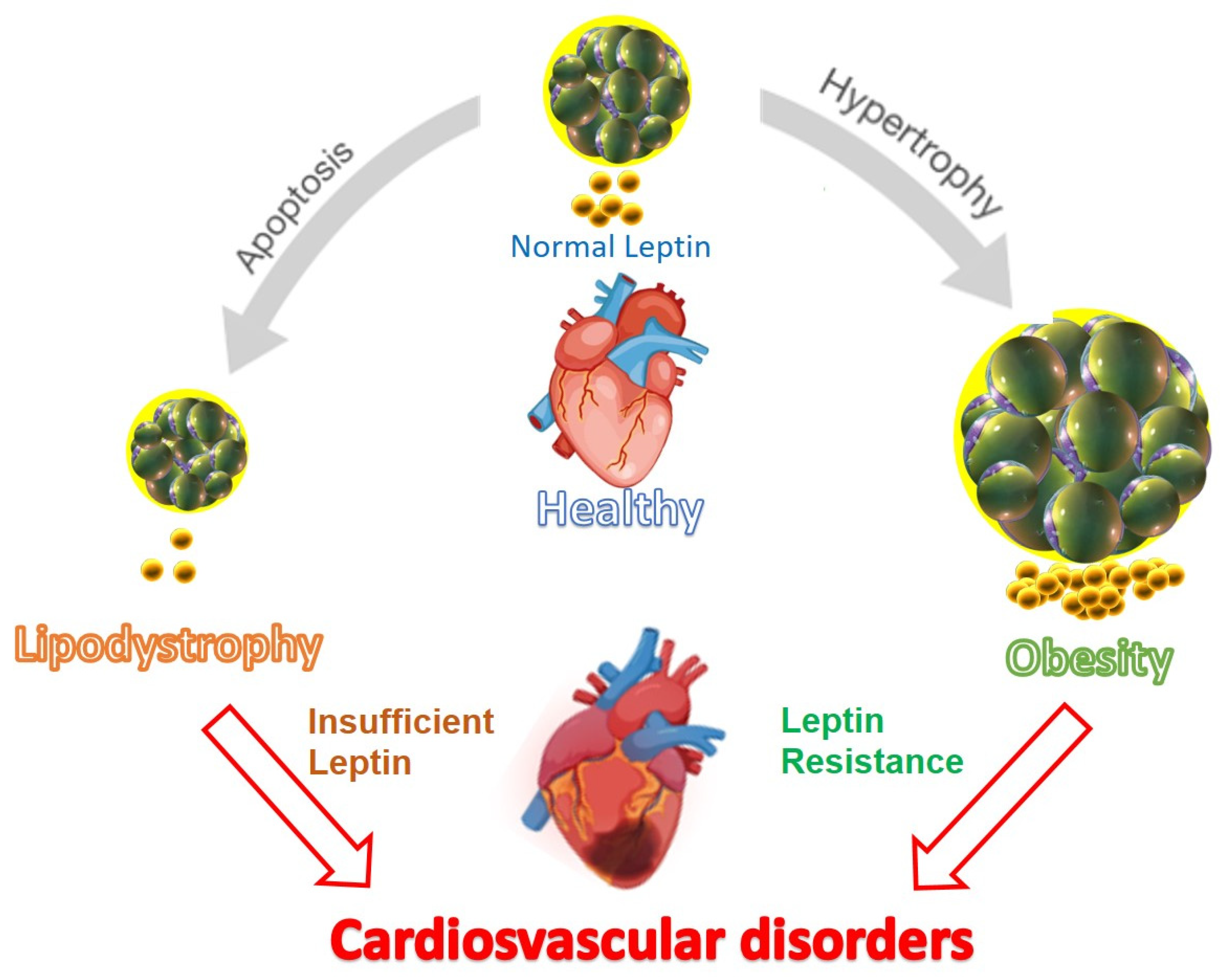{kind=link}
Abstract
:1. Introduction
2. Obesity and CVD
3. Obesity and Diabetes
4. Diabetes and CVD
5. Leptin
6. Leptin and Obesity
7. Leptin and T2DM
8. Leptin and CVD
9. Leptin as a Therapeutic Target
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Correction Statement
References
- Glovaci, D.; Fan, W.; Wong, N.D. Epidemiology of Diabetes Mellitus and Cardiovascular Disease. Curr. Cardiol. Rep. 2019, 21, 21. [Google Scholar] [CrossRef]
- Kerner, W.; Brückel, J. Definition, classification and diagnosis of diabetes mellitus. Exp. Clin. Endocrinol. Diabetes 2014, 122, 384–386. [Google Scholar] [CrossRef]
- Coraci, D.; Giovannini, S.; Loreti, C.; Pecchioli, C.; Piccinini, G.; Padua, L. The past encounters the future: “old” diagnostic methods to check innovative treatments for carpal tunnel syndrome. Comment on: “Treatment of carpal tunnel syndrome: From ultrasonography to ultrasound surgery” by Petrover and Richette. Joint Bone Spine 2017 https://doi.org/10.1016/j.jbspin.2017.11.003. Jt. Bone Spine 2018, 85, 783–784. [Google Scholar] [CrossRef]
- Endalifer, M.L.; Diress, G. Epidemiology, Predisposing Factors, Biomarkers, and Prevention Mechanism of Obesity: A Systematic Review. J. Obes. 2020, 2020, 6134362. [Google Scholar] [CrossRef] [PubMed]
- Chobot, A.; Górowska-Kowolik, K.; Sokołowska, M.; Jarosz-Chobot, P. Obesity and diabetes-Not only a simple link between two epidemics. Diabetes. Metab. Res. Rev. 2018, 34, e3042. [Google Scholar] [CrossRef] [PubMed]
- Bhupathiraju, S.N.; Hu, F.B. Epidemiology of Obesity and Diabetes and Their Cardiovascular Complications. Circ. Res. 2016, 118, 1723–1735. [Google Scholar] [CrossRef] [PubMed]
- La Sala, L.; Pontiroli, A.E. Prevention of Diabetes and Cardiovascular Disease in Obesity. Int. J. Mol. Sci. 2020, 21, 8178. [Google Scholar] [CrossRef]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.M. Leptin, leptin receptors, and the control of body weight. Nutr. Rev. 1998, 56, S38–S46. [Google Scholar] [CrossRef] [PubMed]
- Considine, R.V.; Caro, J.F. Leptin and the regulation of body weight. Int. J. Biochem. Cell Biol. 1997, 29, 1255–1272. [Google Scholar] [CrossRef]
- White, D.W.; Tartaglia, L.A. Leptin and OB-R: Body weight regulation by a cytokine receptor. Cytokine Growth Factor Rev. 1996, 7, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, M.; Sudar-Milovanovic, E.; Soskic, S.; Essack, M.; Arya, S.; Stewart, A.J.; Gojobori, T.; Isenovic, E.R. Leptin and Obesity: Role and Clinical Implication. Front. Endocrinol. 2021, 12, 585887. [Google Scholar] [CrossRef]
- Pérez-Pérez, A.; Sánchez-Jiménez, F.; Vilariño-García, T.; Sánchez-Margalet, V. Role of Leptin in Inflammation and Vice Versa. Int. J. Mol. Sci. 2020, 21, 5887. [Google Scholar] [CrossRef]
- Wang, C.; Chang, L.; Wang, J.; Xia, L.; Cao, L.; Wang, W.; Xu, J.; Gao, H. Leptin and risk factors for atherosclerosis: A review. Medicine 2023, 102, E36076. [Google Scholar] [CrossRef]
- Raman, P.; Khanal, S. Leptin in Atherosclerosis: Focus on Macrophages, Endothelial and Smooth Muscle Cells. Int. J. Mol. Sci. 2021, 22, 5446. [Google Scholar] [CrossRef] [PubMed]
- Neeland, I.J.; Ross, R.; Després, J.P.; Matsuzawa, Y.; Yamashita, S.; Shai, I.; Seidell, J.; Magni, P.; Santos, R.D.; Arsenault, B.; et al. Visceral and ectopic fat, atherosclerosis, and cardiometabolic disease: A position statement. Lancet Diabetes Endocrinol. 2019, 7, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Romero-Corral, A.; Montori, V.M.; Somers, V.K.; Korinek, J.; Thomas, R.J.; Allison, T.G.; Mookadam, F.; Lopez-Jimenez, F. Association of bodyweight with total mortality and with cardiovascular events in coronary artery disease: A systematic review of cohort studies. Lancet 2006, 368, 666–678. [Google Scholar] [CrossRef]
- Kang, K.W.; Ok, M.; Lee, S.K. Leptin as a Key between Obesity and Cardiovascular Disease. J. Obes. Metab. Syndr. 2020, 29, 248–259. [Google Scholar] [CrossRef]
- Bombelli, M.; Facchetti, R.; Sega, R.; Carugo, S.; Fodri, D.; Brambilla, G.; Giannattasio, C.; Grassi, G.; Mancia, G. Impact of body mass index and waist circumference on the long-term risk of diabetes mellitus, hypertension, and cardiac organ damage. Hypertension 2011, 58, 1029–1035. [Google Scholar] [CrossRef]
