

Mechanistic Insights about Sorafenib-, Valproic Acid- and Metformin-Induced Cell Death in Hepatocellular Carcinoma
 , and
, and
Abstract
:1. Introduction
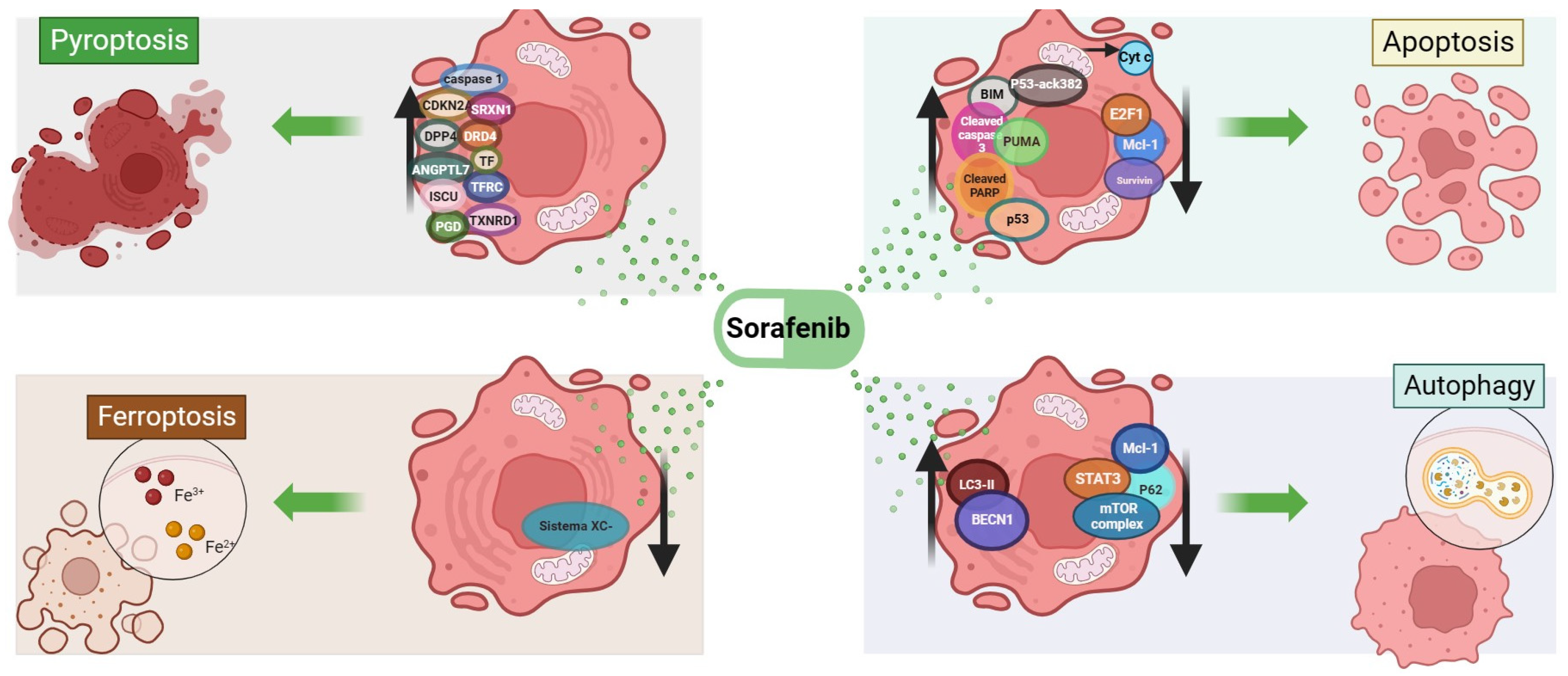
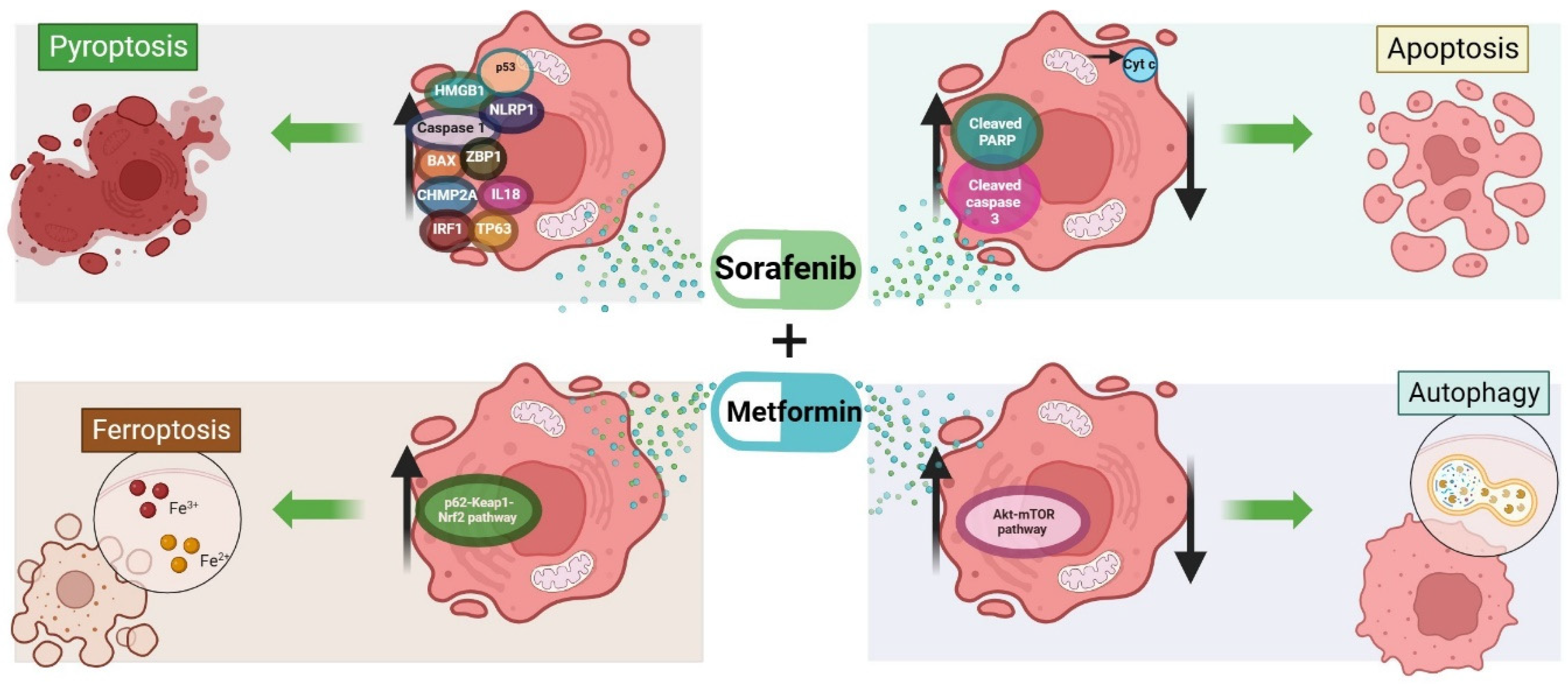
2. Cell Death Mechanisms Related to Sorafenib
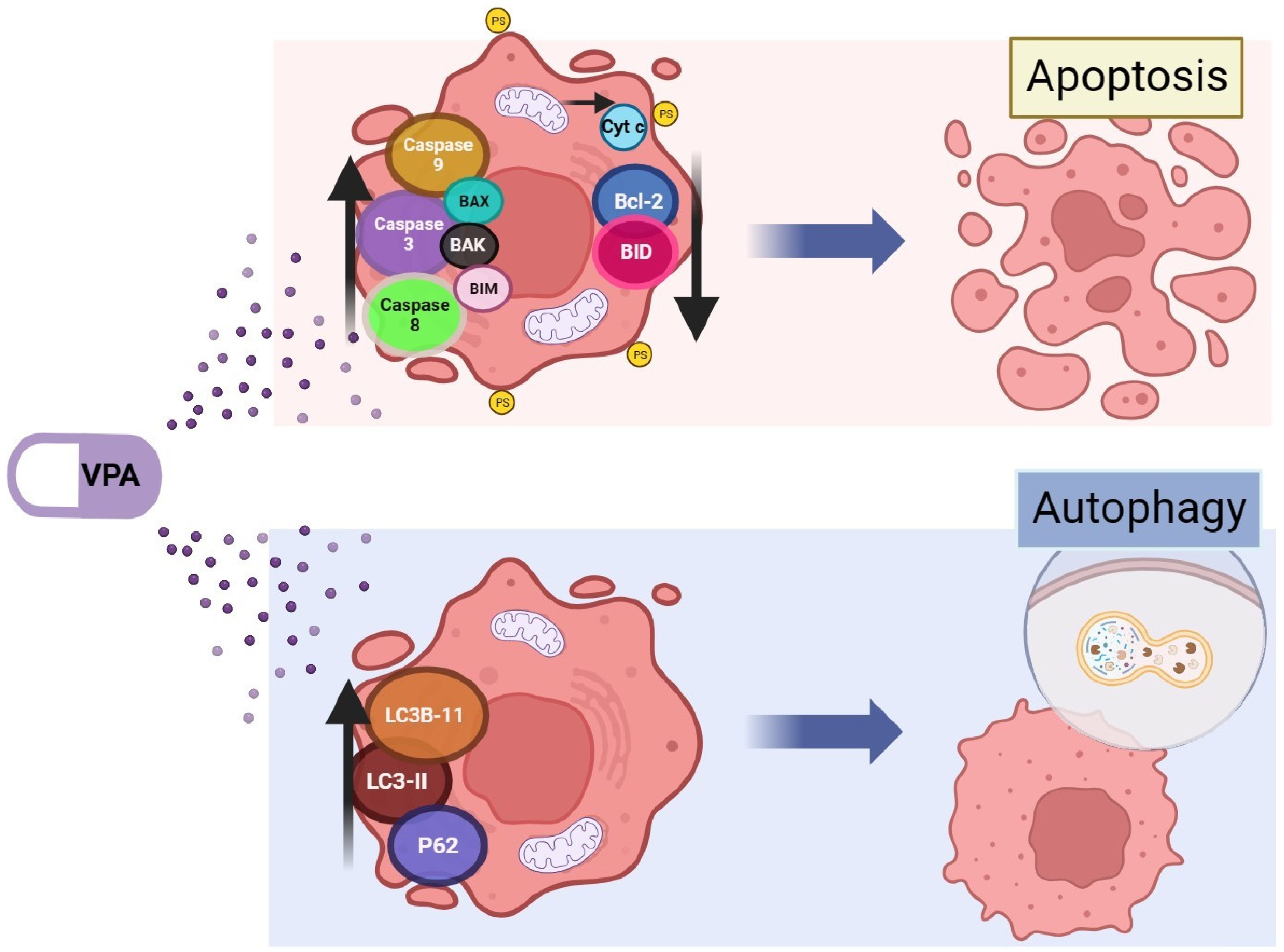
3. Cell Death Mechanisms Related to Valproic Acid
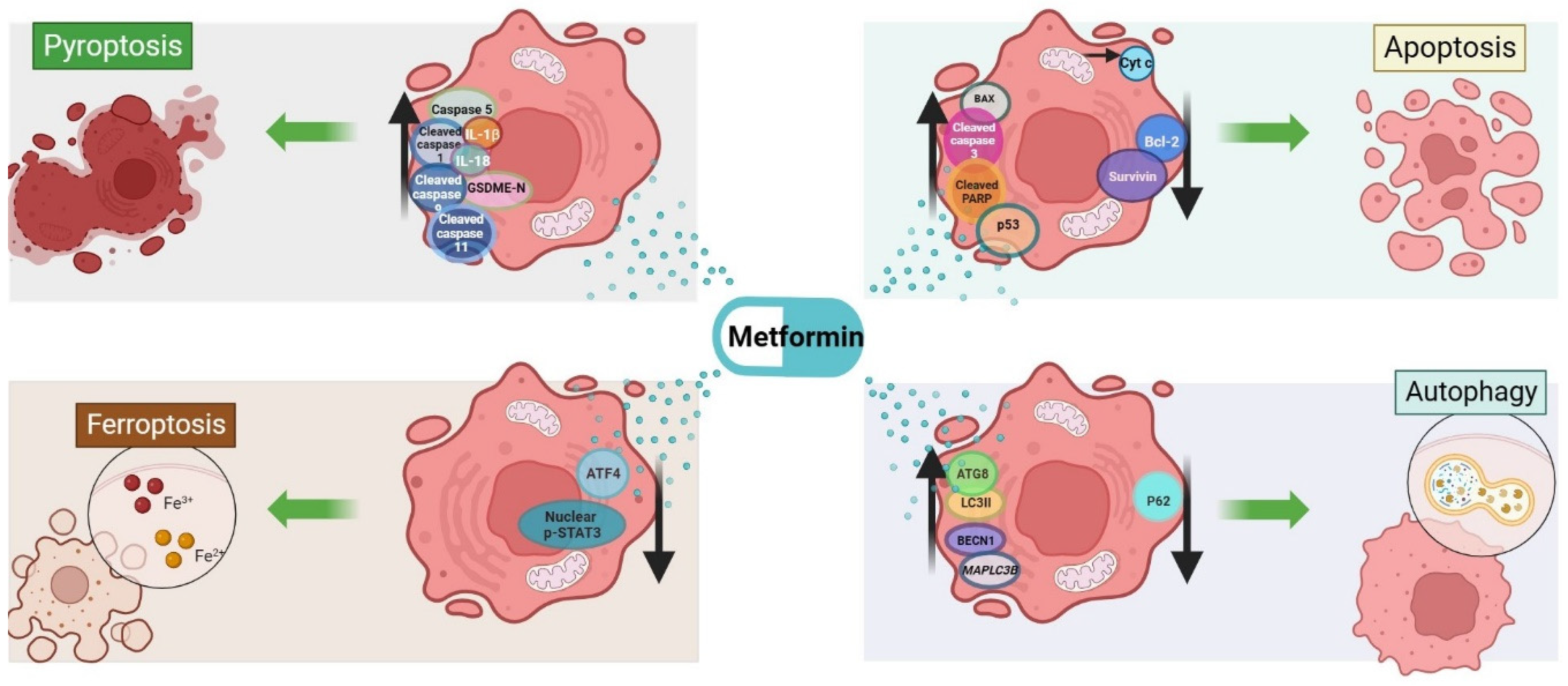
4. Cell Death Mechanisms Related to Metformin
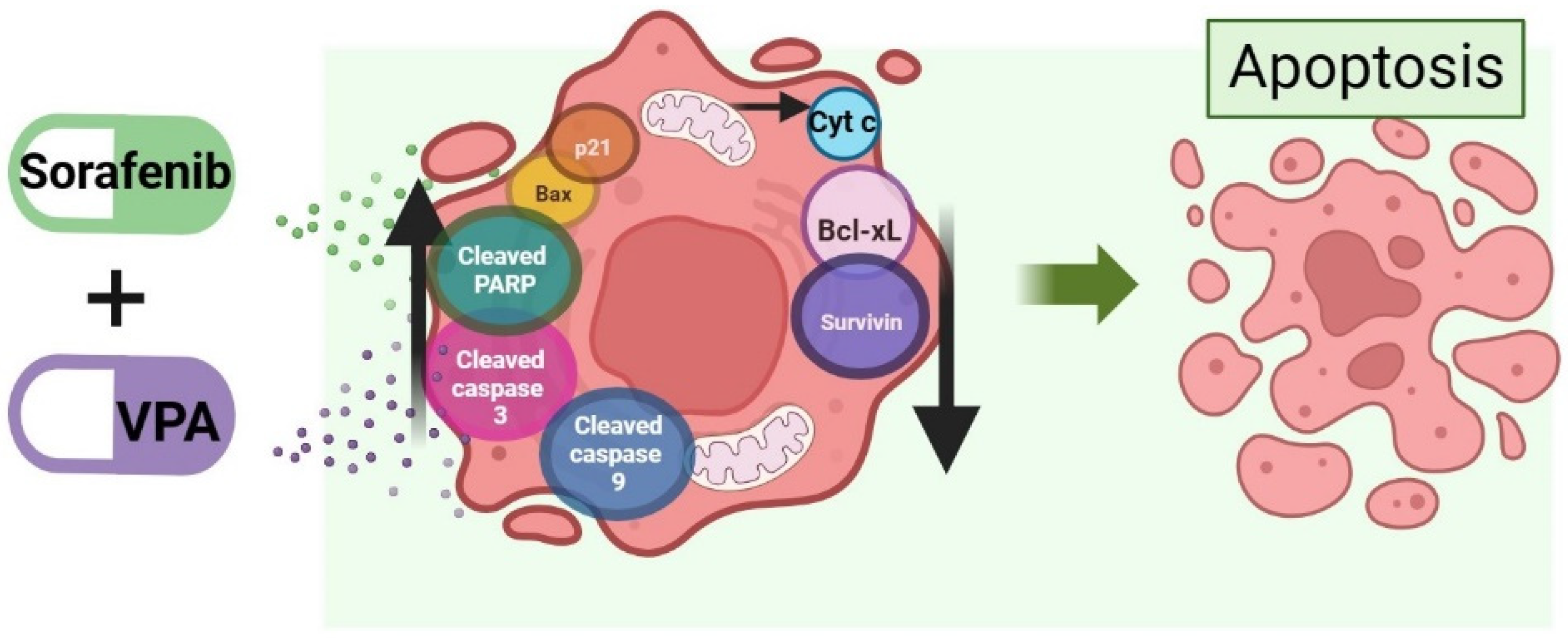
5. Cell Death Mechanism Activated by the Combination of Valproic Acid and Sorafenib in HCC
6. Cell Death Mechanism Activated by the Combination of Metformin and Sorafenib in HCC
7. Future Directions and Challenges
8. Limitations
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Akbulut, S.; Garzali, I.U.; Hargura, A.S.; Aloun, A.; Yilmaz, S. Screening, Surveillance, and Management of Hepatocellular Carcinoma during the COVID-19 Pandemic: A Narrative Review. J. Gastrointest. Cancer 2023, 54, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Viatour, P. Hepatocellular carcinoma: Old friends and new tricks. Exp. Mol. Med. 2020, 52, 1898–1907. Available online: https://pubmed.ncbi.nlm.nih.gov/33268834 (accessed on 23 September 2023). [CrossRef] [PubMed]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [PubMed]
