
Del-1 Plays a Protective Role against COPD Development by Inhibiting Inflammation and Apoptosis
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
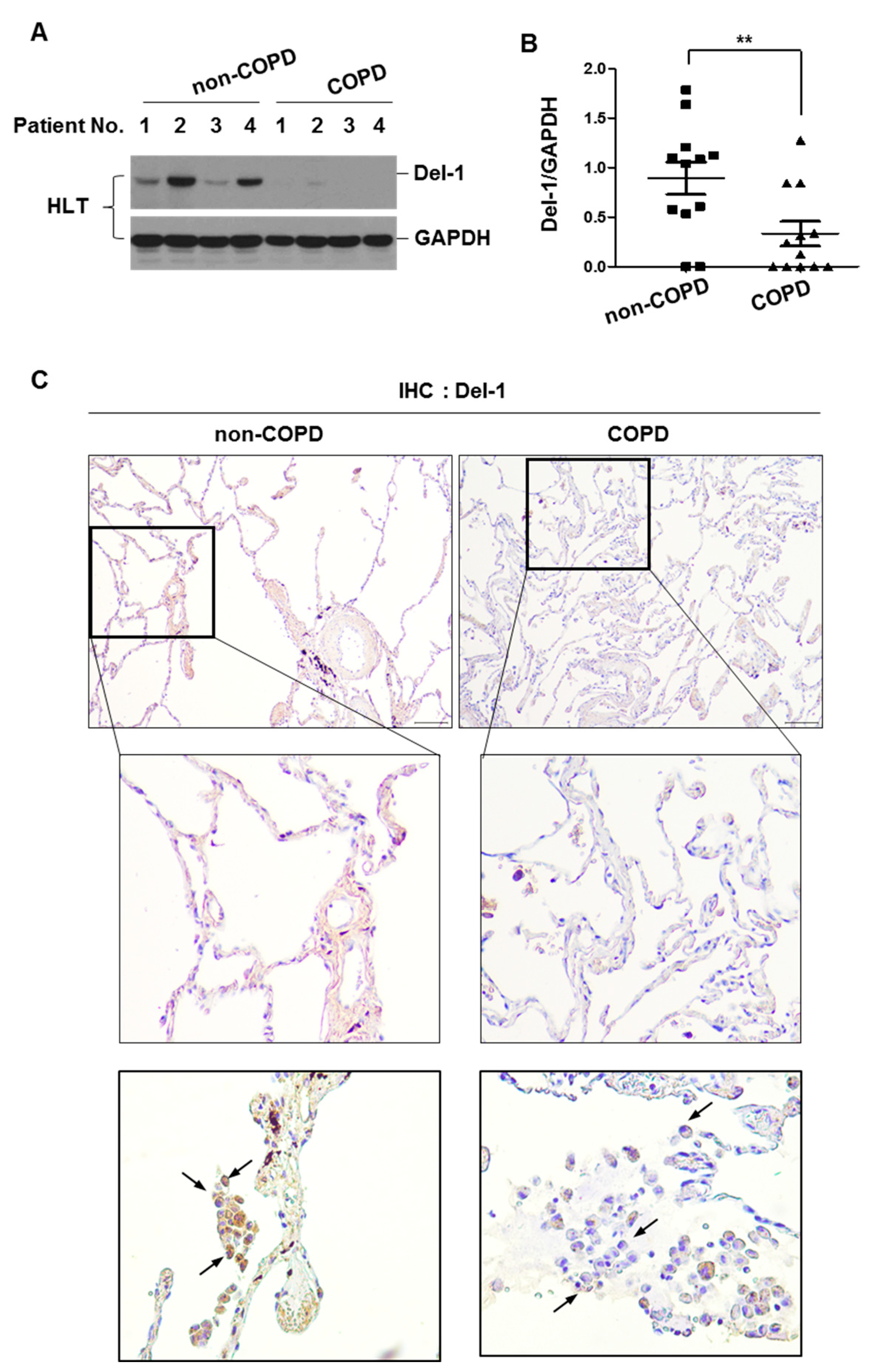
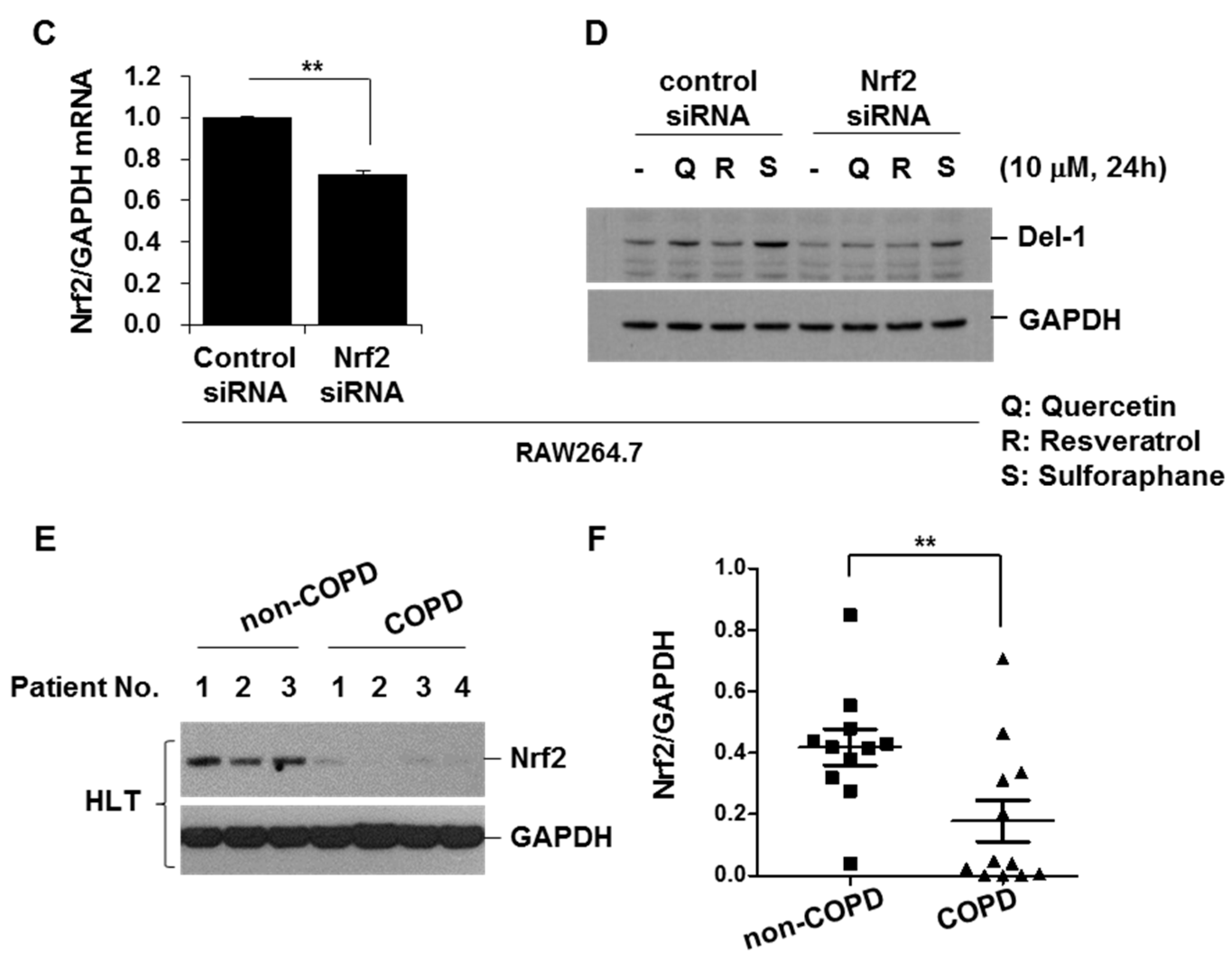
2.1. The Expression Level of Del-1 in Human Lung Tissues
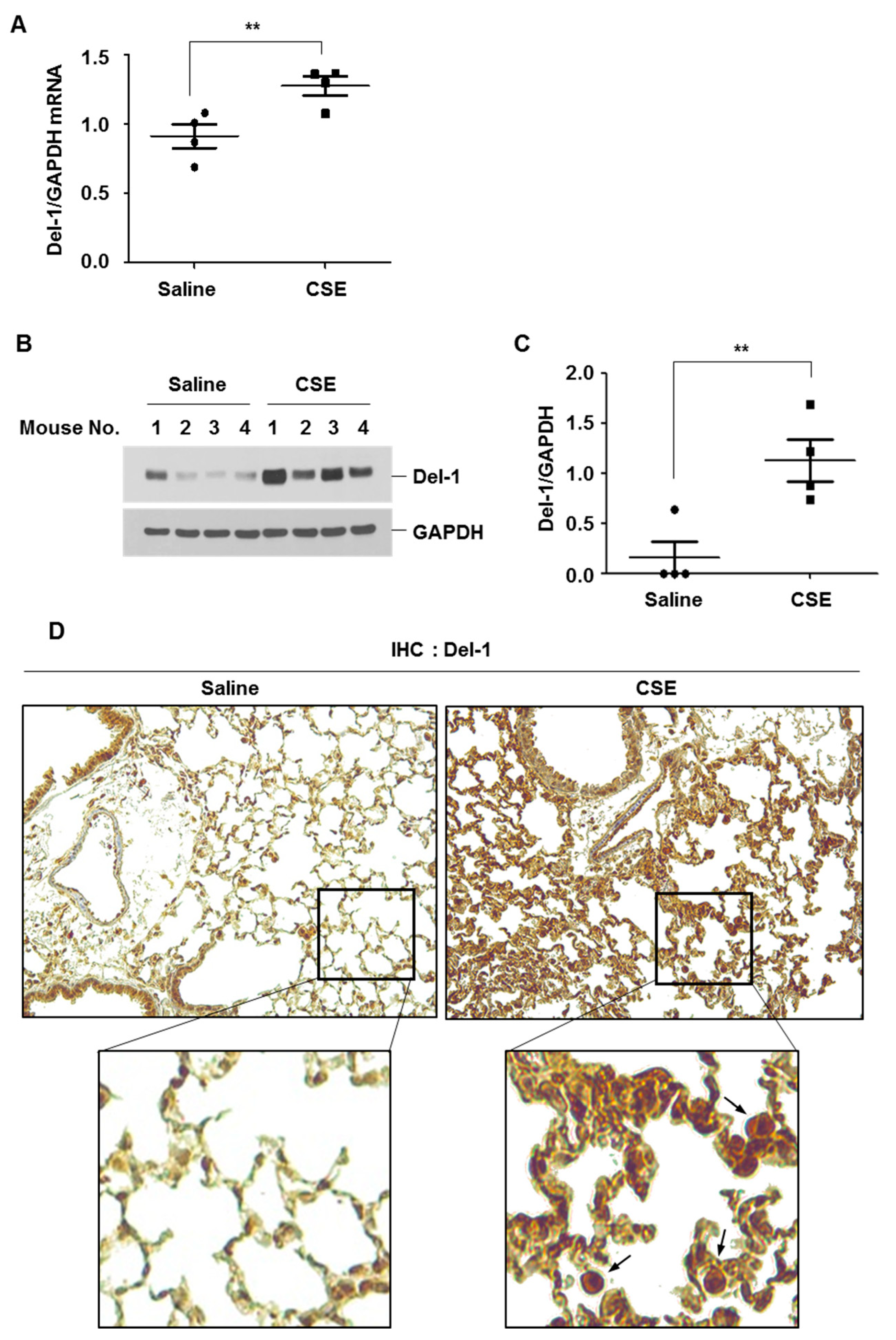
2.2. The Expression Level of Del-1 in Lungs of CSE-Instilled Mice and Its Impact on Oxidative Stress
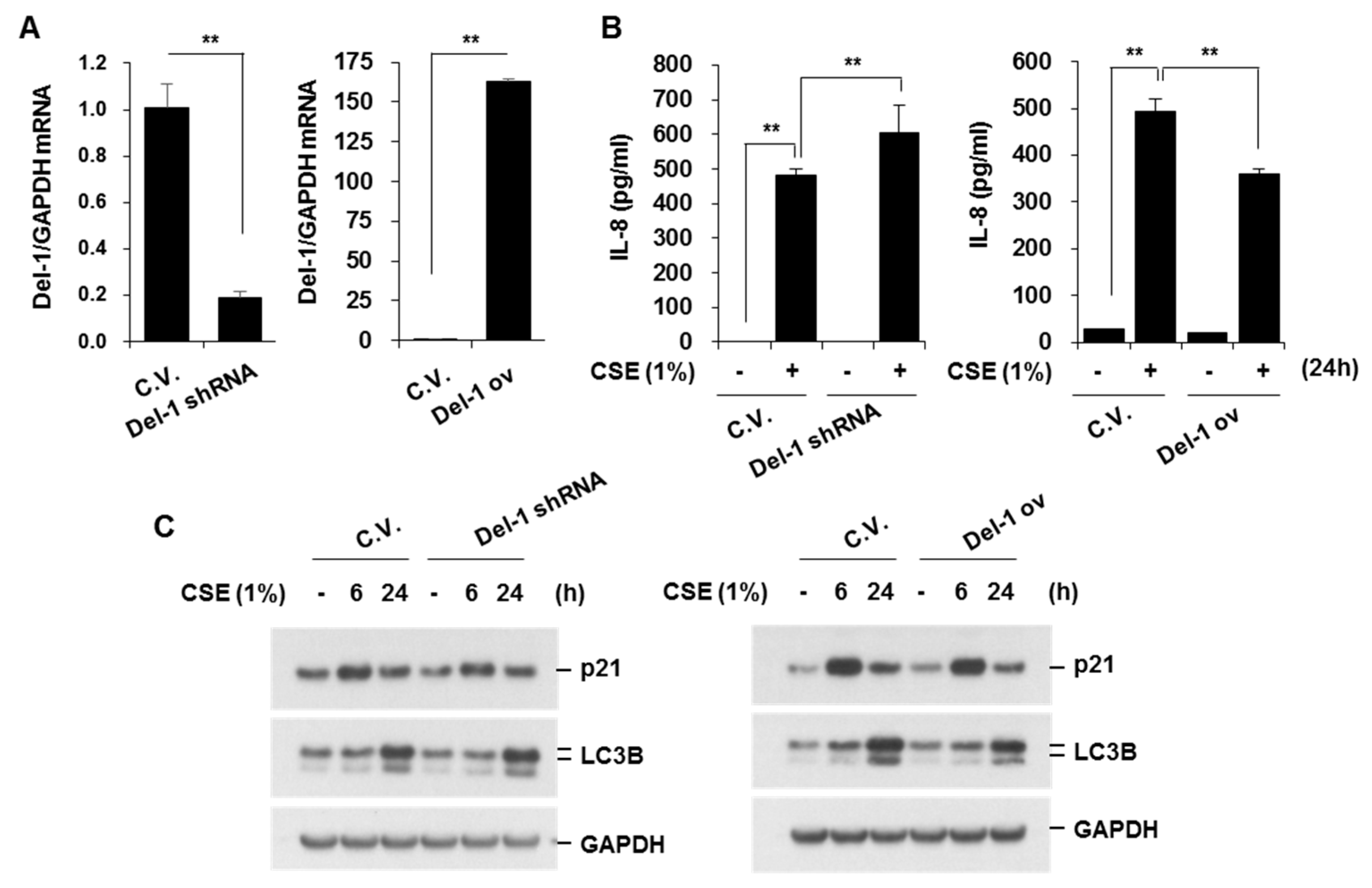
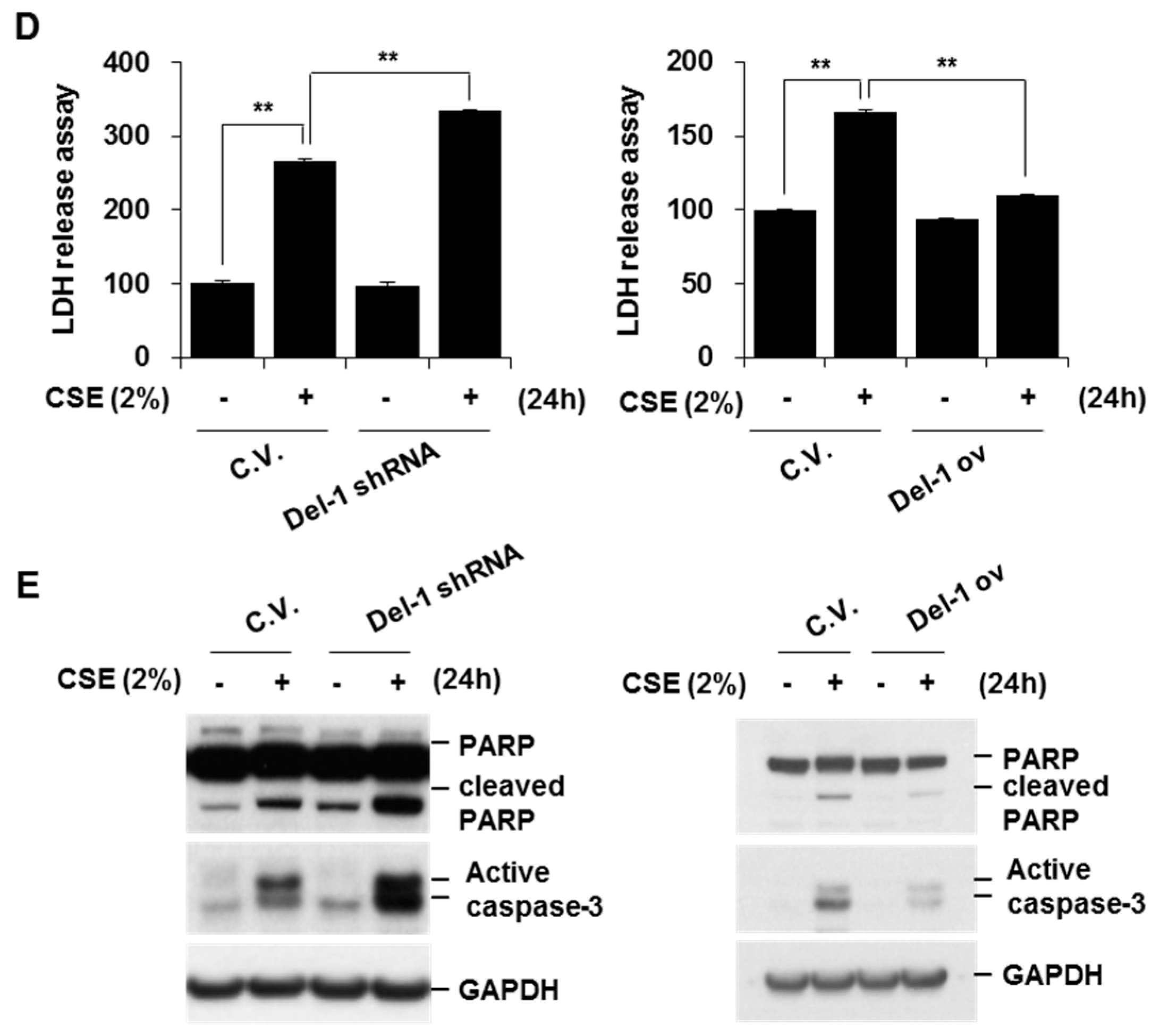
2.3. Del-1 Suppressed CSE-Induced IL-8 Production and Apoptosis in Lung Epithelial Cells
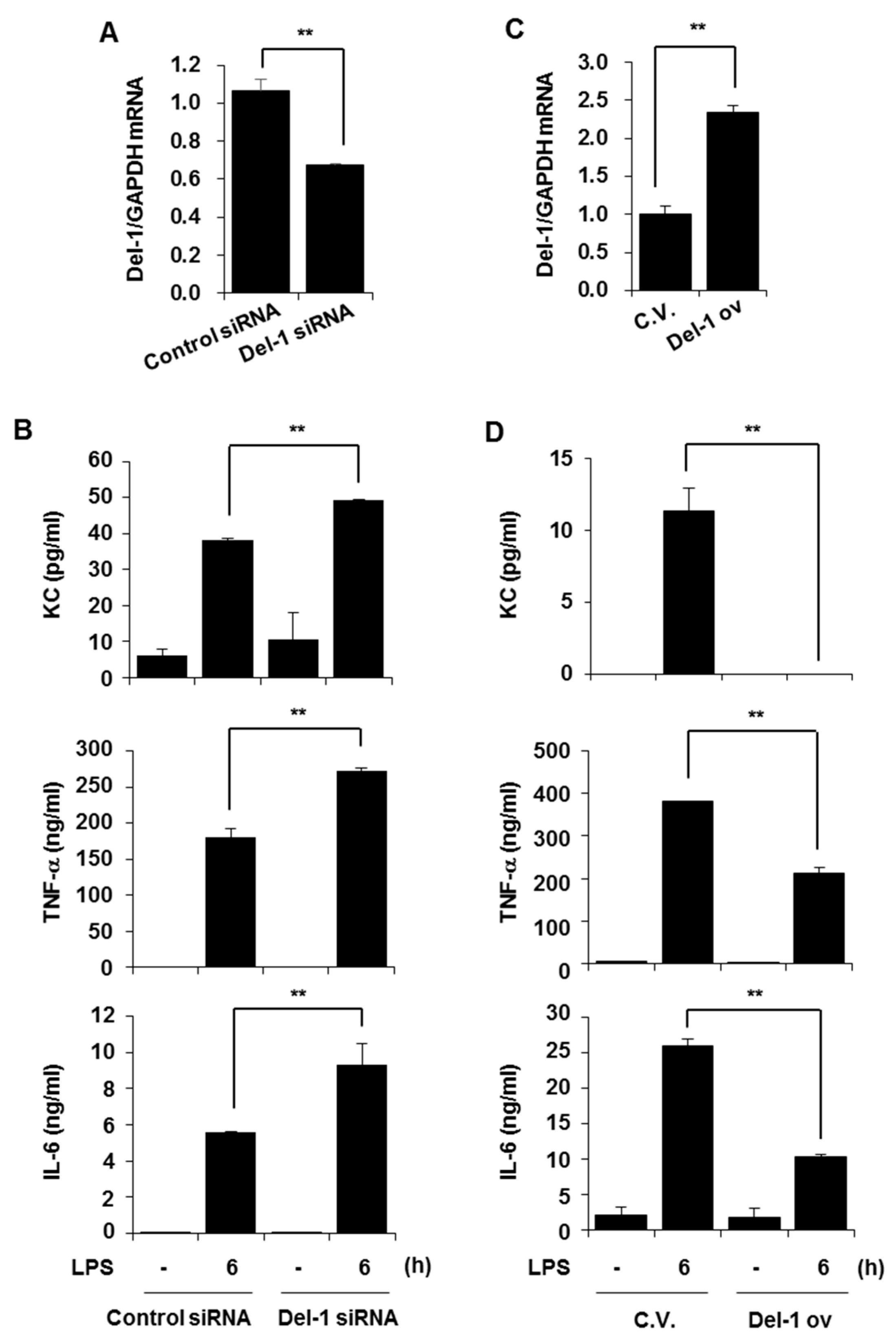
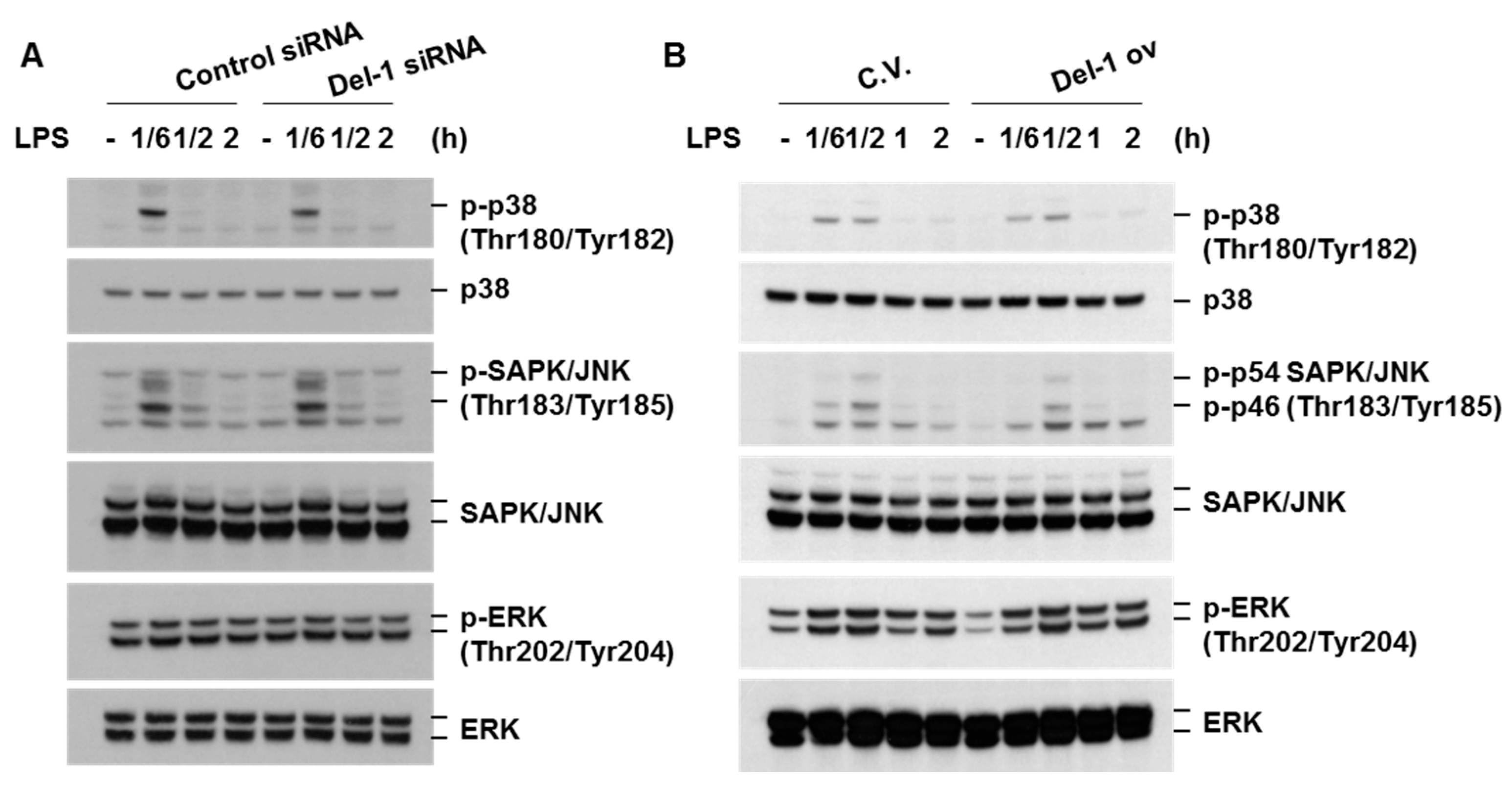
2.4. Del-1 Suppressed LPS-Induced Pro-Inflammatory Chemokine/Cytokines in Macrophages
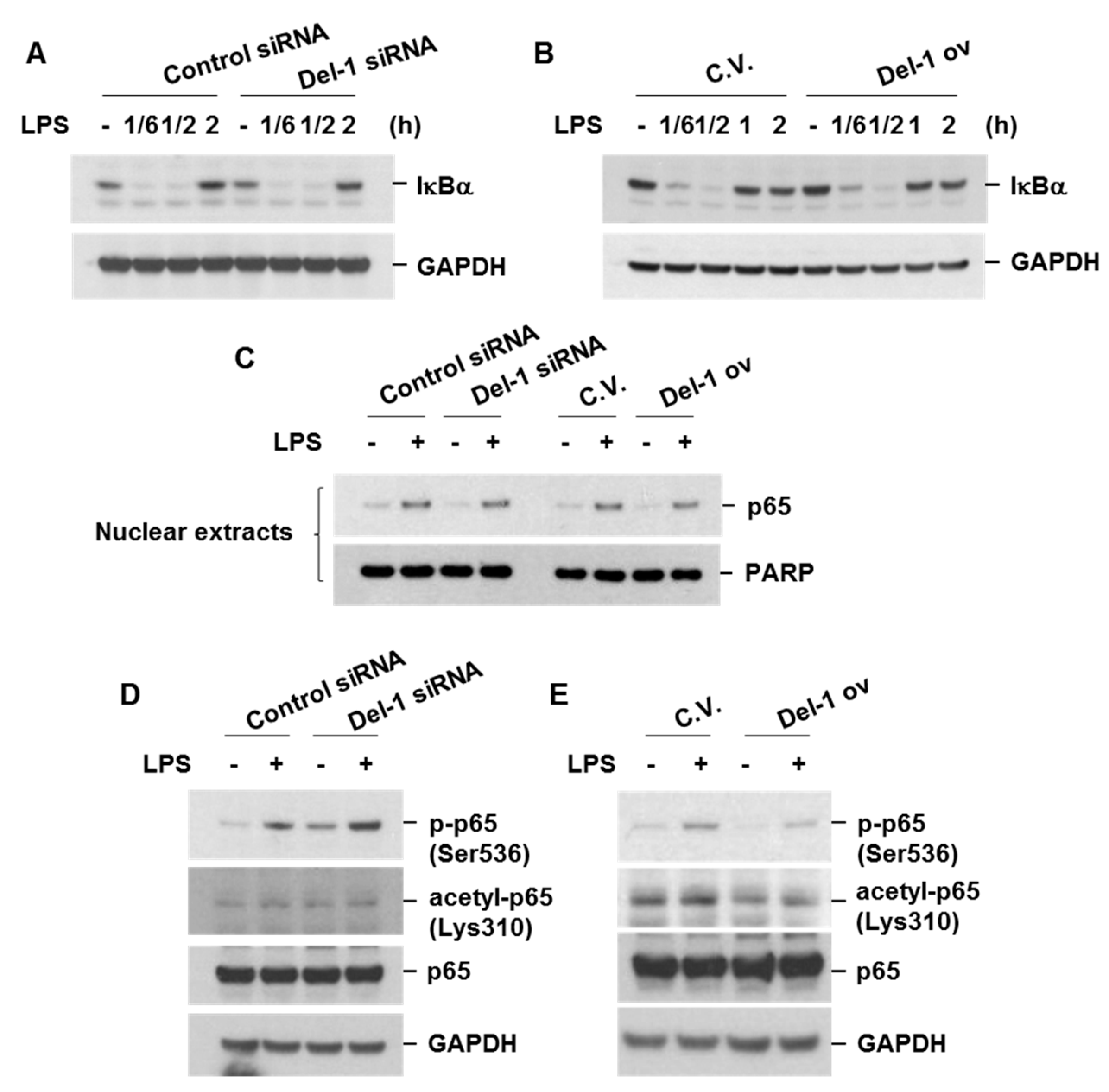
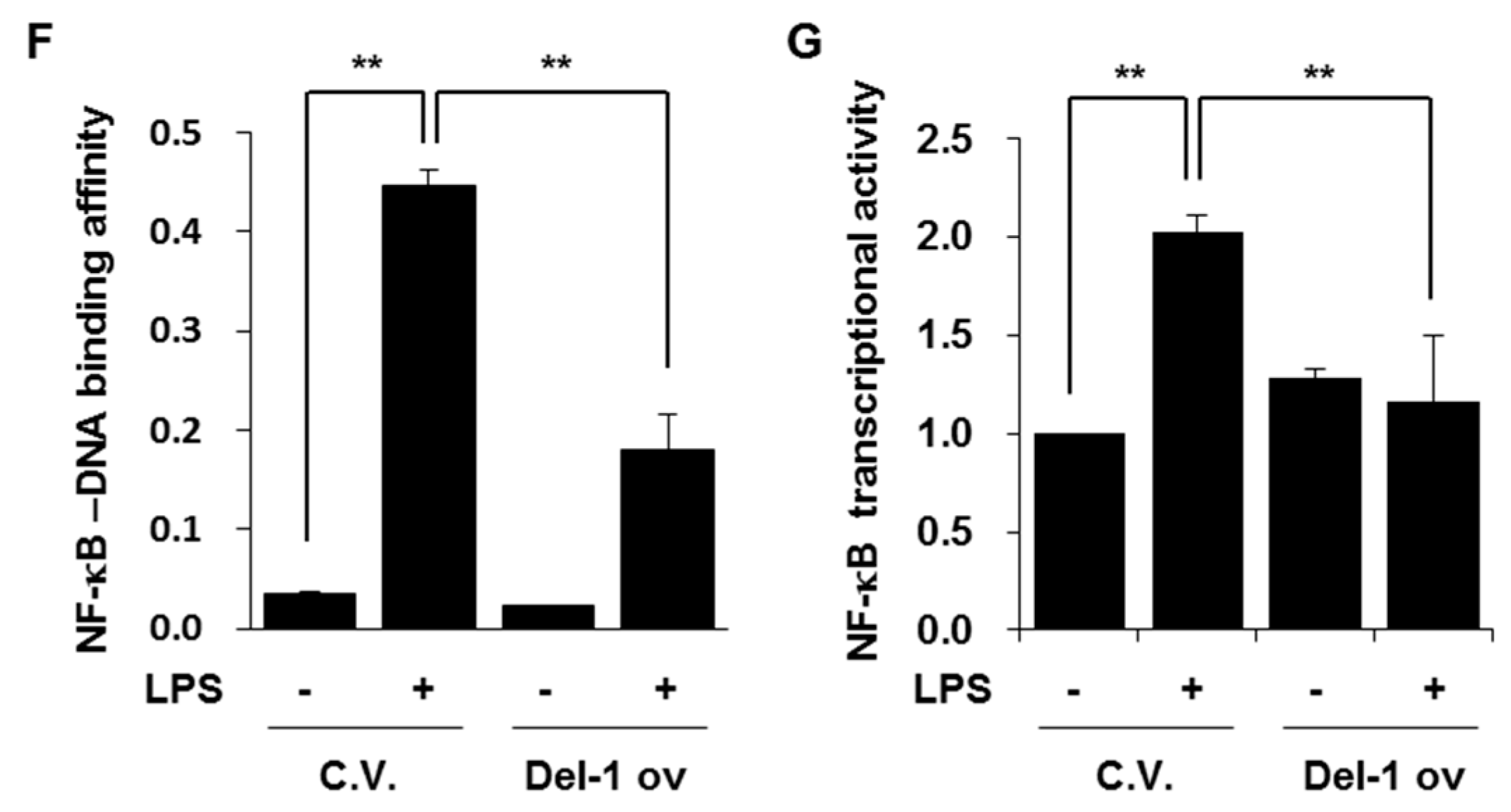
2.5. Del-1 Decreased the Levels of Phospho-p65, NF-kB-DNA Binding Affinity, and NF-κB Transcriptional Activity
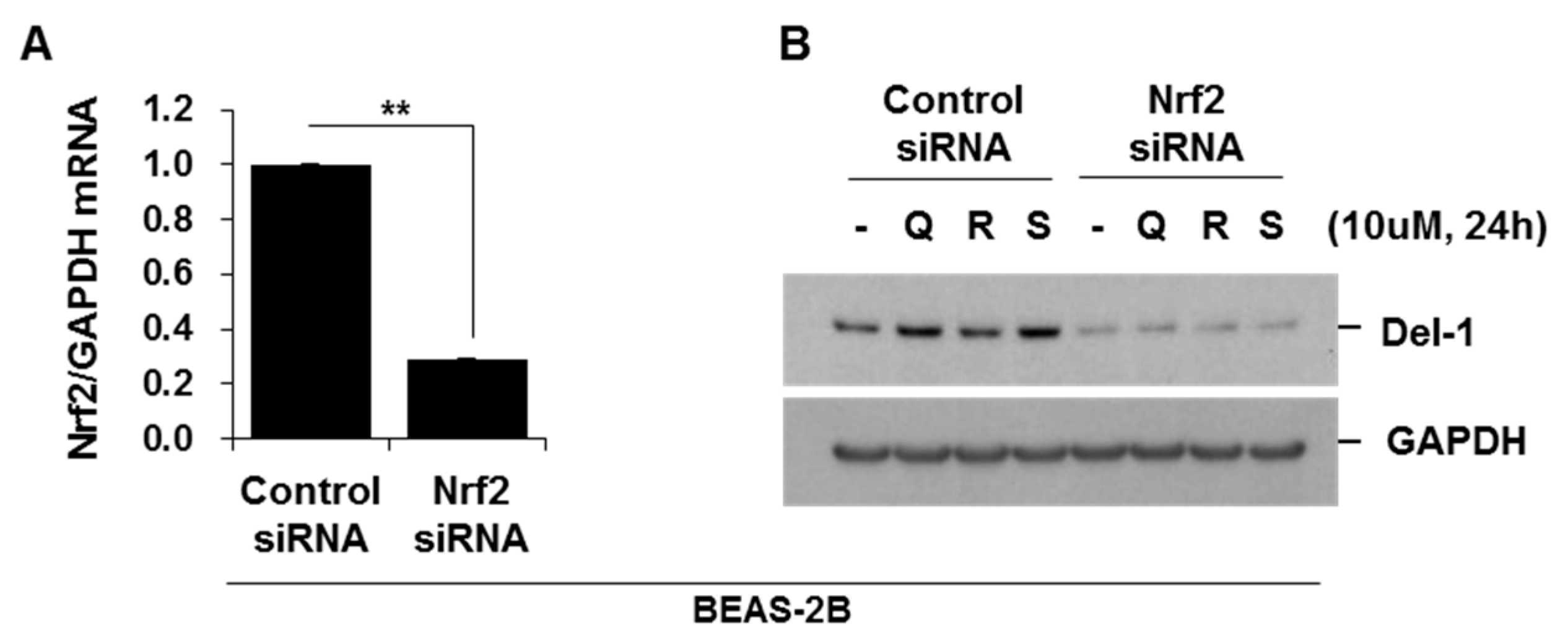
2.6. Activators of Nrf2 Increased Del-1 in Lung Epithelial Cells and Macrophages