- Wensveen, F.M.; Valentić, S.; Šestan, M.; Turk Wensveen, T.; Polić, B. The “Big Bang” in obese fat: Events initiating obesity-induced adipose tissue inflammation. Eur. J. Immunol. 2015, 45, 2446–2456. [Google Scholar] [CrossRef]
- Martin, S.S.; Qasim, A.; Reilly, M.P. Leptin resistance: A possible interface of inflammation and metabolism in obesity-related cardiovascular disease. J. Am. Coll. Cardiol. 2008, 52, 1201–1210. [Google Scholar] [CrossRef]
- Recinella, L.; Orlando, G.; Ferrante, C.; Chiavaroli, A.; Brunetti, L.; Leone, S. Adipokines: New Potential Therapeutic Target for Obesity and Metabolic, Rheumatic, and Cardiovascular Diseases. Front. Physiol. 2020, 11, 578966. [Google Scholar] [CrossRef]
- Carbone, S.; Canada, J.M.; Billingsley, H.E.; Siddiqui, M.S.; Elagizi, A.; Lavie, C.J. Obesity paradox in cardiovascular disease: Where do we stand? Vasc. Health Risk Manag. 2019, 15, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Blüher, M. Metabolically Healthy Obesity. Endocr. Rev. 2020, 41, 405–420. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Huang, R.; Jiang, M.; Wang, W.; Chai, Y.; Liu, Q.; Zhang, W.; Han, Y.; Yan, F.; Lu, Q.; et al. Myocardial Tissue-Level Characteristics of Adults with Metabolically Healthy Obesity. JACC Cardiovasc. Imaging 2023, 16, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, P.; Henriksson, H.; Tynelius, P.; Berglind, D.; Löf, M.; Lee, I.M.; Shiroma, E.J.; Ortega, F.B. Fitness and Body Mass Index During Adolescence and Disability Later in Life: A Cohort Study. Ann. Intern. Med. 2019, 170, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, H.; Henriksson, P.; Tynelius, P.; Ekstedt, M.; Berglind, D.; Labayen, I.; Ruiz, J.R.; Lavie, C.J.; Ortega, F.B. Cardiorespiratory fitness, muscular strength, and obesity in adolescence and later chronic disability due to cardiovascular disease: A cohort study of 1 million men. Eur. Heart J. 2020, 41, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Boutari, C.; DeMarsilis, A.; Mantzoros, C.S. Obesity and diabetes. Diabetes Res. Clin. Pract. 2023, 202, 110773. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Gastaldelli, A.; Yki-Järvinen, H.; Scherer, P.E. Why does obesity cause diabetes? Cell Metab. 2022, 34, 11–20. [Google Scholar] [CrossRef]
- Hocking, S.; Samocha-Bonet, D.; Milner, K.L.; Greenfield, J.R.; Chisholm, D.J. Adiposity and insulin resistance in humans: The role of the different tissue and cellular lipid depots. Endocr. Rev. 2013, 34, 463–500. [Google Scholar] [CrossRef]
- Sinha, R.; Fisch, G.; Teague, B.; Tamborlane, W.V.; Banyas, B.; Allen, K.; Savoye, M.; Rieger, V.; Taksali, S.; Barbetta, G.; et al. Prevalence of impaired glucose tolerance among children and adolescents with marked obesity. N. Engl. J. Med. 2002, 346, 802–810. [Google Scholar] [CrossRef]
- D’Adamo, E.; Cali, A.M.G.; Weiss, R.; Santoro, N.; Pierpont, B.; Northrup, V.; Caprio, S. Central role of fatty liver in the pathogenesis of insulin resistance in obese adolescents. Diabetes Care 2010, 33, 1817–1822. [Google Scholar] [CrossRef]
- Kahkoska, A.R.; Pokaprakarn, T.; Rumay Alexander, G.; Crume, T.L.; Dabelea, D.; Divers, J.; Dolan, L.M.; Jensen, E.T.; Lawrence, J.M.; Marcovina, S.; et al. The Impact of Racial and Ethnic Health Disparities in Diabetes Management on Clinical Outcomes: A Reinforcement Learning Analysis of Health Inequity Among Youth and Young Adults in the SEARCH for Diabetes in Youth Study. Diabetes Care 2022, 45, 108–118. [Google Scholar] [CrossRef]
- Al Amiri, E.; Abdullatif, M.; Abdulle, A.; Al Bitar, N.; Afandi, E.Z.; Parish, M.; Darwiche, G. The prevalence, risk factors, and screening measure for prediabetes and diabetes among Emirati overweight/obese children and adolescents. BMC Public Health 2015, 15, 1298. [Google Scholar] [CrossRef] [PubMed]
- Deeb, A.; Salima, A.; Samia, M.; Ghada, E.; Abubaker, E. Insulin Resistance, Impaired fasting, Glucose Intolerance and Type II Diabetes Mellitus in Overweight and Obese Children in Abu Dhabi. J. Diabetes Obes. 2017, 4, 1–8. [Google Scholar] [CrossRef]