- Petrick, J.L.; McGlynn, K.A. The changing epidemiology of primary liver cancer. Curr. Epidemiol. Rep. 2019, 6, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-W.; Chen, M.; Colombo, M.; Roberts, L.R.; Schwartz, M.; Chen, P.-J.; Kudo, M.; Johnson, P.; Wagner, S.; Orsini, L.S.; et al. Global patterns of hepatocellular carcinoma management from diagnosis to death: The BRIDGE Study. Liver Int. 2015, 35, 2155–2166. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Sandhu, S.; Lai, J.P.; Sandhu, D.S. Hepatocellular carcinoma in non-cirrhotic liver: A comprehensive review. World J. Hepatol. 2019, 11, 1–18. [Google Scholar] [CrossRef]
- Singal, A.G.; Parikh, N.D.; Rich, N.E.; John, B.V.; Pillai, A. Hepatocellular Carcinoma Surveillance and Staging. In Hepatocellular carcinoma: Translational precision medicine approaches; Hoshida, Y., Ed.; Springer: Cham, Switzerland, 2019; pp. 27–51. [Google Scholar]
- Reig, M.; Forner, A.; Rimola, J.; Ferrer-Fàbrega, J.; Burrel, M.; Garcia-Criado, Á.; Kelley, R.K.; Galle, P.R.; Mazzaferro, V.; Salem, R.; et al. BCLC strategy for prognosis prediction and treatment recommendation: The 2022 update. J. Hepatol. 2022, 76, 681–693. [Google Scholar] [CrossRef]
- Matsuki, E.; Younes, A. Checkpoint Inhibitors and Other Immune Therapies for Hodgkin and Non-Hodgkin Lymphoma. Curr. Treat. Options Oncol. 2016, 17, 31. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Callahan, M.K.; Awad, M.M.; Calvo, E.; Ascierto, P.A.; Atmaca, A.; Rizvi, N.A.; Hirsch, F.R.; Selvaggi, G.; Szustakowski, J.D.; et al. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer Cell 2018, 33, 853–861.e4. Available online: https://www.sciencedirect.com/science/article/pii/S1535610818301727 (accessed on 14 July 2023). [CrossRef]