3. Discussion
4. Materials and Methods
4.1. Human Lung Tissues and Cells
4.2. Reagents
4.3. Cigarette Smoke Extract (CSE) Preparation
4.4. Emphysema Mouse Model
4.5. Establishing a Stable Cell Line
4.6. Transfection of Plasmid Vectors or siRNA
4.7. Lactate Dehydrogenase (LDH) Release Assay
4.8. Quantitative Real-Time Polymerase Chain Reaction (PCR)
4.9. Protein Extraction and Western Blot Analysis
4.10. Cytokine Secretion Determination
4.11. Immunohistochemistry
4.12. NF-κB p65 Transcription Factor Assay
4.13. NF-κB Luciferase Activity Assay
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lortet-Tieulent, J.; Soerjomataram, I.; López-Campos, J.L.; Ancochea, J.; Coebergh, J.W.; Soriano, J.B. International trends in COPD mortality, 1995–2017. Eur. Respir. J. 2019, 54, 1901791. [Google Scholar] [CrossRef]
- Adeloye, D.; Song, P.; Zhu, Y.; Campbell, H.; Sheikh, A.; Rudan, I. Global, regional, and national prevalence of, and risk factors for, chronic obstructive pulmonary disease (COPD) in 2019: A systematic review and modelling analysis. Lancet Respir. Med. 2022, 10, 447–458. [Google Scholar] [CrossRef]