- Dilworth, L.; Facey, A.; Omoruyi, F. Diabetes Mellitus and Its Metabolic Complications: The Role of Adipose Tissues. Int. J. Mol. Sci. 2021, 22, 7644. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Mclaughlin, T.; Ackerman, S.E.; Shen, L.; Engleman, E. Role of innate and adaptive immunity in obesity-associated metabolic disease. J. Clin. Investig. 2017, 127, 5–13. [Google Scholar] [CrossRef]
- Pérez-Pérez, A.; Vilariño-García, T.; Fernández-Riejos, P.; Martín-González, J.; Segura-Egea, J.J.; Sánchez-Margalet, V. Role of leptin as a link between metabolism and the immune system. Cytokine Growth Factor Rev. 2017, 35, 71–84. [Google Scholar] [CrossRef]
- Sánchez-Margalet, V.; Martín-Romero, C.; Santos-Alvarez, J.; Goberna, R.; Najib, S.; Gonzalez-Yanes, C. Role of leptin as an immunomodulator of blood mononuclear cells: Mechanisms of action. Clin. Exp. Immunol. 2003, 133, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Margalet, V.; Fernández-Riejos, P.; Najib, S.; Santos-Alvarez, J.; Martín-Romero, C.; Pérez-Pérez, A.; González-Yanes, C. Role of leptin in the activation of immune cells. Mediat. Inflamm. 2010, 2010, 568343. [Google Scholar] [CrossRef]
- Bonamichi, B.D.S.F.; Lee, J. Unusual Suspects in the Development of Obesity-Induced Inflammation and Insulin Resistance: NK cells, iNKT cells, and ILCs. Diabetes Metab. J. 2017, 41, 229–250. [Google Scholar] [CrossRef]
- Wouters, K.; Kusters, Y.H.A.M.; Bijnen, M.; Wetzels, S.; Zhang, X.; Linssen, P.B.C.; Gaens, K.; Houben, A.J.H.M.; Joris, P.J.; Plat, J.; et al. NK cells in human visceral adipose tissue contribute to obesity-associated insulin resistance through low-grade inflammation. Clin. Transl. Med. 2020, 10, e192. [Google Scholar] [CrossRef]
- Caussy, C.; Aubin, A.; Loomba, R. The Relationship Between Type 2 Diabetes, NAFLD, and Cardiovascular Risk. Curr. Diab. Rep. 2021, 21, 15. [Google Scholar] [CrossRef]
- Naito, R.; Kasai, T. Coronary artery disease in type 2 diabetes mellitus: Recent treatment strategies and future perspectives. World J. Cardiol. 2015, 7, 119. [Google Scholar] [CrossRef]
- Shah, A.D.; Langenberg, C.; Rapsomaniki, E.; Denaxas, S.; Pujades-Rodriguez, M.; Gale, C.P.; Deanfield, J.; Smeeth, L.; Timmis, A.; Hemingway, H. Type 2 diabetes and incidence of cardiovascular diseases: A cohort study in 1·9 million people. Lancet Diabetes Endocrinol. 2015, 3, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Punthakee, Z.; Goldenberg, R.; Katz, P. Definition, Classification and Diagnosis of Diabetes, Prediabetes and Metabolic Syndrome. Can. J. Diabetes 2018, 42 (Suppl. S1), S10–S15. [Google Scholar] [CrossRef] [PubMed]
- Prandi, F.R.; Evangelista, I.; Sergi, D.; Palazzuoli, A.; Romeo, F. Mechanisms of cardiac dysfunction in diabetic cardiomyopathy: Molecular abnormalities and phenotypical variants. Heart Fail. Rev. 2023, 28, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.L.; Pietz, K.; Battleman, D.S.; Beyth, R.J. Prevalence of Comorbid Hypertension and Dyslipidemia and Associated Cardiovascular Disease. Heart Dis. 2004, 2, 3. [Google Scholar]
- Piché, M.E.; Tchernof, A.; Després, J.P. Obesity Phenotypes, Diabetes, and Cardiovascular Diseases. Circ. Res. 2020, 126, 1477–1500. [Google Scholar] [CrossRef]
- Barrett-Connor, E. Hypercholesterolemia predicts early death from coronary heart disease in elderly men but not women. The Rancho Bernardo Study. Ann. Epidemiol. 1992, 2, 77–83. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, C.; Hong, J.; Zeng, J.; Lai, S.; Lv, A.; Su, Q.; Dong, Y.; Zhou, Z.; Tang, W.; et al. Lipid profiling reveals different therapeutic effects of metformin and glipizide in patients with type 2 diabetes and coronary artery disease. Diabetes Care 2014, 37, 2804–2812. [Google Scholar] [CrossRef]
- Nelson, A.J.; Pagidipati, N.J.; Aroda, V.R.; Cavender, M.A.; Green, J.B.; Lopes, R.D.; Al-Khalidi, H.; Gaynor, T.; Kaltenbach, L.A.; Kirk, J.K.; et al. Incorporating SGLT2i and GLP-1RA for Cardiovascular and Kidney Disease Risk Reduction: Call for Action to the Cardiology Community. Circulation 2021, 144, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Strain, W.D.; Paldánius, P.M. Diabetes, cardiovascular disease and the microcirculation. Cardiovasc. Diabetol. 2018, 17, 57. [Google Scholar] [CrossRef]
- Halaas, J.L.; Gajiwala, K.S.; Maffei, M.; Cohen, S.L.; Chait, B.T.; Rabinowitz, D.; Lallone, R.L.; Burley, S.K.; Friedman, J.M. Weight-reducing effects of the plasma protein encoded by the obese gene. Science 1995, 269, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L.; et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef]