- Oh, A.; Tran, D.M.; McDowell, L.C.; Keyvani, D.; Barcelon, J.A.; Merino, O.; Wilson, L. Cost-Effectiveness of Nivolumab-Ipilimumab Combination Therapy Compared with Monotherapy for First-Line Treatment of Metastatic Melanoma in the United States. J. Manag. Care Spec. Pharm. 2017, 23, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Park, J.-W.; Finn, R.S.; Cheng, A.-L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2022, 23, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Chan, L.S.; Gu, S.; Bai, Y.; Ren, Z.; Lin, X.; Chen, Z.; Jia, W.; Jin, Y.; Guo, Y.; et al. LBA35 Camrelizumab (C) plus rivoceranib (R) vs. sorafenib (S) as first-line therapy for unresectable hepatocellular carcinoma (uHCC): A randomized, phase III trial. Ann. Oncol. 2022, 33, S1401–S1402. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Chan, S.L.; Kudo, M.; Lau, G.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; Dao, T.V.; De Toni, E.N.; et al. Phase 3 randomized, open-label, multicenter study of tremelimumab (T) and durvalumab (D) as first-line therapy in patients (pts) with unresectable hepatocellular carcinoma (uHCC): HIMALAYA. J. Clin. Oncol. 2022, 40 (Suppl. S4), 379. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves Atezolizumab Plus Bevacizumab for Unresectable Hepatocellular Carcinoma | FDA [Internet]. [cited 2023 Nov 15]. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-atezolizumab-plus-bevacizumab-unresectable-hepatocellular-carcinoma (accessed on 15 November 2023).