- Safiri, S.; Carson-Chahhoud, K.; Noori, M.; Nejadghaderi, S.A.; Sullman, M.J.; Heris, J.A.; Ansarin, K.; Mansournia, M.A.; Collins, G.S.; Kolahi, A.-A. Burden of chronic obstructive pulmonary disease and its attributable risk factors in 204 countries and territories, 1990–2019: Results from the Global Burden of Disease Study 2019. BMJ 2022, 378, e069679. [Google Scholar] [CrossRef] [PubMed]
- Agustí, A.; Hogg, J.C. Update on the pathogenesis of chronic obstructive pulmonary disease. N. Engl. J. Med. 2019, 381, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, R.; Breen, D.; Wilson, S.; Djukanovic, R. Inflammatory cells in the airways in COPD. Thorax 2006, 61, 448–454. [Google Scholar] [CrossRef] [PubMed]
- MacNee, W.; Tuder, R.M. New paradigms in the pathogenesis of chronic obstructive pulmonary disease I. Proc. Am. Thorac. Soc. 2009, 6, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Celli, B.R.; Fabbri, L.M.; Aaron, S.D.; Agusti, A.; Brook, R.; Criner, G.J.; Franssen, F.M.; Humbert, M.; Hurst, J.R.; O’Donnell, D. An updated