
Identification of New EGFR Inhibitors by Structure-Based Virtual Screening and Biological Evaluation

Abstract
:1. Introduction
2. Results and Discussions
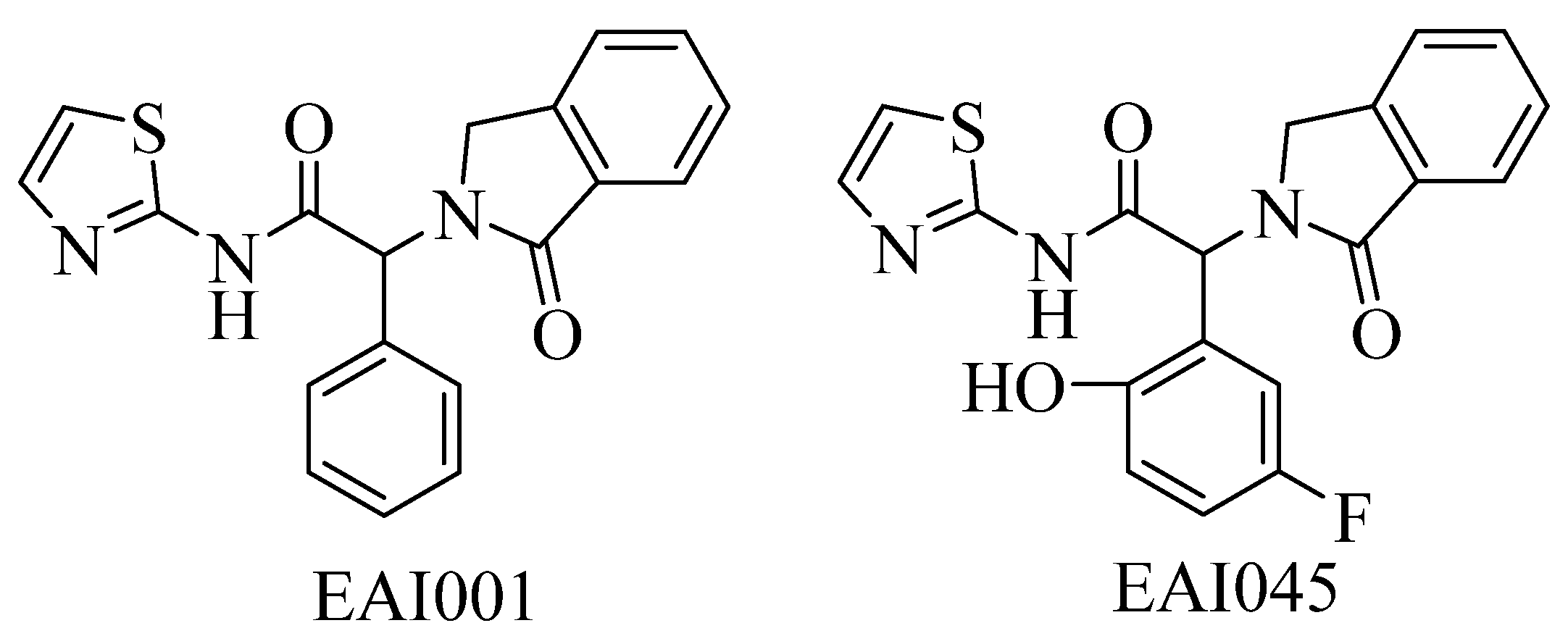
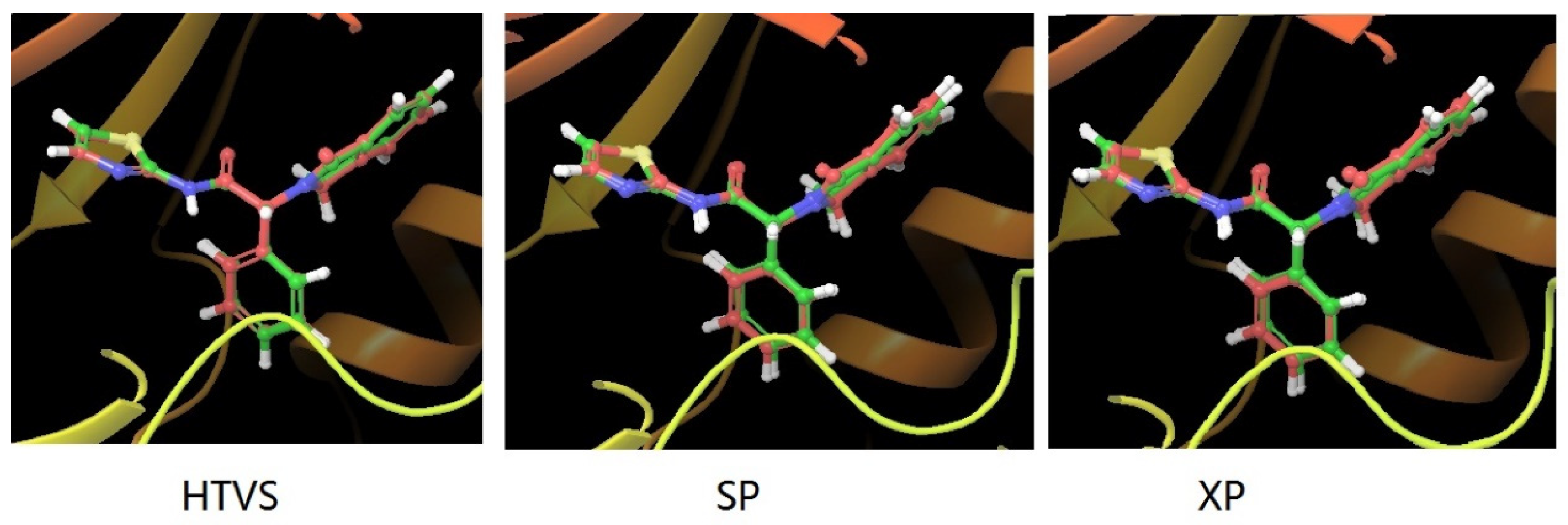
2.1. Molecular Docking Protocol Assessment
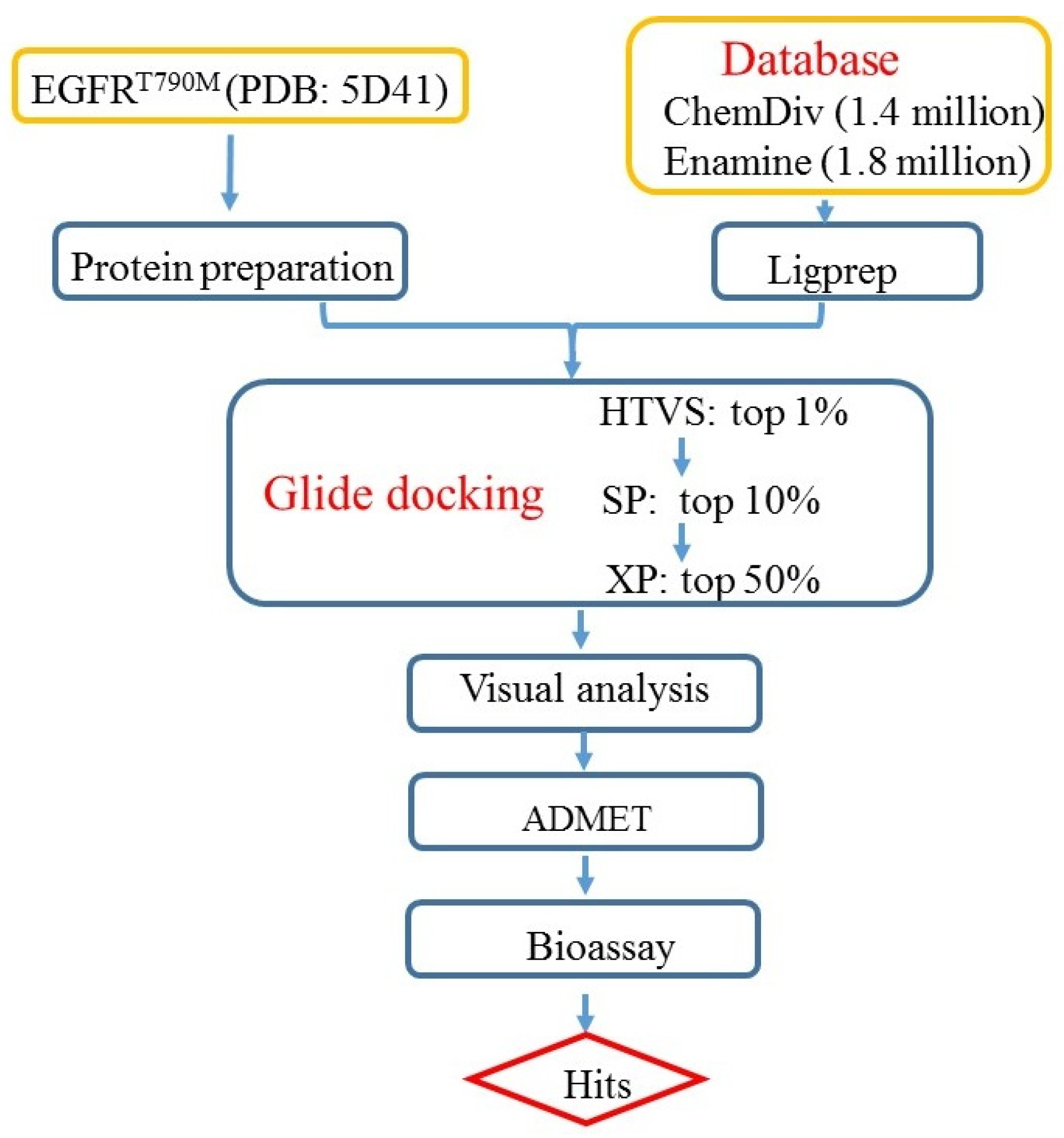
2.2. Structure-Based Virtual Screening
2.3. ADME and Toxicity Prediction
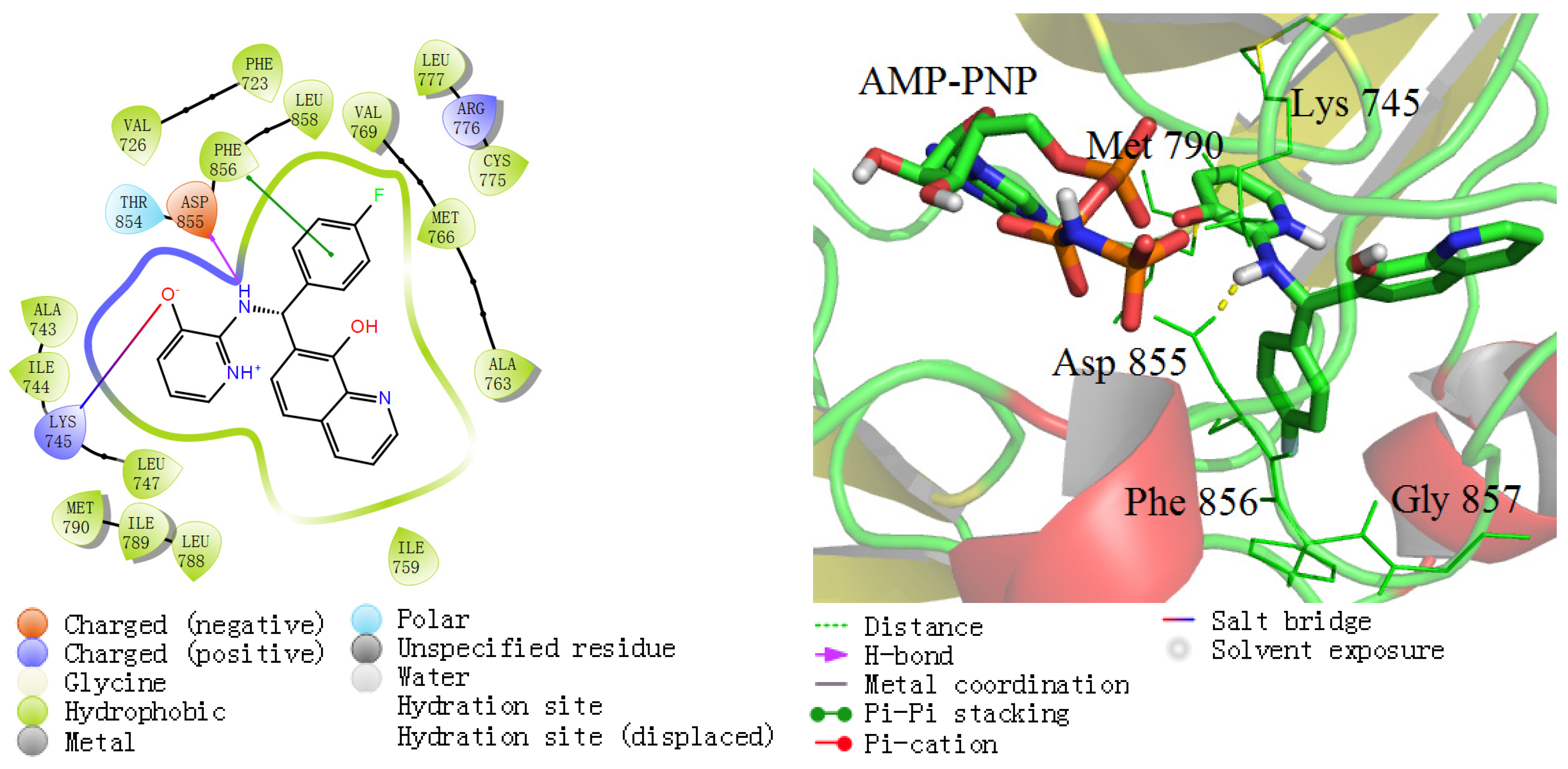
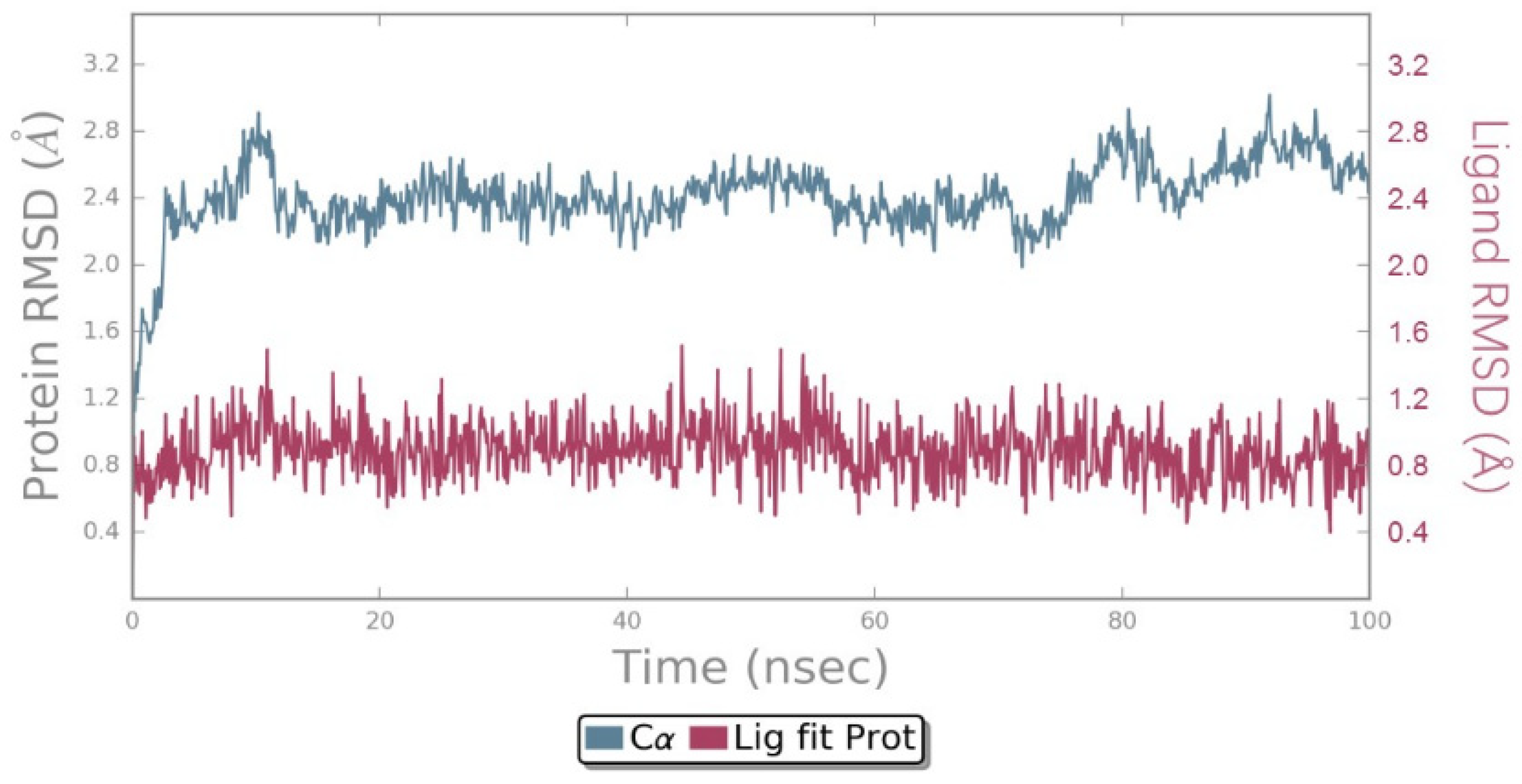
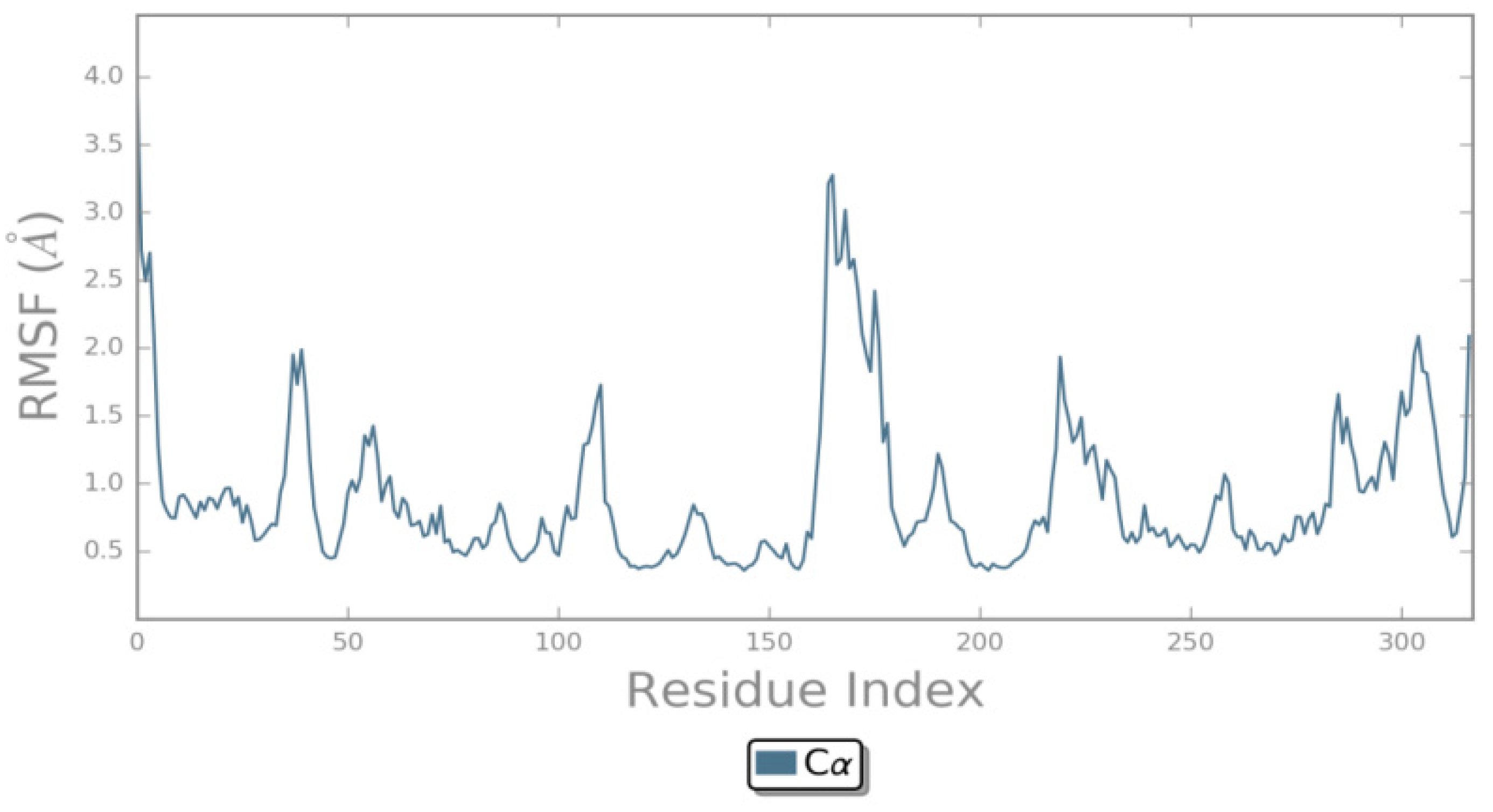
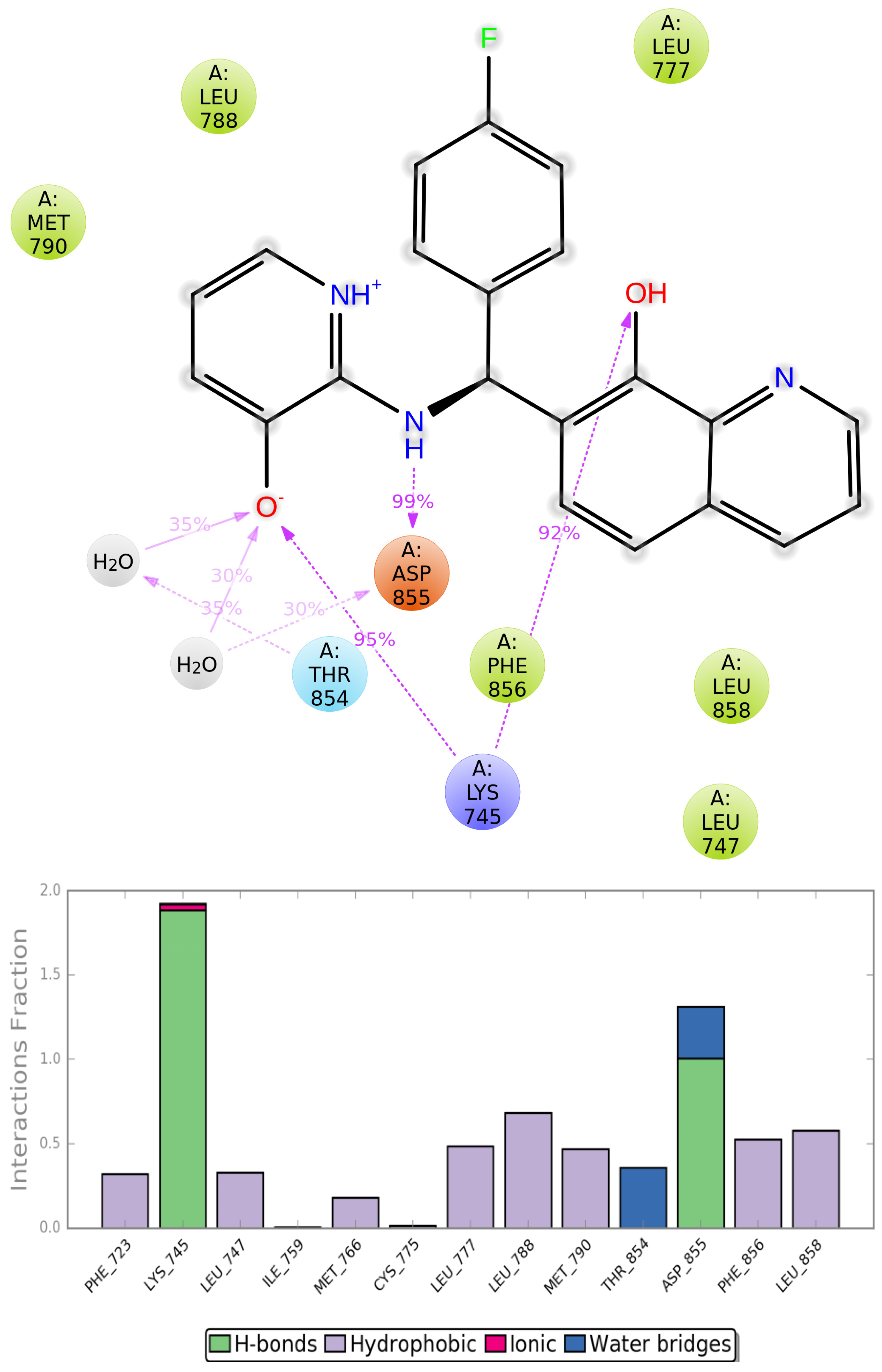
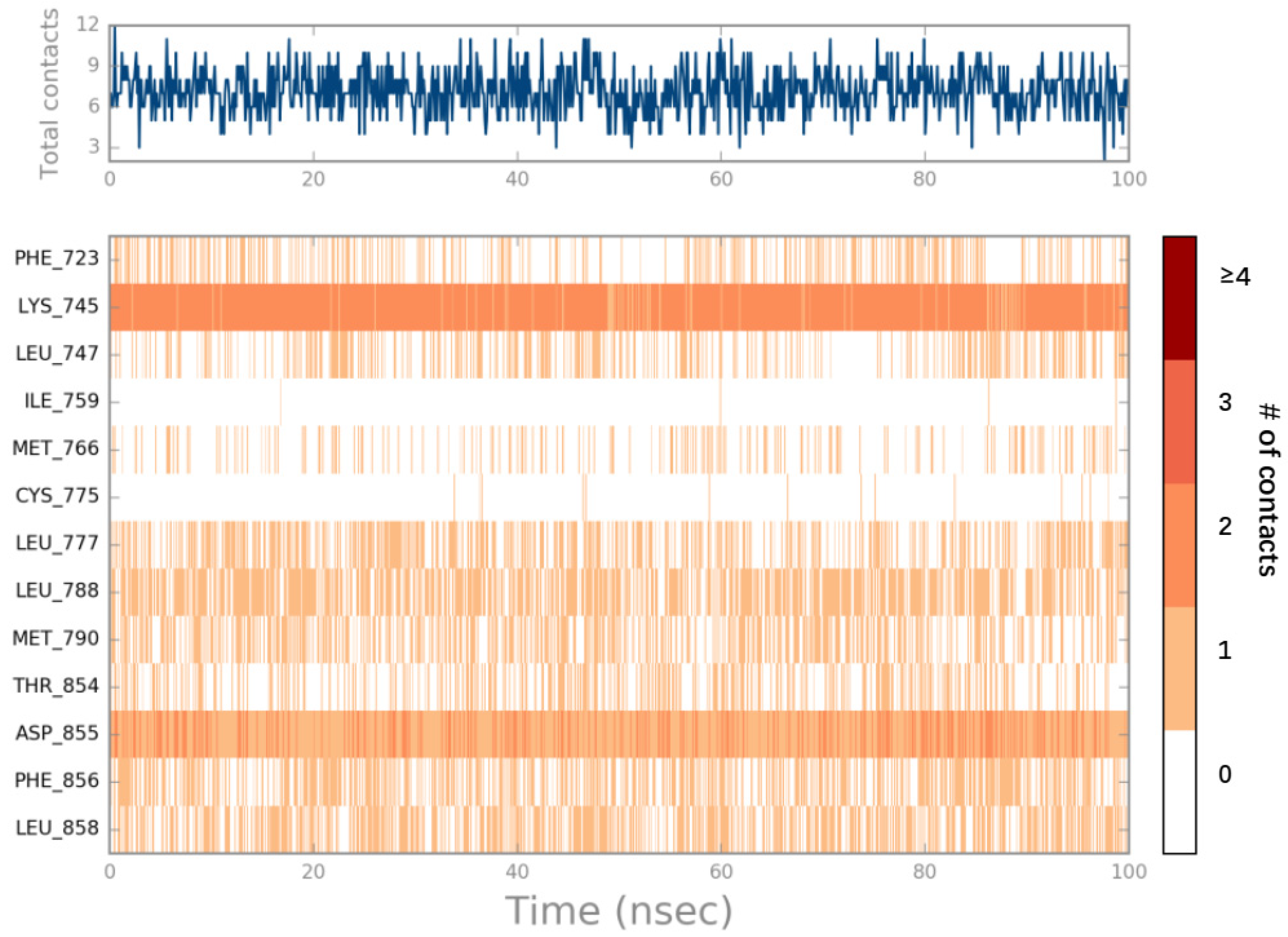
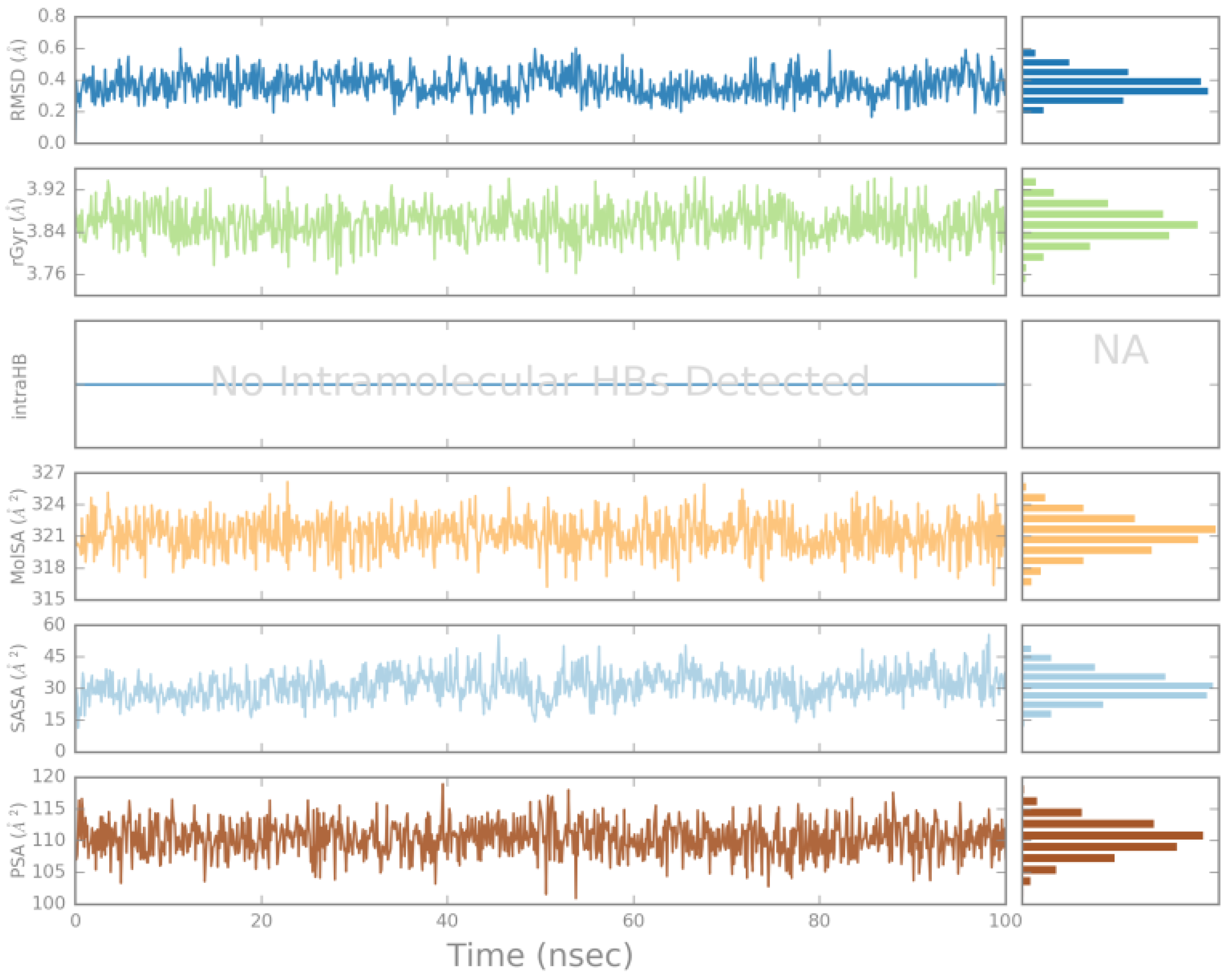
2.4. MD Simulations Analysis
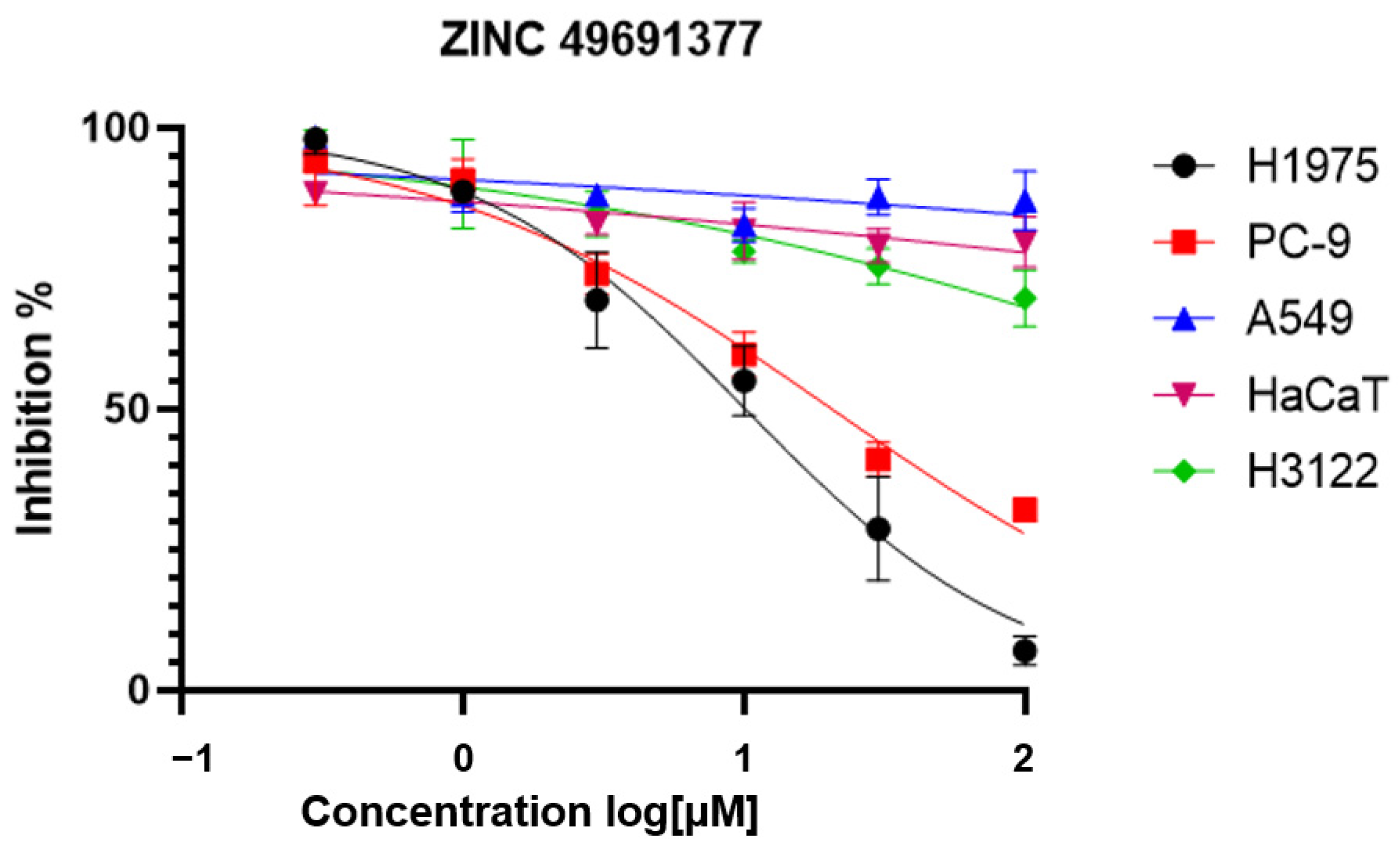
2.5. The Anti-Proliferation Activity of the Selected Compounds
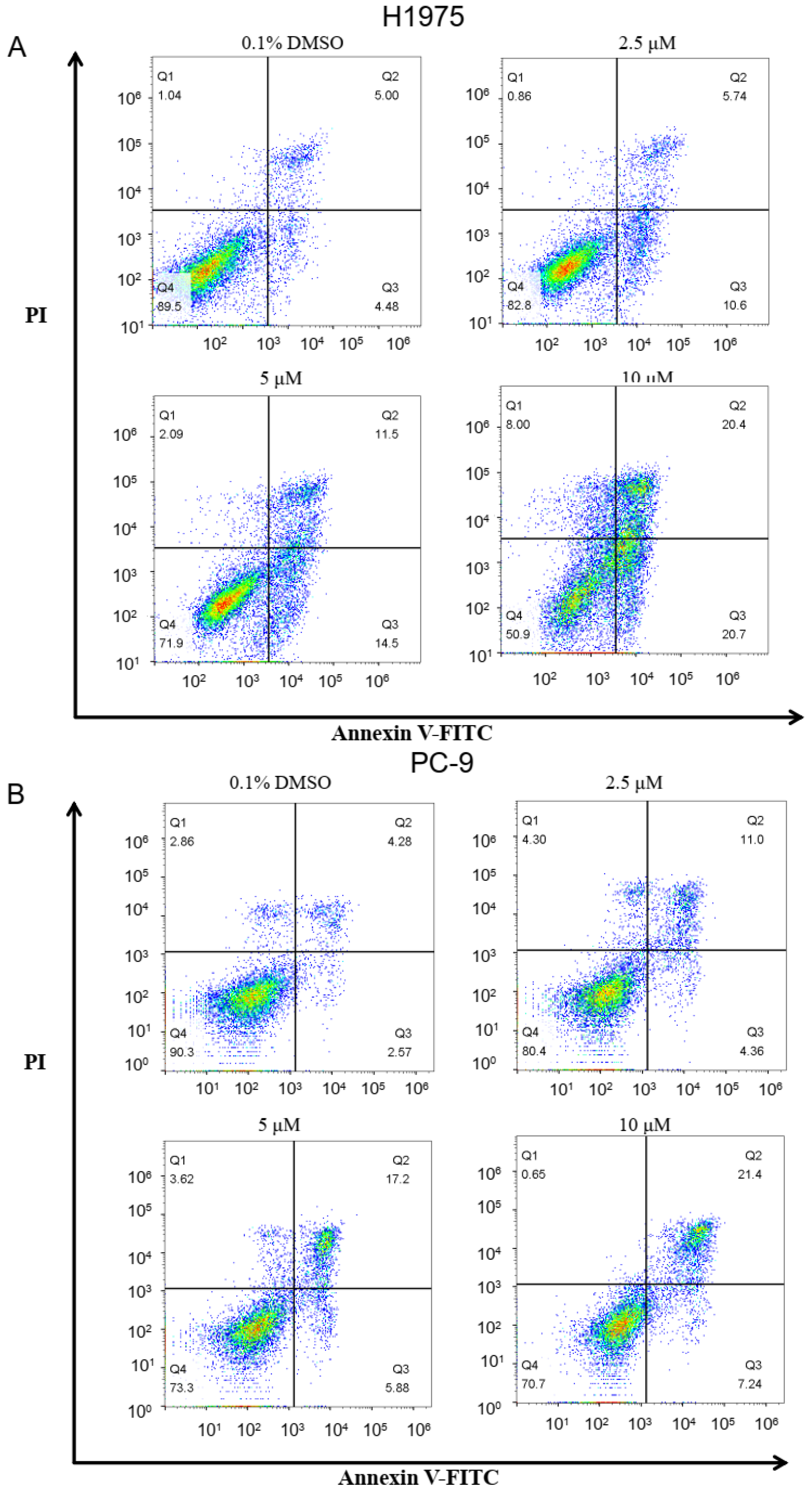
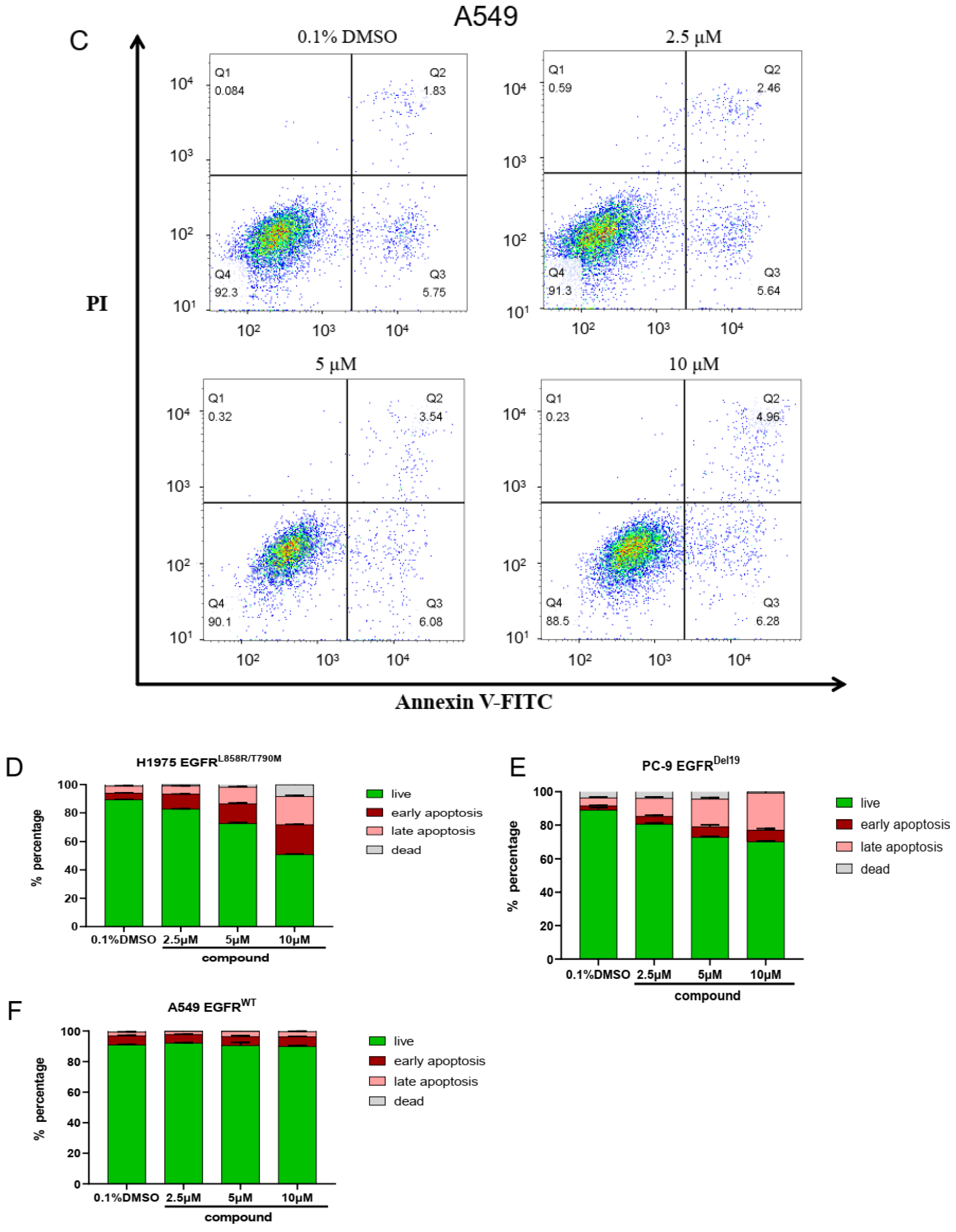
2.6. The Cell Apoptosis of Compound ZINC49691377
3. Materials and Methods
3.1. Protein Preparation
3.2. Preparation of the Databases
3.3. Docking-Based Virtual Screening
3.4. Prediction of ADMET Properties
3.5. Molecular Dynamics Simulations
3.6. Cancer Cell Proliferation Inhibition Assay