- Jain, A.; Chitturi, S.; Peters, G.; Yip, D. Atezolizumab and bevacizumab as first line therapy in advanced hepatocellular carcinoma: Practical considerations in routine clinical practice. World J. Hepatol. 2021, 13, 1132–1142. [Google Scholar] [CrossRef] [PubMed]
- Lang, L. FDA approves sorafenib for patients with inoperable liver cancer. Gastroenterology 2008, 134, 379. [Google Scholar] [CrossRef]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; McNabola, A.; Wilkie, D.; Wilhelm, S.; Lynch, M.; Carter, C. Sorafenib Blocks the RAF/MEK/ERK Pathway, Inhibits Tumor Angiogenesis, and Induces Tumor Cell Apoptosis in Hepatocellular Carcinoma Model PLC/PRF/5. Cancer Res. 2006, 66, 11851–11858. [Google Scholar] [CrossRef]
- Vogel, A.; Cervantes, A.; Chau, I.; Daniele, B.; Llovet, J.M.; Meyer, T.; Nault, J.-C.; Neumann, U.; Ricke, J.; Sangro, B.; et al. Hepatocellular carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv238–iv255. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.-L.; Kang, Y.-K.; Chen, Z.; Tsao, C.-J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.-S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Fernando, J.; Sancho, P.; Fernández-Rodriguez, C.M.; Lledó, J.L.; Caja, L.; Campbell, J.S.; Fausto, N.; Fabregat, I. Sorafenib sensitizes hepatocellular carcinoma cells to physiological apoptotic stimuli. J. Cell Physiol. 2012, 227, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Sonntag, R.; Gassler, N.; Bangen, J.M.; Trautwein, C.; Liedtke, C. Pro-apoptotic Sorafenib signaling in murine hepatocytes depends on malignancy and is associated with PUMA expression in vitro and in vivo. Cell Death Dis. 2014, 5, e1030. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.-T.; Shiau, C.-W.; Chen, H.-L.; Liu, C.-Y.; Lin, C.-S.; Cheng, A.-L.; Chen, P.-J.; Chen, K.-F. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell Death Dis. 2013, 4, e485. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Takehara, T.; Hikita, H.; Kodama, T.; Tsunematsu, H.; Miyagi, T.; Hosui, A.; Ishida, H.; Tatsumi, T.; Kanto, T.; et al. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int. J. Cancer 2012, 131, 548–557. [Google Scholar] [CrossRef]
- Louandre, C.; Ezzoukhry, Z.; Godin, C.; Barbare, J.-C.; Mazière, J.-C.; Chauffert, B.; Galmiche, A. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int. J. Cancer 2013, 133, 1732–1742. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 20, 3. [Google Scholar] [CrossRef]
- Hage, C.; Hoves, S.; Strauss, L.; Bissinger, S.; Prinz, Y.; Pöschinger, T.; Kiessling, F.; Ries, C.H. Sorafenib Induces Pyroptosis in Macrophages and Triggers Natural Killer Cell–Mediated Cytotoxicity Against Hepatocellular Carcinoma. Hepatology 2019, 70, 1280–1297. [Google Scholar] [CrossRef]
- El-Sewedy, T.; Salama, A.F.; Mohamed, A.E.; Elbaioumy, N.M.; El-Far, A.H.; Albalawi, A.N.; Elmetwalli, A. Hepatocellular Carcinoma cells: Activity of Amygdalin and Sorafenib in Targeting AMPK/mTOR and BCL-2 for anti-angiogenesis and apoptosis cell death. BMC Complement Med. Ther. 2023, 23, 329. [Google Scholar] [CrossRef]
- Zhu, W.; Liang, Q.; Yang, X.; Yu, Y.; Shen, X.; Sun, G. Combination of sorafenib and Valproic acid synergistically induces cell apoptosis and inhibits hepatocellular carcinoma growth via down-regulating Notch3 and pAkt. Am. J. Cancer Res. 2017, 7, 2503–2514. [Google Scholar] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Vakkila, J.; Lotze, M.T. Inflammation and necrosis promote tumour growth. Vol. 4, Nature reviews. Immunology 2004, 4, 641–648. [Google Scholar] [PubMed]
- Eum, K.H.; Ahn, S.K.; Kang, H.; Lee, M. Differential inhibitory effects of two Raf-targeting drugs, sorafenib and PLX4720, on the growth of multidrug-resistant cells. Mol. Cell Biochem. 2013, 372, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Hu, C.; Yao, M.; Han, G. Mechanism of sorafenib resistance associated with ferroptosis in HCC. Front Pharmacol. 2023, 14, 1207496. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Yang, Y.; Pan, D.; Ding, Y.; Zhang, H.; Ye, Y.; Li, J.; Zhao, L. HSP90α Mediates Sorafenib Resistance in Human Hepatocellular Carcinoma by Necroptosis Inhibition under Hypoxia. Cancers 2021, 13, 243. [Google Scholar] [CrossRef] [PubMed]
- Garten, A.; Grohmann, T.; Kluckova, K.; Lavery, G.G.; Kiess, W.; Penke, M. Sorafenib-Induced Apoptosis in Hepatocellular Carcinoma Is Reversed by SIRT1. Int. J. Mol. Sci. 2019, 20, 4048. [Google Scholar] [CrossRef]
- Ling, S.; Song, L.; Fan, N.; Feng, T.; Liu, L.; Yang, X.; Wang, M.; Li, Y.; Tian, Y.; Zhao, F.; et al. Combination of metformin and sorafenib suppresses proliferation and induces autophagy of hepatocellular carcinoma via targeting the mTOR pathway. Int. J. Oncol. 2017, 50, 297–309. [Google Scholar] [CrossRef]
- Schult, C.; Dahlhaus, M.; Ruck, S.; Sawitzky, M.; Amoroso, F.; Lange, S.; Etro, D.; Glass, A.; Fuellen, G.; Boldt, S.; et al. The multikinase inhibitor Sorafenib displays significant antiproliferative effects and induces apoptosis via caspase 3, 7 and PARP in B- and T-lymphoblastic cells. BMC Cancer 2010, 10, 560. [Google Scholar] [CrossRef]
- Zhao, X.; Tian, C.; Puszyk, W.M.; Ogunwobi, O.O.; Cao, M.; Wang, T.; Cabrera, R.; Nelson, D.R.; Liu, C. OPA1 downregulation is involved in sorafenib-induced apoptosis in hepatocellular carcinoma. Lab. Investig. 2013, 93, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Liao, M.; Qin, R.; Zhu, S.; Peng, C.; Fu, L.; Chen, Y.; Han, B. Regulated cell death (RCD) in cancer: Key pathways and targeted therapies. Signal. Transduct. Target Ther. 2022, 7, 286. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef] [PubMed]