definition and severity classification of chronic obstructive pulmonary disease exacerbations: The Rome proposal. Am. J. Respir. Crit. Care Med. 2021, 204, 1251–1258. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Chavakis, T. DEL-1-regulated immune plasticity and inflammatory disorders. Trends Mol. Med. 2019, 25, 444–459. [Google Scholar] [CrossRef] [PubMed]
- Eskan, M.A.; Jotwani, R.; Abe, T.; Chmelar, J.; Lim, J.-H.; Liang, S.; Ciero, P.A.; Krauss, J.L.; Li, F.; Rauner, M. The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat. Immunol. 2012, 13, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Kourtzelis, I.; Li, X.; Mitroulis, I.; Grosser, D.; Kajikawa, T.; Wang, B.; Grzybek, M.; Von Renesse, J.; Czogalla, A.; Troullinaki, M. DEL-1 promotes macrophage efferocytosis and clearance of inflammation. Nat. Immunol. 2019, 20, 40–49. [Google Scholar] [CrossRef]
- Joo, D.-H.; Lee, K.-H.; Lee, C.-H.; Woo, J.; Kim, J.; Park, S.J.; Rhee, C.K.; Lee, W.-Y.; Park, D.; Lee, J.S. Developmental endothelial locus-1 as a potential biomarker for the incidence of acute exacerbation in patients with chronic obstructive pulmonary disease. Respir. Res. 2021, 22, 1–9. [Google Scholar] [CrossRef]