- Fain, J.N.; Madan, A.K.; Hiler, M.L.; Cheema, P.; Bahouth, S.W. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 2004, 145, 2273–2282. [Google Scholar] [CrossRef]
- Wajchenberg, B.L. Subcutaneous and visceral adipose tissue: Their relation to the metabolic syndrome. Endocr. Rev. 2000, 21, 697–738. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pérez, A.; Maymó, J.; Gambino, Y.; Guadix, P.; Dueñas, J.L.; Varone, C.; Sánchez-Margalet, V. Insulin enhances leptin expression in human trophoblastic cells. Biol. Reprod. 2013, 89, 20. [Google Scholar] [CrossRef] [PubMed]
- Miell, J.P.; Englaro, P.; Blum, W.F. Dexamethasone induces an acute and sustained rise in circulating leptin levels in normal human subjects. Horm. Metab. Res. 1996, 28, 704–707. [Google Scholar] [CrossRef]
- De Vos, P.; Saladin, R.; Auwerx, J.; Staels, B. Induction of ob gene expression by corticosteroids is accompanied by body weight loss and reduced food intake. J. Biol. Chem. 1995, 270, 15958–15961. [Google Scholar] [CrossRef]
- Ahima, R.S.; Flier, J.S. Leptin. Annu. Rev. Physiol. 2000, 62, 413–437. [Google Scholar] [CrossRef]
- Margetic, S.; Gazzola, C.; Pegg, G.G.; Hill, R.A. Leptin: A review of its peripheral actions and interactions. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 1407–1433. [Google Scholar] [CrossRef]
- Mantzoros, C.S.; Magkos, F.; Brinkoetter, M.; Sienkiewicz, E.; Dardeno, T.A.; Kim, S.Y.; Hamnvik, O.P.R.; Koniaris, A. Leptin in human physiology and pathophysiology. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E567–E584. [Google Scholar] [CrossRef]
- Hickey, M.S.; Israel, R.G.; Gardiner, S.N.; Considine, R.V.; McCammon, M.R.; Tyndall, G.L.; Houmard, J.A.; Marks, R.H.L.; Caro, J.F. Gender Differences in Serum Leptin Levels in Humans. Biochem. Mol. Med. 1996, 59, 1–6. [Google Scholar] [CrossRef]
- Arch, J.R.S.; Stock, M.J.; Trayhurn, P. Leptin resistance in obese humans: Does it exist and what does it mean? Int. J. Obes. Relat. Metab. Disord. 1998, 22, 1159–1163. [Google Scholar] [CrossRef]
- Knight, Z.A.; Hannan, K.S.; Greenberg, M.L.; Friedman, J.M. Hyperleptinemia is required for the development of leptin resistance. PLoS ONE 2010, 5, e11376. [Google Scholar] [CrossRef] [PubMed]
- Poetsch, M.S.; Strano, A.; Guan, K. Role of Leptin in Cardiovascular Diseases. Front. Endocrinol. 2020, 11, 354. [Google Scholar] [CrossRef] [PubMed]
- Ren, J. Leptin and hyperleptinemia—From friend to foe for cardiovascular function. J. Endocrinol. 2004, 181, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Kusminski, C.M.; Scherer, P.E. Adiponectin, Leptin and Cardiovascular Disorders. Circ. Res. 2021, 128, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.J.; Clifton, D.K.; Steiner, R.A. Leptin’s actions on the reproductive axis: Perspectives and mechanisms. Biol. Reprod. 1999, 60, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Bray, G.A. Obesity and reproduction. Hum. Reprod. 1997, 12 (Suppl. S1), 26–32. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.L.; Nagatani, S. Physiological perspectives on leptin as a regulator of reproduction: Role in timing puberty. Biol. Reprod. 1999, 60, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pérez, A.; Sánchez-Jiménez, F.; Maymó, J.; Dueñas, J.L.; Varone, C.; Sánchez-Margalet, V. Role of leptin in female reproduction. Clin. Chem. Lab. Med. 2015, 53, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pérez, A.; Toro, A.; Vilariño-García, T.; Maymó, J.; Guadix, P.; Dueñas, J.L.; Fernández-Sánchez, M.; Varone, C.; Sánchez-Margalet, V. Leptin action in normal and pathological pregnancies. J. Cell. Mol. Med. 2018, 22, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.B.S. Leptin—Much more than a satiety signal. Annu. Rev. Nutr. 2000, 20, 45–75. [Google Scholar] [CrossRef]
- Meister, B. Control of food intake via leptin receptors in the hypothalamus. Vitam. Horm. 2000, 59, 265–304. [Google Scholar] [CrossRef]
- Sanchez-Margalet, V.; Martin-Romero, C. Human leptin signaling in human peripheral blood mononuclear cells: Activation of the JAK-STAT pathway. Cell. Immunol. 2001, 211, 30–36. [Google Scholar] [CrossRef]