3.7. Cell Apoptosis Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shepherd, F.A.; Rodrigues, P.J.; Ciuleanu, T.; Tan, E.H.; Hirsh, V.; Thongprasert, S.; Campos, D.; Maoleekoonpiroj, S.; Smylie, M.; Martins, R.; et al. Erlotinib in previously treated non-small-cell lung cancer. N. Engl. J. Med. 2005, 353, 123–132. [Google Scholar] [CrossRef]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef]
- Yusuf, S.W.; Kim, P.; Durand, J.B. Erlotinib or gefitinib for non-small-cell lung cancer. N. Engl. J. Med. 2011, 364, 2367–2368. [Google Scholar] [PubMed]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Pao, W.; Sequist, L.V. Acquired resistance to TKIs in solid tumours: Learning from lung cancer. Nat. Rev. Clin. Oncol. 2014, 11, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non–small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, Y.; Yan, F.; Liu, D. Mechanisms of resistance to third-generation EGFR tyrosine kinase inhibitors. Front. Med.-Prc. 2016, 10, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Minari, R.; Bordi, P.; Tiseo, M. Third-generation epidermal growth factor receptor-tyrosine kinase inhibitors in T790M-positive non-small cell lung cancer: Review on emerged mechanisms of resistance. Transl. Lung Cancer Res. 2016, 5, 695–708. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. Small-molecule kinase inhibitors: An analysis of FDA-approved drugs. Drug Discov. Today 2016, 21, 5–10. [Google Scholar] [CrossRef]