- Pihán, P.; Carreras-Sureda, A.; Hetz, C. BCL-2 family: Integrating stress responses at the ER to control cell demise. Cell Death Differ. 2017, 24, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Berger, M.; Goldberg, Z.; Haupt, Y. Apoptosis—The p53 network. J. Cell Sci. 2003, 116 Pt 20, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Hernández, M.A.; de la Cruz-Ojeda, P.; Gallego, P.; Navarro-Villarán, E.; Staňková, P.; Del Campo, J.A.; Kučera, O.; Elkalaf, M.; Maseko, T.E.; Červinková, Z.; et al. Dose-dependent regulation of mitochondrial function and cell death pathway by sorafenib in liver cancer cells. Biochem. Pharmacol. 2020, 176, 113902. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.; Ngo, V.C.; Koong, H.N.; Poon, D.; Choo, S.P.; Thng, C.H.; Chow, P.; Ong, H.S.; Chung, A.; Soo, K.C. Sorafenib and rapamycin induce growth suppression in mouse models of hepatocellular carcinoma. J. Cell Mol. Med. 2009, 13, 2673–2683. [Google Scholar] [CrossRef]
- Wei, J.C.; Meng, F.D.; Qu, K.; Wang, Z.X.; Wu, Q.F.; Zhang, L.Q.; Pang, Q.; Liu, C. Sorafenib inhibits proliferation and invasion of human hepatocellular carcinoma cells via up-regulation of p53 and suppressing FoxM1. Acta Pharmacol. Sin. 2015, 36, 241–251. [Google Scholar] [CrossRef]
- Zhai, J.-M.; Yin, X.-Y.; Lai, Y.-R.; Hou, X.; Cai, J.-P.; Hao, X.-Y.; Liang, L.-J.; Zhang, L.-J. Sorafenib enhances the chemotherapeutic efficacy of S-1 against hepatocellular carcinoma through downregulation of transcription factor E2F-1. Cancer Chemother. Pharmacol. 2013, 71, 1255–1264. [Google Scholar] [CrossRef]
- Pietsch, E.C.; Sykes, S.M.; McMahon, S.B.; Murphy, M.E. The p53 family and programmed cell death. Oncogene 2008, 27, 6507–6521. [Google Scholar] [CrossRef] [PubMed]
- Marouco, D.; Garabadgiu, A.V.; Melino, G.; Barlev, N.A. Lysine-specific modifications of p53: A matter of life and death? Oncotarget 2013, 4, 1556–1571. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Barsotti, A.M.; Prives, C. Pro-proliferative FoxM1 is a target of p53-mediated repression. Oncogene 2009, 28, 4295–4305. [Google Scholar] [CrossRef] [PubMed]
- Brochier, C.; Dennis, G.; Rivieccio, M.A.; McLaughlin, K.; Coppola, G.; Ratan, R.R.; Langley, B. Specific acetylation of p53 by HDAC inhibition prevents DNA damage-induced apoptosis in neurons. J. Neurosci. 2013, 33, 8621–8632. [Google Scholar] [CrossRef] [PubMed]
- Sykes, S.M.; Mellert, H.S.; Holbert, M.A.; Li, K.; Marmorstein, R.; Lane, W.S.; McMahon, S.B. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol. Cell. 2006, 24, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, K.; Herrera, J.E.; Saito, S.; Miki, T.; Bustin, M.; Vassilev, A.; Anderson, C.W.; Appella, E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998, 12, 2831–2841. [Google Scholar] [CrossRef]
- van den Heuvel, S.; Dyson, N.J. Conserved functions of the pRB and E2F families. Nat. Rev. Mol. Cell Biol. 2008, 9, 713–724. [Google Scholar] [CrossRef]
- Palaiologou, M.; Koskinas, J.; Karanikolas, M.; Fatourou, E.; Tiniakos, D.G. E2F-1 is overexpressed and pro-apoptotic in human hepatocellular carcinoma. Virchows. Arch. 2012, 460, 439–446. [Google Scholar] [CrossRef]
- Teufel, A.; Staib, F.; Kanzler, S.; Weinmann, A.; Schulze-Bergkamen, H.; Galle, P.R. Genetics of hepatocellular carcinoma. World J. Gastroenterol. 2007, 13, 2271–2282. [Google Scholar] [CrossRef]
- Stanelle, J.; Pützer, B.M. E2F1-induced apoptosis: Turning killers into therapeutics. Trends Mol. Med. 2006, 12, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Bell, L.A.; O’Prey, J.; Ryan, K.M. DNA-binding independent cell death from a minimal proapoptotic region of E2F-1. Oncogene 2006, 25, 5656–5663. [Google Scholar] [CrossRef] [PubMed]
- Iaquinta, P.J.; Lees, J.A. Life and death decisions by the E2F transcription factors. Curr. Opin. Cell Biol. 2007, 19, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Ciro, M.; Cocito, A.; Helin, K. E2F target genes: Unraveling the biology. Trends Biochem. Sci. 2004, 29, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Hershko, T.; Ginsberg, D. Up-regulation of Bcl-2 homology 3 (BH3)-only proteins by E2F1 mediates apoptosis. J. Biol. Chem. 2004, 279, 8627–8634. [Google Scholar] [CrossRef] [PubMed]
- Moroni, M.C.; Hickman, E.S.; Lazzerini Denchi, E.; Caprara, G.; Colli, E.; Cecconi, F.; Müller, H.; Helin, K. Apaf-1 is a transcriptional target for E2F and p53. Nat. Cell Biol. 2001, 3, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Henley, S.A.; Dick, F.A. The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div. 2012, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Louandre, C.; Marcq, I.; Bouhlal, H.; Lachaier, E.; Godin, C.; Saidak, Z.; François, C.; Chatelain, D.; Debuysscher, V.; Barbare, J.-C.; et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015, 356 Pt B, 971–977. [Google Scholar] [CrossRef]
- Heqing, Y.; Bin, L.; Xuemei, Y.; Linfa, L. The role and mechanism of autophagy in sorafenib targeted cancer therapy. Crit. Rev. Oncol. Hematol. 2016, 100, 137–140. [Google Scholar] [CrossRef]
- Fischer, T.D. Role of autophagy in differential sensitivity of hepatocarcinoma cells to sorafenib. World J. Hepatol. 2014, 6, 752. [Google Scholar] [CrossRef]