- Villanueva, E.C.; Myers, M.G. Leptin receptor signaling and the regulation of mammalian physiology. Int. J. Obes. 2008, 32 (Suppl. S7), S8–S12. [Google Scholar] [CrossRef]
- Bates, S.H.; Myers, M.G. The role of leptin receptor signaling in feeding and neuroendocrine function. Trends Endocrinol. Metab. 2003, 14, 447–452. [Google Scholar] [CrossRef]
- Bjørbaek, C.; Kahn, B.B. Leptin signaling in the central nervous system and the periphery. Recent Prog. Horm. Res. 2004, 59, 305–331. [Google Scholar] [CrossRef] [PubMed]
- Jéquier, E. Leptin signaling, adiposity, and energy balance. Ann. N. Y. Acad. Sci. 2002, 967, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Murakami, T.; Otani, S.; Kuwajima, M.; Shima, K. Leptin receptor signal transduction: OBRa and OBRb of fa type. Biochem. Biophys. Res. Commun. 1998, 246, 752–759. [Google Scholar] [CrossRef]
- Takahashi, Y.; Okimura, Y.; Mizuno, I.; Iida, K.; Takahashi, T.; Kaji, H.; Abe, H.; Chihara, K. Leptin induces mitogen-activated protein kinase-dependent proliferation of C3H10T1/2 cells. J. Biol. Chem. 1997, 272, 12897–12900. [Google Scholar] [CrossRef] [PubMed]
- Bjørbæk, C.; Uotani, S.; Da Silva, B.; Flier, J.S. Divergent signaling capacities of the long and short isoforms of the leptin receptor. J. Biol. Chem. 1997, 272, 32686–32695. [Google Scholar] [CrossRef] [PubMed]
- Baumann, H.; Morella, K.K.; White, D.W.; Dembski, M.; Bailon, P.S.; Kim, H.; Lai, C.F.; Tartaglia, L.A. The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. Proc. Natl. Acad. Sci. USA 1996, 93, 8374–8378. [Google Scholar] [CrossRef] [PubMed]
- Bjørbæk, C.; El-Haschimi, K.; Frantz, J.D.; Flier, J.S. The role of SOCS-3 in leptin signaling and leptin resistance. J. Biol. Chem. 1999, 274, 30059–30065. [Google Scholar] [CrossRef]
- Zabolotny, J.M.; Bence-Hanulec, K.K.; Stricker-Krongrad, A.; Haj, F.; Wang, Y.; Minokoshi, Y.; Kim, Y.B.; Elmquist, J.K.; Tartaglia, L.A.; Kahn, B.B.; et al. PTP1B regulates leptin signal transduction in vivo. Dev. Cell 2002, 2, 489–495. [Google Scholar] [CrossRef]
- Martín-Romero, C.; Sánchez-Margalet, V. Human leptin activates PI3K and MAPK pathways in human peripheral blood mononuclear cells: Possible role of Sam68. Cell. Immunol. 2001, 212, 83–91. [Google Scholar] [CrossRef]
- Ren, D.; Li, M.; Duan, C.; Rui, L. Identification of SH2-B as a key regulator of leptin sensitivity, energy balance, and body weight in mice. Cell Metab. 2005, 2, 95–104. [Google Scholar] [CrossRef]
- Liu, J.; Lai, F.; Hou, Y.; Zheng, R. Leptin signaling and leptin resistance. Med. Rev. 2022, 2, 363–384. [Google Scholar] [CrossRef]
- Minokoshi, Y.; Alquier, T.; Furukawa, H.; Kim, Y.B.; Lee, A.; Xue, B.; Mu, J.; Foufelle, F.; Ferré, P.; Birnbaum, M.J.; et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 2004, 428, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Minokoshi, Y.; Kim, Y.B.; Peroni, O.D.; Fryer, L.G.D.; Müller, C.; Carling, D.; Kahn, B.B. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002, 415, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMP-activated protein kinase: A key regulator of energy balance with many roles in human disease. J. Intern. Med. 2014, 276, 543–559. [Google Scholar] [CrossRef] [PubMed]
- Tomaszuk, A.; Simpson, C.; Williams, G. Neuropeptide Y, the hypothalamus and the regulation of energy homeostasis. Horm. Res. 1996, 46, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Seeley, R.J.; Yagaloff, K.A.; Fisher, S.L.; Burn, P.; Thiele, T.E.; Van Dijk, G.; Baskin, D.G.; Schwartz, M.W. Melanocortin receptors in leptin effects. Nature 1997, 390, 349. [Google Scholar] [CrossRef] [PubMed]
- Matson, C.A.; Wiater, M.F.; Kuijper, J.L.; Weigle, D.S. Synergy between leptin and cholecystokinin (CCK) to control daily caloric intake. Peptides 1997, 18, 1275–1278. [Google Scholar] [CrossRef] [PubMed]
- Ellacott, K.L.J.; Halatchev, I.G.; Cone, R.D. Interactions between gut peptides and the central melanocortin system in the regulation of energy homeostasis. Peptides 2006, 27, 340–349. [Google Scholar] [CrossRef]
- Klein, S.; Coppack, S.W.; Mohamed-Ali, V.; Landt, M. Adipose tissue leptin production and plasma leptin kinetics in humans. Diabetes 1996, 45, 984–987. [Google Scholar] [CrossRef]