- Jia, Y.; Yun, C.H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef]
- Meng, X.; Zhang, H.; Mezei, M.; Cui, M. Molecular Docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Wan, S.; Yan, R.; Jiang, Y.; Li, Z.; Zhang, J.; Wu, X. Insight into binding mechanisms of EGFR allosteric inhibitors using molecular dynamics simulations and free energy calculations. J. Biomol. Struct. Dyn. 2019, 37, 4384–4394. [Google Scholar] [CrossRef]
- Hevener, K.E.; Zhao, W.; Ball, D.M.; Babaoglu, K.; Qi, J.; White, S.W.; Lee, R.E. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J. Chem. Inf. Model. 2009, 49, 444–460. [Google Scholar] [CrossRef]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Guo, Q.; Li, Q.; Wan, S.; Li, Z.; Zhang, J. Molecular mechanism study of EGFR allosteric inhibitors using molecular dynamics simulations and free energy calculations. J. Biomol. Struct. Dyn. 2022, 40, 5848–5857. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Artursson, P.; Palm, K.; Luthman, K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv. Drug Deliv. Rev. 2001, 46, 27–43. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties that Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, J.I.; Walker, B.D.; Campbell, T.J. HERG K+ channels: Friend and foe. Trends Pharmacol. Sci. 2001, 22, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Aronov, A.M. Predictive in silico modeling for hERG channel blockers. Drug Discov. Today 2005, 10, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Shaw, D.E. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. IEEE Sci. Conf. 2006, 2006, 43. [Google Scholar]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Friesner, R.A. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2015, 12, 281–296. [Google Scholar] [CrossRef]
- Jorgensen, W.; Chandrasekhar, J.; Madura, J. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ikeguchi, M. Partial rigid-body dynamics in NPT, NPAT and NPT ensembles for proteins and membranes. J. Comput. Chem. 2004, 25, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Stuart, S.J.; Zhou, R.; Berne, B.J. Molecular dynamics with multiple time scales: The selection of efficient reference system propagators. J. Chem. Phys. 1996, 105, 1426–1436. [Google Scholar] [CrossRef]
- Li, Q.; Guo, Q.; Wang, S.; Wan, S.; Li, Z.; Zhang, J.; Wu, X. Design and synthesis of proteolysis targeting chimeras (PROTACs) as an EGFR degrader based on CO-1686. Eur. J. Med. Chem. 2022, 238, 114455. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HTVS | SP | XP | |