- Lee, K.H.; Woo, J.; Kim, J.; Lee, C.H.; Yoo, C.G. YPL-001 Shows Various Beneficial Effects against Cigarette Smoke Extract-Induced Emphysema Formation: Anti-Inflammatory, Anti-Oxidative, and Anti-Apoptotic Effects. Antioxidants 2022, 12, 15. [Google Scholar] [CrossRef]
- Park, J.W.; Ryter, S.W.; Choi, A.M. Functional significance of apoptosis in chronic obstructive pulmonary disease. COPD 2007, 4, 347–353. [Google Scholar] [CrossRef]
- Duan, T.; Du, Y.; Xing, C.; Wang, H.Y.; Wang, R.F. Toll-Like Receptor Signaling and Its Role in Cell-Mediated Immunity. Front. Immunol. 2022, 13, 812774. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig. 2004, 114, 1248–1259. [Google Scholar] [CrossRef]
- Boutten, A.; Goven, D.; Artaud-Macari, E.; Boczkowski, J.; Bonay, M. NRF2 targeting: A promising therapeutic strategy in chronic obstructive pulmonary disease. Trends Mol. Med. 2011, 17, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Petrache, I. Pathogenesis of chronic obstructive pulmonary disease. J. Clin. Investig. 2012, 122, 2749–2755. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.; Peinado, V.I.; Ramirez, J.; Morales-Blanhir, J.; Bastos, R.; Roca, J.; Rodriguez-Roisin, R.; Barbera, J.A. Enhanced expression of vascular endothelial growth factor in pulmonary arteries of smokers and patients with moderate chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2003, 167, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, H. Role of vascular endothelial growth factor in the pathogenesis of chronic obstructive pulmonary disease. Med. Sci. Monit. 2007, 13, RA189–RA195. [Google Scholar] [PubMed]
- Yamamoto, C.; Yoneda, T.; Yoshikawa, M.; Fu, A.; Tokuyama, T.; Tsukaguchi, K.; Narita, N. Airway inflammation in COPD assessed by sputum levels of interleukin-8. Chest 1997, 112, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, M.; Di Vincenzo, S.; Sangiorgi, C.; Leto Barone, S.; Gangemi, S.; Lanata, L.; Pace, E. Carbocysteine Modifies Circulating miR-21, IL-8, sRAGE, and fAGEs Levels in Mild Acute Exacerbated COPD Patients: A Pilot Study. Pharmaceuticals 2022, 11, 218. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zeng, J.; Ren, T. Expression of DEL-1 in alveolar epithelial cells prevents lipopolysaccharide-induced inflammation, oxidative stress, and eosinophil recruitment in acute lung injury. Int. Immunopharmacol. 2022, 110, 108961. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kundu, R.K.; Longaker, M.T.; Quertermous, T.; Yang, G.P. The angiogenic factor Del1 prevents apoptosis of endothelial cells through integrin binding. Surgery 2012, 151, 296–305. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, D.Y.; Kang, Y.Y.; Kim, H.; Jang, J.; Lee, M.N.; Oh, G.T.; Kang, S.W.; Choi, E.Y. Developmental endothelial locus-1 inhibits MIF production through suppression of NF-κB in macrophages. Int. J. Mol. Med. 2014, 33, 919–924. [Google Scholar] [CrossRef]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Niture, S.K.; Jaiswal, A.K. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J. Biol. Chem. 2012, 287, 9873–9886. [Google Scholar] [CrossRef]
- Jeong, Y.J.; Lee, K.-H.; Woo, J.; Kim, J.Y.; Lee, C.-H.; Yoo, C.G. Downregulation of Lysosome-Associated Membrane Protein-2A Contributes to the Pathogenesis of COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 2023, 18, 289–303. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwak, N.; Lee, K.-H.; Woo, J.; Kim, J.; Park, J.; Lee, C.-H.; Yoo, C.-G. Del-1 Plays a Protective Role against COPD Development by Inhibiting Inflammation and Apoptosis. Int. J. Mol. Sci. 2024, 25, 1955. https://doi.org/10.3390/ijms25041955
Kwak N, Lee K-H, Woo J, Kim J, Park J, Lee C-H, Yoo C-G. Del-1 Plays a Protective Role against COPD Development by Inhibiting Inflammation and Apoptosis. International Journal of Molecular Sciences. 2024; 25(4):1955. https://doi.org/10.3390/ijms25041955
Chicago/Turabian StyleKwak, Nakwon, Kyoung-Hee Lee, Jisu Woo, Jiyeon Kim, Jimyung Park, Chang-Hoon Lee, and Chul-Gyu Yoo. 2024. "Del-1 Plays a Protective Role against COPD Development by Inhibiting Inflammation and Apoptosis" International Journal of Molecular Sciences 25, no. 4: 1955. https://doi.org/10.3390/ijms25041955