- Niskanen, L.; Haffner, S.; Karhunen, L.J.; Turpeinen, A.K.; Miettinen, H.; Uusitupa, M.I.J. Serum leptin in relation to resting energy expenditure and fuel metabolism in obese subjects. Int. J. Obes. Relat. Metab. Disord. 1997, 21, 309–313. [Google Scholar] [CrossRef]
- Frederich, R.C.; Hamann, A.; Anderson, S.; Löllmann, B.; Lowell, B.B.; Flier, J.S. Leptin levels reflect body lipid content in mice: Evidence for diet-induced resistance to leptin action. Nat. Med. 1995, 1, 1311–1314. [Google Scholar] [CrossRef]
- Myers, M.G.; Heymsfield, S.B.; Haft, C.; Kahn, B.B.; Laughlin, M.; Leibel, R.L.; Tschöp, M.H.; Yanovski, J.A. Challenges and opportunities of defining clinical leptin resistance. Cell Metab. 2012, 15, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Wabitsch, M.; Funcke, J.-B.; Lennerz, B.; Kuhnle-Krahl, U.; Lahr, G.; Debatin, K.-M.; Vatter, P.; Gierschik, P.; Moepps, B.; Fischer-Posovszky, P. Biologically inactive leptin and early-onset extreme obesity. N. Engl. J. Med. 2015, 372, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Mantzoros, C.S. The role of leptin in human obesity and disease: A review of current evidence. Ann. Intern. Med. 1999, 130, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Mark, A.L. Selective leptin resistance revisited. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R566–R581. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J. 20 years of leptin: Leptin at 20: An overview. J. Endocrinol. 2014, 223, T1–T8. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Wong, J.; Brooks, V.L. Obesity: Sex and sympathetics. Biol. Sex Differ. 2020, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Oliveira, V.; Câmara, N.O.S.; Moraes-Vieira, P.M. Adipokines as drug targets in diabetes and underlying disturbances. J. Diabetes Res. 2015, 2015, 681612. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Seeley, R.J.; Campfield, L.A.; Burn, P.; Baskin, D.G. Identification of targets of leptin action in rat hypothalamus. J. Clin. Investig. 1996, 98, 1101–1106. [Google Scholar] [CrossRef]
- German, J.P.; Thaler, J.P.; Wisse, B.E.; Oh-I, S.; Sarruf, D.A.; Matsen, M.E.; Fischer, J.D.; Taborsky, G.J.; Schwartz, M.W.; Morton, G.J. Leptin activates a novel CNS mechanism for insulin-independent normalization of severe diabetic hyperglycemia. Endocrinology 2011, 152, 394–404. [Google Scholar] [CrossRef]
- Li, S.; Li, H.; Wang, R.; Zhang, J.P. The Effect of Sitagliptin on Obese Patients with Insulin Treatment-Induced Diabetes Mellitus. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3490–3495. [Google Scholar] [PubMed]
- Tang, X.; Li, J.; Xiang, W.; Cui, Y.; Xie, B.; Wang, X.; Xu, Z.; Gan, L. Metformin increases hepatic leptin receptor and decreases steatosis in mice. J. Endocrinol. 2016, 230, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Farooq, R.; Amin, S.; Hayat Bhat, M.; Malik, R.; Wani, H.A.; Majid, S. Type 2 diabetes and metabolic syndrome—Adipokine levels and effect of drugs. Gynecol. Endocrinol. 2017, 33, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Greer, J.J.M.; Ware, D.P.; Lefer, D.J. Myocardial infarction and heart failure in the db/db diabetic mouse. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H146–H153. [Google Scholar] [CrossRef] [PubMed]
- Belke, D.D.; Larsen, T.S.; Gibbs, E.M.; Severson, D.L. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E1104–E1113. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Rodrigues, B. Role of changes in cardiac metabolism in development of diabetic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1489–H1506. [Google Scholar] [CrossRef] [PubMed]
- Karmazyn, M.; Purdham, D.M.; Rajapurohitam, V.; Zeidan, A. Leptin as a cardiac hypertrophic factor: A potential target for therapeutics. Trends Cardiovasc. Med. 2007, 17, 206–211. [Google Scholar] [CrossRef]
- Katsiki, N.; Mikhailidis, D.P.; Banach, M. Leptin, cardiovascular diseases and type 2 diabetes mellitus. Acta Pharmacol. Sin. 2018, 39, 1176–1188. [Google Scholar] [CrossRef]
- Lieb, W.; Sullivan, L.M.; Harris, T.B.; Roubenoff, R.; Benjamin, E.; Levy, D.; Fox, C.S.; Wang, T.J.; Wilson, P.W.; Kannel, W.B.; et al. Plasma leptin levels and incidence of heart failure, cardiovascular disease, and total mortality in elderly individuals. Diabetes Care 2009, 32, 612–616. [Google Scholar] [CrossRef]