|---|---|---|---|
| G-score (Kcal/mol) | −11.472 | −11.532 | −11.005 |
| RMSD (Å) | 0.048 | 0.178 | 0.163 |
| G-Score (kcal/mol) | MW a | logPo/w b | logS c | PSA d | PCaco e | PMDCK f | logBB g | logHERG h | Violation of Ro5 i | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| EAI045 | −11.96 | 383.38 | 2.93 | −4.67 | 86.8 | 877.17 | 1538.77 | −0.60 | −6.32 | 0 | |
| 1 | ZINC49691377 | −14.03 | 361.38 | 4.14 | −5.06 | 71.00 | 1072.92 | 966.13 | −0.67 | −6.56 | 0 |
| 2 | ZINC09616958 | −13.03 | 356.42 | 4.60 | −5.38 | 69.31 | 907.73 | 445.56 | −0.93 | −6.68 | 0 |
| 3 | ZINC09775243 | −12.33 | 361.83 | 4.98 | −5.49 | 48.91 | 2785.90 | 3693.51 | −0.11 | −6.37 | 0 |
| 4 | ZINC10910059 | −12.19 | 355.40 | 3.70 | −4.70 | 74.03 | 1303.15 | 658.64 | −0.63 | −6.73 | 0 |
| 5 | ZINC89827617 | −12.07 | 329.79 | 3.47 | −4.33 | 91.10 | 259.21 | 543.26 | −0.80 | −4.02 | 0 |
| 6 | ZINC22017635 | −11.99 | 342.40 | 4.95 | −5.86 | 53.87 | 2283.11 | 1946.38 | −0.18 | −6.47 | 0 |
| 7 | ZINC53674458 | −11.93 | 328.37 | 4.37 | −5.43 | 55.48 | 2342.50 | 1241.45 | −0.35 | −7.23 | 0 |
| 8 | ZINC69419433 | −11.89 | 348.40 | 3.66 | −5.49 | 96.22 | 758.32 | 366.85 | −0.97 | −6.61 | 0 |
| 9 | ZINC15778674 | −11.53 | 363.35 | 4.10 | −5.50 | 82.31 | 1408.00 | 1056.08 | −0.52 | −7.12 | 0 |