- Blechacz, B.R.A.; Smoot, R.L.; Bronk, S.F.; Werneburg, N.W.; Sirica, A.E.; Gores, G.J. Sorafenib inhibits signal transducer and activator of transcription-3 signaling in cholangiocarcinoma cells by activating the phosphatase shatterproof 2. Hepatology 2009, 50, 1861–1870. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wei, H.; Huang, Y.; Chen, D.; Zeng, G.; Lian, Y.; Huang, Y. The combination of lonafarnib and sorafenib induces cyclin D1 degradation via ATG3-mediated autophagic flux in hepatocellular carcinoma cells. Aging 2019, 11, 5769–5785. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Hah, Y.J.; Kang, Y.N.; Kang, K.J.; Hwang, J.S.; Chung, W.J.; Cho, K.B.; Park, K.S.; Kim, E.S.; Seo, H.-Y.; et al. The autophagy-related marker LC3 can predict prognosis in human hepatocellular carcinoma. PLoS ONE 2013, 8, e81540. [Google Scholar] [CrossRef]
- Wu, W.Y.; Kim, H.; Zhang, C.L.; Meng, X.L.; Wu, Z.S. Clinical significance of autophagic protein LC3 levels and its correlation with XIAP expression in hepatocellular carcinoma. Med. Oncol. 2014, 31, 108. [Google Scholar] [CrossRef] [PubMed]
- Ajoolabady, A.; Tang, D.; Kroemer, G.; Ren, J. Ferroptosis in hepatocellular carcinoma: Mechanisms and targeted therapy. Br. J. Cancer 2023, 128, 190–205. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Chen, L.J.; Chen, G.; Chao , T.I.; Wang, C.Y. SHP-1/STAT3-Signaling-Axis-Regulated Coupling between BECN1 and SLC7A11 Contributes to Sorafenib-Induced Ferroptosis in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2022, 23, 11092. [Google Scholar] [CrossRef]
- Zheng, J.; Sato, M.; Mishima, E.; Sato, H.; Proneth, B.; Conrad, M. Sorafenib fails to trigger ferroptosis across a wide range of cancer cell lines. Cell Death Dis. 2021, 12, 698. [Google Scholar] [CrossRef]
- Liu, X.; Xia, S.; Zhang, Z.; Wu, H.; Lieberman, J. Channelling inflammation: Gasdermins in physiology and disease. Nat. Rev. Drug Discov. 2021, 20, 384–405. [Google Scholar] [CrossRef]
- Shi, J.; Wu, P.; Sheng, L.; Sun, W.; Zhang, H. Ferroptosis-related gene signature predicts the prognosis of papillary thyroid carcinoma. Cancer Cell Int. 2021, 21, 669. [Google Scholar] [CrossRef] [PubMed]
- Khalid, N.; Azimpouran, M. Necrosis; StatPearls Publishing LLC: Treasure Island, FL, USA, 2023. [Google Scholar]
- Abdu, S.; Juaid, N.; Amin, A.; Moulay, M.; Miled, N. Effects of Sorafenib and Quercetin Alone or in Combination in Treating Hepatocellular Carcinoma: In Vitro and In Vivo Approaches. Molecules 2022, 27, 8082. [Google Scholar] [CrossRef] [PubMed]
- Armeanu, S.; Pathil, A.; Venturelli, S.; Mascagni, P.; Weiss, T.S.; Göttlicher, M.; Gregor, M.; Lauer, U.M.; Bitzer, M. Apoptosis on hepatoma cells but not on primary hepatocytes by histone deacetylase inhibitors valproate and ITF2357. J. Hepatol. 2005, 42, 210–217. [Google Scholar] [CrossRef]
- Wolinska, E.; Skrzypczak, M. Epigenetic Changes Affecting the Development of Hepatocellular Carcinoma. Cancers 2021, 13, 4237. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, Y.; He, H.; Lou, L.; Ye, W.; Chen, Y.; Wang, J. Synergistically killing activity of aspirin and histone deacetylase inhibitor valproic acid (VPA) on hepatocellular cancer cells. Biochem. Biophys. Res. Commun. 2013, 436, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zhao, X.; Pei, F.; Ji, M.; Ma, W.; Wang, Y.; Jiang, G. Activation of the intrinsic apoptosis pathway contributes to the induction of apoptosis in hepatocellular carcinoma cells by valproic acid. Oncol. Lett. 2015, 9, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Insinga, A.; Monestiroli, S.; Ronzoni, S.; Gelmetti, V.; Marchesi, F.; Viale, A.; Altucci, L.; Nervi, C.; Minucci, S.; Pelicci, P.G. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat. Med. 2005, 11, 71–76. [Google Scholar] [CrossRef]
- Korsmeyer, S.J.; Wei, M.C.; Saito, M.; Weiler, S.; Oh, K.J.; Schlesinger, P.H. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000, 7, 1166–1173. [Google Scholar] [CrossRef]
- Kawagoe, R.; Kawagoe, H.; Sano, K. Valproic acid induces apoptosis in human leukemia cells by stimulating both caspase-dependent and -independent apoptotic signaling pathways. Leuk Res. 2002, 26, 495–502. [Google Scholar] [CrossRef]
- Yagi, Y.; Fushida, S.; Harada, S.; Kinoshita, J.; Makino, I.; Oyama, K.; Tajima, H.; Fujita, H.; Takamura, H.; Ninomiya, I.; et al. Effects of valproic acid on the cell cycle and apoptosis through acetylation of histone and tubulin in a scirrhous gastric cancer cell line. J. Exp. Clin. Cancer Res. 2010, 29, 149. [Google Scholar] [CrossRef]
- Gilardini Montani, M.S.; Granato, M.; Santoni, C.; Del Porto, P.; Merendino, N.; D’Orazi, G.; Faggioni, A.; Cirone, M. Histone deacetylase inhibitors VPA and TSA induce apoptosis and autophagy in pancreatic cancer cells. Cell. Oncol. 2017, 40, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xu, D.; Zhu, S.-J.; Ye, B.; Dong, J.-D.; Zhang, Y.-L.; Zhang, Y. Induction of apoptosis and autophagy in metastatic thyroid cancer cells by valproic acid (VPA). Int. J. Clin. Exp. Pathol. 2015, 8, 8291–8297. [Google Scholar] [PubMed]
- Liu, Y.; Gu, W. The complexity of p53-mediated metabolic regulation in tumor suppression. Semin. Cancer Biol. 2022, 85, 4–32. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.M.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. Br. Med. J. 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed]
- Papadakos, S.P.; Ferraro, D.; Carbone, G.; Frampton, A.E.; Vennarecci, G.; Kykalos, S.; Schizas, D.; Theocharis, S.; Machairas, N. The Emerging Role of Metformin in the Treatment of Hepatocellular Carcinoma: Is There Any Value in Repurposing Metformin for HCC Immunotherapy? Cancers 2023, 15, 3161. [Google Scholar] [CrossRef] [PubMed]