- Rajapurohitam, V.; Izaddoustdar, F.; Martinez-Abundis, E.; Karmazyn, M. Leptin-induced cardiomyocyte hypertrophy reveals both calcium-dependent and calcium-independent/RhoA-dependent calcineurin activation and NFAT nuclear translocation. Cell. Signal. 2012, 24, 2283–2290. [Google Scholar] [CrossRef]
- Hall, M.E.; Harmancey, R.; Stec, D.E. Lean heart: Role of leptin in cardiac hypertrophy and metabolism. World J. Cardiol. 2015, 7, 511. [Google Scholar] [CrossRef] [PubMed]
- Leifheit-Nestler, M.; Wagner, N.M.; Gogiraju, R.; Didié, M.; Konstantinides, S.; Hasenfuss, G.; Schäfer, K. Importance of leptin signaling and signal transducer and activator of transcription-3 activation in mediating the cardiac hypertrophy associated with obesity. J. Transl. Med. 2013, 11, 170. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Matthes, J.; Schuster, I.; Valdivia, H.H.; Herzig, S.; Richard, S.; Gómez, A.M. Mechanisms of [Ca2+]i transient decrease in cardiomyopathy of db/db type 2 diabetic mice. Diabetes 2006, 55, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Kho, C.; Lee, A.; Hajjar, R.J. Altered sarcoplasmic reticulum calcium cycling—targets for heart failure therapy. Nat. Rev. Cardiol. 2012, 9, 717–733. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Gustafsson, Å.B. Role of apoptosis in cardiovascular disease. Apoptosis 2009, 14, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Zhao, Y.; Xu, S.; Jin, C.; Wang, M.; Fu, G. Leptin confers protection against TNF-α-induced apoptosis in rat cardiomyocytes. Biochem. Biophys. Res. Commun. 2014, 455, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Abundis, E.; Rajapurohitam, V.; Haist, J.V.; Gan, X.T.; Karmazyn, M. The obesity-related peptide leptin sensitizes cardiac mitochondria to calcium-induced permeability transition pore opening and apoptosis. PLoS ONE 2012, 7, e41612. [Google Scholar] [CrossRef]
- Dixon, R.A.; Davidson, S.M.; Wynne, A.M.; Yellon, D.M.; Smith, C.C.T. The cardioprotective actions of leptin are lost in the Zucker obese (fa/fa) rat. J. Cardiovasc. Pharmacol. 2009, 53, 311–317. [Google Scholar] [CrossRef]
- McGaffin, K.R.; Witham, W.G.; Yester, K.A.; Romano, L.C.; Odoherty, R.M.; McTiernan, C.F.; Odonnell, C.P. Cardiac-specific leptin receptor deletion exacerbates ischaemic heart failure in mice. Cardiovasc. Res. 2011, 89, 60–71. [Google Scholar] [CrossRef]
- Procopio, C.; Andreozzi, F.; Laratta, E.; Cassese, A.; Beguinot, F.; Arturi, F.; Hribal, M.L.; Perticone, F.; Sesti, G. Leptin-stimulated endothelial nitric-oxide synthase via an adenosine 5’-monophosphate-activated protein kinase/Akt signaling pathway is attenuated by interaction with C-reactive protein. Endocrinology 2009, 150, 3584–3593. [Google Scholar] [CrossRef]
- Bełtowski, J.; Wójcicka, G.; Jamroz-Wiśniewska, A.; Borkowska, E. Role of PI3K and PKB/Akt in acute natriuretic and NO-mimetic effects of leptin. Regul. Pept. 2007, 140, 168–177. [Google Scholar] [CrossRef]
- Jamroz-Wiśniewska, A.; Gertler, A.; Solomon, G.; Wood, M.E.; Whiteman, M.; Beltowski, J. Leptin-induced endothelium-dependent vasorelaxation of peripheral arteries in lean and obese rats: Role of nitric oxide and hydrogen sulfide. PLoS ONE 2014, 9, e86744. [Google Scholar] [CrossRef]
- Rahmouni, K.; Morgan, D.A.; Morgan, G.M.; Mark, A.L.; Haynes, W.G. Role of selective leptin resistance in diet-induced obesity hypertension. Diabetes 2005, 54, 2012–2018. [Google Scholar] [CrossRef]
- Bell, B.B.; Rahmouni, K. Leptin as a Mediator of Obesity-Induced Hypertension. Curr. Obes. Rep. 2016, 5, 397–404. [Google Scholar] [CrossRef]
- Faulkner, J.L.; Bruder-Nascimento, T.; Belin De Chantemèle, E.J. The regulation of aldosterone secretion by leptin: Implications in obesity-related cardiovascular disease. Curr. Opin. Nephrol. Hypertens. 2018, 27, 63–69. [Google Scholar] [CrossRef]
- Beldhuis, I.E.; Myhre, P.L.; Bristow, M.; Claggett, B.; Damman, K.; Fang, J.C.; Fleg, J.L.; McKinlay, S.; Lewis, E.F.; O’Meara, E.; et al. Spironolactone in Patients With Heart Failure, Preserved Ejection Fraction, and Worsening Renal Function. J. Am. Coll. Cardiol. 2021, 77, 1211–1221. [Google Scholar] [CrossRef]
- Beltowski, J. Leptin and atherosclerosis. Atherosclerosis 2006, 189, 47–60. [Google Scholar] [CrossRef] [PubMed]