| 10 | ZINC29507326 | −11.52 | 365.38 | 3.72 | −5.02 | 66.11 | 1657.03 | 1656.87 | −0.20 | −6.36 | 0 |
| 11 | ZINC05577262 | −11.52 | 363.35 | 3.80 | −5.18 | 78.59 | 1483.36 | 1371.80 | −0.35 | −6.52 | 0 |
| 12 | ZINC03876430 | −11.42 | 341.41 | 4.58 | −6.18 | 58.56 | 2121.51 | 1115.36 | −0.27 | −6.47 | 0 |
| 13 | ZINC18205922 | −11.41 | 347.80 | 4.92 | −5.84 | 47.30 | 3375.47 | 4550.21 | 0.01 | −6.88 | 0 |
| 14 | ZINC00036286 | −11.36 | 343.42 | 4.85 | −5.17 | 48.57 | 3155.77 | 1713.26 | −0.22 | −6.19 | 0 |
| 15 | ZINC20531081 | −11.31 | 342.40 | 4.21 | −5.18 | 61.33 | 2484.77 | 1323.15 | −0.30 | −7.00 | 0 |
| 16 | ZINC89756684 | −11.30 | 331.38 | 4.22 | −5.47 | 65.15 | 2241.61 | 1183.76 | −0.37 | −6.89 | 0 |
| 17 | ZINC01201194 | −11.28 | 382.25 | 4.87 | −6.31 | 57.21 | 2291.39 | 6343.36 | 0.11 | −6.39 | 0 |
| 18 | ZINC01108543 | −11.27 | 368.43 | 4.49 | −5.03 | 56.21 | 1887.49 | 982.99 | −0.27 | −5.35 | 0 |
| 19 | ZINC13351329 | −11.08 | 355.44 | 4.77 | −5.87 | 54.80 | 1767.73 | 1368.58 | −0.17 | −5.27 | 0 |
| 20 | ZINC04918676 | −11.06 | 356.42 | 3.55 | −4.44 | 70.31 | 1237.96 | 623.09 | −0.45 | −5.44 | 0 |
| 21 | ZINC04784223 | −11.06 | 371.44 | 4.75 | −5.43 | 70.92 | 1544.54 | 791.45 | −0.65 | −6.62 | 0 |