- Azit, N.A.; Sahran, S.; Leow, V.M.; Subramaniam, M.K.; Mokhitar, S.; Nawi, A.M. The survival outcomes and prognostic factors of hepatocellular carcinoma among type 2 diabetes patients: A two-centre retrospective cohort study. Turk. J. Med. Sci. 2022, 52, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, K.A.; Petrick, J.L.; London, W.T. Global epidemiology of hepatocellular carcinoma: An emphasis on demographic and regional variability. Clin. Liver Dis. 2015, 19, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Liu, X.; Zhang, Y.; Reske, J.; Bahal, D.; Gohl, T.G.; Hollern, D.; Ensink, E.; Kiupel, M.; Luo, R.; et al. NCOA5 deficiency promotes a unique liver protumorigenic microenvironment through p21WAF1/CIP1 overexpression, which is reversed by metformin. Oncogene 2020, 39, 3821–3836. [Google Scholar] [CrossRef]
- Shahid, A.; Chen, M.; Yeung, S.; Parsa, C.; Orlando, R.; Huang, Y. The medicinal mushroom Ganoderma lucidum prevents lung tumorigenesis induced by tobacco smoke carcinogens. Front. Pharmacol. 2023, 14, 1244150. [Google Scholar] [CrossRef]
- Sun, Y.; Tao, C.; Huang, X.; He, H.; Shi, H.; Zhang, Q.; Wu, H. Metformin induces apoptosis of human hepatocellular carcinoma HepG2 cells by activating an AMPK/p53/miR-23a/FOXA1 pathway. Onco. Targets Ther. 2016, 12, 2845. [Google Scholar]
- Tawfik, S.M.; Abdollah, M.R.A.; Elmazar, M.M.; El-Fawal, H.A.N.; Abdelnaser, A. Effects of Metformin Combined with Antifolates on HepG2 Cell Metabolism and Cellular Proliferation. Front. Oncol. 2022, 12, 828988. [Google Scholar] [CrossRef] [PubMed]
- S Shen, Z.; Zhou, H.; Li, A.; Wu, T.; Ji, X.; Guo, L.; Zhu, X.; Zhang, D.; He, X. Metformin inhibits hepatocellular carcinoma development by inducing apoptosis and pyroptosis through regulating FOXO3. Aging 2021, 13, 22120–22133. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, H.; Kato, K.; Iwama, H.; Maeda, E.; Sakamoto, T.; Fujita, K.; Toyota, Y.; Tani, J.; Nomura, T.; Mimura, S.; et al. Effect of the anti-diabetic drug metformin in hepatocellular carcinoma in vitro and in vivo. Int. J. Oncol. 2014, 45, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Xue, D.; Wang, K.; Zhang, F.; Shi, J.; Jia, B.; Yang, D.; Zhang, Q.; Zhang, S.; Jiang, H.; et al. Metformin exerts an antitumor effect by inhibiting bladder cancer cell migration and growth, and promoting apoptosis through the PI3K/AKT/mTOR pathway. BMC Urol. 2022, 22, 79. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhou, P.; Xu, K.; Chen, T.; Jiao, J.; Wei, H.; Yang, X.; Xu, W.; Wan, W.; Xiao, J. Metformin induces cell cycle arrest, apoptosis and autophagy through ROS/JNK signaling pathway in human osteosarcoma. Int. J. Biol. Sci. 2020, 16, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Gong, H.; Wang, Y.; Guo, S.; Liu, K. AMPK/mTOR-mediated inhibition of survivin partly contributes to metformin-induced apoptosis in human gastric cancer cell. Cancer Biol. Ther. 2015, 16, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sun, H.; Feng, M.; Zhao, J.; Zhao, X.; Wan, Q.; Cai, D. Metformin is associated with reduced cell proliferation in human endometrial cancer by inbibiting PI3K/AKT/mTOR signaling. Gynecol. Endocrinol. 2018, 34, 428–432. [Google Scholar] [CrossRef]
- Gao, C.; Fang, L.; Zhang, H.; Zhang, W.S.; Li, X.O.; Du, S.Y. Metformin Induces Autophagy via the AMPK-mTOR Signaling Pathway in Human Hepatocellular Carcinoma Cells. Cancer Manag. Res. 2020, 12, 5803–5811. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Yang, X.; Liu, Y.; Li, M.; Wu, H.; Wang, Y.; You, Y.; Li, P.; Ding, X.; Liu, C.; Gong, J. Predictive and preventive significance of AMPK activation on hepatocarcinogenesis in patients with liver cirrhosis. Cell Death Dis. 2018, 9, 264. [Google Scholar] [CrossRef]
- Liu, S.; Yue, C.; Chen, H.; Chen, Y.; Li, G. Metformin Promotes Beclin1-Dependent Autophagy to Inhibit the Progression of Gastric Cancer. Onco. Targets Ther. 2020, 13, 4445–4455. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Peng, Y.-F.; Ni, H.-M.; Li, Y.; Shi, Y.-H.; Ding, W.-X.; Fan, J. Basal Autophagy and Feedback Activation of Akt Are Associated with Resistance to Metformin-Induced Inhibition of Hepatic Tumor Cell Growth. PLoS ONE 2015, 10, e0130953. [Google Scholar] [CrossRef] [PubMed]
- Jo, W.; Yu, E.-S.; Chang, M.; Park, H.-K.; Choi, H.-J.; Ryu, J.-E.; Jang, S.; Lee, H.-J.; Jang, J.-J.; Son, W.-C. Metformin inhibits early stage diethylnitrosamine-induced hepatocarcinogenesis in rats. Mol. Med. Rep. 2016, 13, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, J.A.; García-Valdecasas, M.; Gil-Gómez, A.; Rojas, Á.; Gallego, P.; Ampuero, J.; Gallego-Durán, R.; Pastor, H.; Grande, L.; Padillo, F.J.; et al. Simvastatin and metformin inhibit cell growth in hepatitis C virus infected cells via mTOR increasing PTEN and autophagy. PLoS ONE 2018, 13, e0191805. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-H.; Lai, H.-Y.; Chen, Y.-C.; Li, C.-F.; Huang, H.-S.; Liu, H.-S.; Tsai, Y.-S.; Wang, J.-M. Metformin promotes apoptosis in hepatocellular carcinoma through the CEBPD-induced autophagy pathway. Oncotarget 2017, 8, 13832–13845. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Zhai, R.; Ma, C.; Miao, W. Combination of aloin and metformin enhances the antitumor effect by inhibiting the growth and invasion and inducing apoptosis and autophagy in hepatocellular carcinoma through PI3K/AKT/mTOR pathway. Cancer Med. 2020, 9, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhao, Y.; Li, L.; Jiang, J.; Li, W.; Mang, Y.; Gao, Y.; Dong, Y.; Zhu, J.; Yang, C.; et al. Metformin promotes ferroptosis and sensitivity to sorafenib in hepatocellular carcinoma cells via ATF4/STAT3. Mol. Biol. Rep. 2023, 50, 6399–6413. [Google Scholar] [CrossRef]
- Hsu, S.-K.; Cheng, K.-C.; Mgbeahuruike, M.O.; Lin, Y.-H.; Wu, C.-Y.; Wang, H.-M.D.; Yen, C.-H.; Chiu, C.-C.; Sheu, S.-J. New Insight into the Effects of Metformin on Diabetic Retinopathy, Aging and Cancer: Nonapoptotic Cell Death, Immunosuppression, and Effects beyond the AMPK Pathway. Int. J. Mol. Sci. 2021, 22, 9453. [Google Scholar] [CrossRef]