- VanPatten, S.; Karkanias, G.B.; Rossetti, L.; Cohen, D.E. Intracerebroventricular leptin regulates hepatic cholesterol metabolism. Biochem. J. 2004, 379, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Townsend, L.K.; Steinberg, G.R. AMPK and the Endocrine Control of Metabolism. Endocr. Rev. 2023, 44, 910–933. [Google Scholar] [CrossRef] [PubMed]
- Tommerdahl, K.L.; Baumgartner, K.; Schäfer, M.; Bjornstad, P.; Melena, I.; Hegemann, S.; Baumgartner, A.D.; Pyle, L.; Cree-Green, M.; Truong, U.; et al. Impact of Obesity on Measures of Cardiovascular and Kidney Health in Youth with Type 1 Diabetes as Compared with Youth with Type 2 Diabetes. Diabetes Care 2021, 44, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Bruder-Nascimento, T.; Kress, T.C.; Belin De Chantemele, E.J. Recent advances in understanding lipodystrophy: A focus on lipodystrophy-associated cardiovascular disease and potential effects of leptin therapy on cardiovascular function. F1000Research 2019, 8, F1000. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, A.G.; Crujeiras, A.B.; Casanueva, F.F.; Carreira, M.C. Leptin, Obesity, and Leptin Resistance: Where Are We 25 Years Later? Nutrients 2019, 11, 2704. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Mattace Raso, G.; Meli, R. Drug targeting of leptin resistance. Life Sci. 2015, 140, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.D.; Roland, B.L.; Cole, R.L.; Trevaskis, J.L.; Weyer, C.; Koda, J.E.; Anderson, C.M.; Parkes, D.G.; Baron, A.D. Leptin responsiveness restored by amylin agonism in diet-induced obesity: Evidence from nonclinical and clinical studies. Proc. Natl. Acad. Sci. USA 2008, 105, 7257–7262. [Google Scholar] [CrossRef]
- Trevaskis, J.L.; Coffey, T.; Cole, R.; Lei, C.; Wittmer, C.; Walsh, B.; Weyer, C.; Koda, J.; Baron, A.D.; Parkes, D.G.; et al. Amylin-mediated restoration of leptin responsiveness in diet-induced obesity: Magnitude and mechanisms. Endocrinology 2008, 149, 5679–5687. [Google Scholar] [CrossRef]
- Liu, J.; Lee, J.; Hernandez, M.A.S.; Mazitschek, R.; Ozcan, U. Treatment of obesity with celastrol. Cell 2015, 161, 999–1011. [Google Scholar] [CrossRef]
- Feng, X.; Guan, D.; Auen, T.; Choi, J.W.; Salazar Hernández, M.A.; Lee, J.; Chun, H.; Faruk, F.; Kaplun, E.; Herbert, Z.; et al. IL1R1 is required for celastrol’s leptin-sensitization and antiobesity effects. Nat. Med. 2019, 25, 575–582. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, J.; Duan, X.; Zhao, G.; Zhang, M. Celastrol: A Promising Agent Fighting against Cardiovascular Diseases. Antioxidants 2022, 11, 1597. [Google Scholar] [CrossRef]
- Berglund, E.D.; Grobe, J.L.; Rahmouni, K.; Cui, H.; Saito, K.; Davis, K.C.; Morgan, D.A.; Toth, B.A.; Jiang, J.; Singh, U. Celastrol Reduces Obesity in MC4R Deficiency and Stimulates Sympathetic Nerve Activity Affecting Metabolic and Cardiovascular Functions. Diabetes 2019, 68, 1210–1220. [Google Scholar] [CrossRef]
- Chakhtoura, M.; Haber, R.; Ghezzawi, M.; Rhayem, C.; Tcheroyan, R.; Mantzoros, C.S. Pharmacotherapy of obesity: An update on the available medications and drugs under investigation. eClinicalMedicine 2023, 58, 101882. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vilariño-García, T.; Polonio-González, M.L.; Pérez-Pérez, A.; Ribalta, J.; Arrieta, F.; Aguilar, M.; Obaya, J.C.; Gimeno-Orna, J.A.; Iglesias, P.; Navarro, J.; et al. Role of Leptin in Obesity, Cardiovascular Disease, and Type 2 Diabetes. Int. J. Mol. Sci. 2024, 25, 2338. https://doi.org/10.3390/ijms25042338
Vilariño-García T, Polonio-González ML, Pérez-Pérez A, Ribalta J, Arrieta F, Aguilar M, Obaya JC, Gimeno-Orna JA, Iglesias P, Navarro J, et al. Role of Leptin in Obesity, Cardiovascular Disease, and Type 2 Diabetes. International Journal of Molecular Sciences. 2024; 25(4):2338. https://doi.org/10.3390/ijms25042338
Chicago/Turabian StyleVilariño-García, Teresa, María L. Polonio-González, Antonio Pérez-Pérez, Josep Ribalta, Francisco Arrieta, Manuel Aguilar, Juan C. Obaya, José A. Gimeno-Orna, Pedro Iglesias, Jorge Navarro, and et al. 2024. "Role of Leptin in Obesity, Cardiovascular Disease, and Type 2 Diabetes" International Journal of Molecular Sciences 25, no. 4: 2338. https://doi.org/10.3390/ijms25042338