| 22 | ZINC43232082 | −10.99 | 374.36 | 4.35 | −5.30 | 62.00 | 2051.67 | 5859.35 | −0.05 | −4.64 | 0 |
| 23 | ZINC00178936 | −10.70 | 345.42 | 4.33 | −5.97 | 56.46 | 2085.50 | 2130.51 | −0.23 | −6.85 | 0 |
| Title | Antiproliferative Activity (IC50, μM) a | |||
|---|---|---|---|---|
| A549 | PC-9 | H1975 | ||
| EAI045 | >100 | >100 | 23.64 ± 4.78 | |
| 1 | ZINC49691377 | >100 | 20.48 ± 0.03 | 10.02 ± 0.02 |
| 2 | ZINC09616958 | 32.32 ± 1.57 | 82.34 ± 2.20 | 40.36 ± 0.95 |
| 3 | ZINC09775243 | 96.17 ± 2.04 | 82.68 ± 2.47 | 61.10 ± 6.04 |
| 4 | ZINC10910059 | 19.03 ± 0.76 | 13.98 ± 0.18 | 15.06 ± 0.50 |
| 5 | ZINC89827617 | >100 | >100 | >100 |
| 6 | ZINC22017635 | 39.33 ± 3.95 | 18.95 ± 3.60 | 13.60 ± 1.46 |
| 7 | ZINC53674458 | >100 | 90.13 ± 1.75 | 78.74 ± 8.80 |
| 8 | ZINC69419433 | >100 | >100 | 14.21 ± 4.57 |
| 9 | ZINC15778674 | >100 | 67.20 ± 2.20 | 40.13 ± 4.50 |
| 10 | ZINC29507326 | >100 | 85.25 ± 19.00 | 43.91 ± 15.16 |
| 11 | ZINC05577262 | >100 | >100 | 74.26 ± 4.82 |
| 12 | ZINC03876430 | 34.53 ± 0.74 | 38.05 ± 7.36 | 9.69 ± 0.91 |
| 13 | ZINC18205922 | 12.52 ± 0.58 | 33.98 ± 4.50 | 20.24 ± 0.43 |
| 14 | ZINC00036286 | 87.23 ± 0.68 | 70.19 ± 3.65 | 75.78 ± 4.24 |
| 15 | ZINC20531081 | >100 | 93.65 ± 6.67 | 62.98 ± 7.65 |
| 16 | ZINC89756684 | 73.01 ± 10.27 | 73.06 ± 7.68 | 43.12 ± 1.34 |