- Zhao, S.; Li, P.; Wu, W.; Wang, Q.; Qian, B.; Li, X.; Shen, M. Roles of ferroptosis in urologic malignancies. Cancer Cell Int. 2021, 21, 676. [Google Scholar] [CrossRef]
- Liu, B.; Xu, J.; Lu, L.; Gao, L.; Zhu, S.; Sui, Y.; Cao, T.; Yang, T. Metformin induces pyroptosis in leptin receptor-defective hepatocytes via overactivation of the AMPK axis. Cell Death Dis. 2023, 14, 82. [Google Scholar] [CrossRef]
- Gilad, Y.; Gellerman, G.; Lonard, D.M.; O’Malley, B.W. Drug Combination in Cancer Treatment—From Cocktails to Conjugated Combinations. Cancers 2021, 13, 669. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Sanderson, P.E.; Zheng, W. Drug combination therapy increases successful drug repositioning. Drug Discov. Today 2016, 21, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, X.; Liang, Q.; Yu, Y.; Shen, X.; Sun, G. Valproic acid overcomes sorafenib resistance by reducing the migration of Jagged2-mediated Notch1 signaling pathway in hepatocellular carcinoma cells. Int. J. Biochem. Cell Biol. 2020, 126, 105820. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Liu, J.; Liang, Q.; Sun, G. Valproic acid reverses sorafenib resistance through inhibiting activated Notch/Akt signaling pathway in hepatocellular carcinoma. Fundam. Clin. Pharmacol. 2021, 35, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, S.C.; Tsai, J.P.; Yang, S.F.; Tang, M.J.; Hsieh, Y.H. Metformin inhibits the invasion of human hepatocellular carcinoma cells and enhances the chemosensitivity to sorafenib through a downregulation of the ERK/JNK-mediated NF-κB-dependent pathway that reduces uPA and MMP-9 expression. Amino Acids 2014, 46, 2809–2822. [Google Scholar] [CrossRef] [PubMed]
- Siddharth, S.; Kuppusamy, P.; Wu, Q.; Nagalingam, A.; Saxena, N.K.; Sharma, D. Metformin Enhances the Anti-Cancer Efficacy of Sorafenib via Suppressing MAPK/ERK/Stat3 Axis in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2022, 23, 8083. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.-G.; Tak, E.; Hwang, S.; Lee, J.-Y.; Kim, J.-Y.; Kim, Y.-Y.; Song, G.-W.; Lee, K.-J.; Kim, N. Synergistic effect of metformin on sorafenib in in vitro study using hepatocellular carcinoma cell lines. Ann. Hepatobiliary Pancreat Surg. 2018, 22, 179. [Google Scholar] [CrossRef]
- Harati, R.; Vandamme, M.; Blanchet, B.; Bardin, C.; Praz, F.; Hamoudi, R.A.; Desbois-Mouthon, C. Drug-Drug Interaction between Metformin and Sorafenib Alters Antitumor Effect in Hepatocellular Carcinoma Cells. Mol. Pharmacol. 2021, 100, 32–45. [Google Scholar] [CrossRef]
- Guo, Z.; Cao, M.; You, A.; Gao, J.; Zhou, H.; Li, H.; Cui, Y.; Fang, F.; Zhang, W.; Song, T.; et al. Metformin inhibits the prometastatic effect of sorafenib in hepatocellular carcinoma by upregulating the expression of TIP30. Cancer Sci. 2016, 107, 507–513. [Google Scholar] [CrossRef]
- National Institutes of Health US. ClinicalTrials.gov. 2023. Available online: https://clinicaltrials.gov/ (accessed on 14 November 2023).
- Tang, K.; Chen, Q.; Liu, Y.; Wang, L.; Lu, W. Combination of Metformin and Sorafenib Induces Ferroptosis of Hepatocellular Carcinoma Through p62-Keap1-Nrf2 Pathway. J. Cancer 2022, 13, 3234–3243. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, Y.; Dong, Y.; Li, H.; Gao, C.; Zhu, G.; Mi, X.; Li, C.; Xu, Y.; Wang, G.; et al. A pyroptosis-related gene signature for prognosis prediction in hepatocellular carcinoma. Front. Oncol. 2023, 13, 1085188. [Google Scholar] [CrossRef]
- Martina, B.E.E.; Smreczak, M.; Orlowska, A.; Marzec, A.; Trebas, P.; Roose, J.M.; Zmudzinski, J.; Gerhauser, I.; Wohlsein, P.; Baumgärtner, W.; et al. Combination drug treatment prolongs survival of experimentally infected mice with silver-haired bat rabies virus. Vaccine 2019, 37, 4736–4742. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Incidence | Rank | Mortality | Rank | World Population | % of Relative Prevalence |
|---|---|---|---|---|---|---|
| 2008 | 748,000 1 | 6 | 696,000 1 | 3 | 6,789,088,686 2 | 0.011 |
| 2012 | 782,000 1 | 6 | 745,000 1 | 2 | 7,125,828,059 2 | 0.010 |
| 2018 | 841,080 1 | 6 | 781,631 1 | 3 | 7,631,091,040 2 | 0.011 |
| 2020 | 905,677 1 | 6 | 830,180 1 | 4 | 7,794,798,739 2 | 0.011 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco-Juárez, E.X.; González-Villasana, V.; Camacho-Moll, M.E.; Rendón-Garlant, L.; Ramírez-Flores, P.N.; Silva-Ramírez, B.; Peñuelas-Urquides, K.; Cabello-Ruiz, E.D.; Castorena-Torres, F.; Bermúdez de León, M. Mechanistic Insights about Sorafenib-, Valproic Acid- and Metformin-Induced Cell Death in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2024, 25, 1760. https://doi.org/10.3390/ijms25031760
Franco-Juárez EX, González-Villasana V, Camacho-Moll ME, Rendón-Garlant L, Ramírez-Flores PN, Silva-Ramírez B, Peñuelas-Urquides K, Cabello-Ruiz ED, Castorena-Torres F, Bermúdez de León M. Mechanistic Insights about Sorafenib-, Valproic Acid- and Metformin-Induced Cell Death in Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2024; 25(3):1760. https://doi.org/10.3390/ijms25031760
Chicago/Turabian StyleFranco-Juárez, Edgar Xchel, Vianey González-Villasana, María Elena Camacho-Moll, Luisa Rendón-Garlant, Patricia Nefertari Ramírez-Flores, Beatriz Silva-Ramírez, Katia Peñuelas-Urquides, Ethel Daniela Cabello-Ruiz, Fabiola Castorena-Torres, and Mario Bermúdez de León. 2024. "Mechanistic Insights about Sorafenib-, Valproic Acid- and Metformin-Induced Cell Death in Hepatocellular Carcinoma" International Journal of Molecular Sciences 25, no. 3: 1760. https://doi.org/10.3390/ijms25031760