| 17 | ZINC01201194 | 8.20 ± 1.18 | 2.30 ± 0.63 | 4.79 ± 1.11 |
| 18 | ZINC01108543 | 37.23 ± 3.20 | 30.95 ± 0.55 | 38.13 ± 1.22 |
| 19 | ZINC13351329 | 10.80 ± 0.94 | 6.00 ± 0.76 | 16.52 ± 2.55 |
| 20 | ZINC04918676 | >100 | >100 | >100 |
| 21 | ZINC04784223 | >100 | 46.42 ± 1.97 | >100 |
| 22 | ZINC43232082 | 28.04 ± 2.02 | 30.69 ± 0.14 | 23.27 ± 1.62 |
| 23 | ZINC00178936 | >100 | >100 | >100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Xu, X.; Pan, C.; Guo, Q.; Li, Q.; Wan, S.; Li, Z.; Zhang, J.; Wu, X. Identification of New EGFR Inhibitors by Structure-Based Virtual Screening and Biological Evaluation. Int. J. Mol. Sci. 2024, 25, 1887. https://doi.org/10.3390/ijms25031887
Wang S, Xu X, Pan C, Guo Q, Li Q, Wan S, Li Z, Zhang J, Wu X. Identification of New EGFR Inhibitors by Structure-Based Virtual Screening and Biological Evaluation. International Journal of Molecular Sciences. 2024; 25(3):1887. https://doi.org/10.3390/ijms25031887
Chicago/Turabian StyleWang, Shuyi, Xiaotian Xu, Chuxin Pan, Qian Guo, Qinlan Li, Shanhe Wan, Zhonghuang Li, Jiajie Zhang, and Xiaoyun Wu. 2024. "Identification of New EGFR Inhibitors by Structure-Based Virtual Screening and Biological Evaluation" International Journal of Molecular Sciences 25, no. 3: 1887. https://doi.org/10.3390/ijms25031887