
Dysfunctional Postnatal Mitochondrial Energy Metabolism in a Patient with Neurodevelopmental Defects Caused by Intrauterine Growth Restriction Due to Idiopathic Placental Insufficiency
 , ,
, , 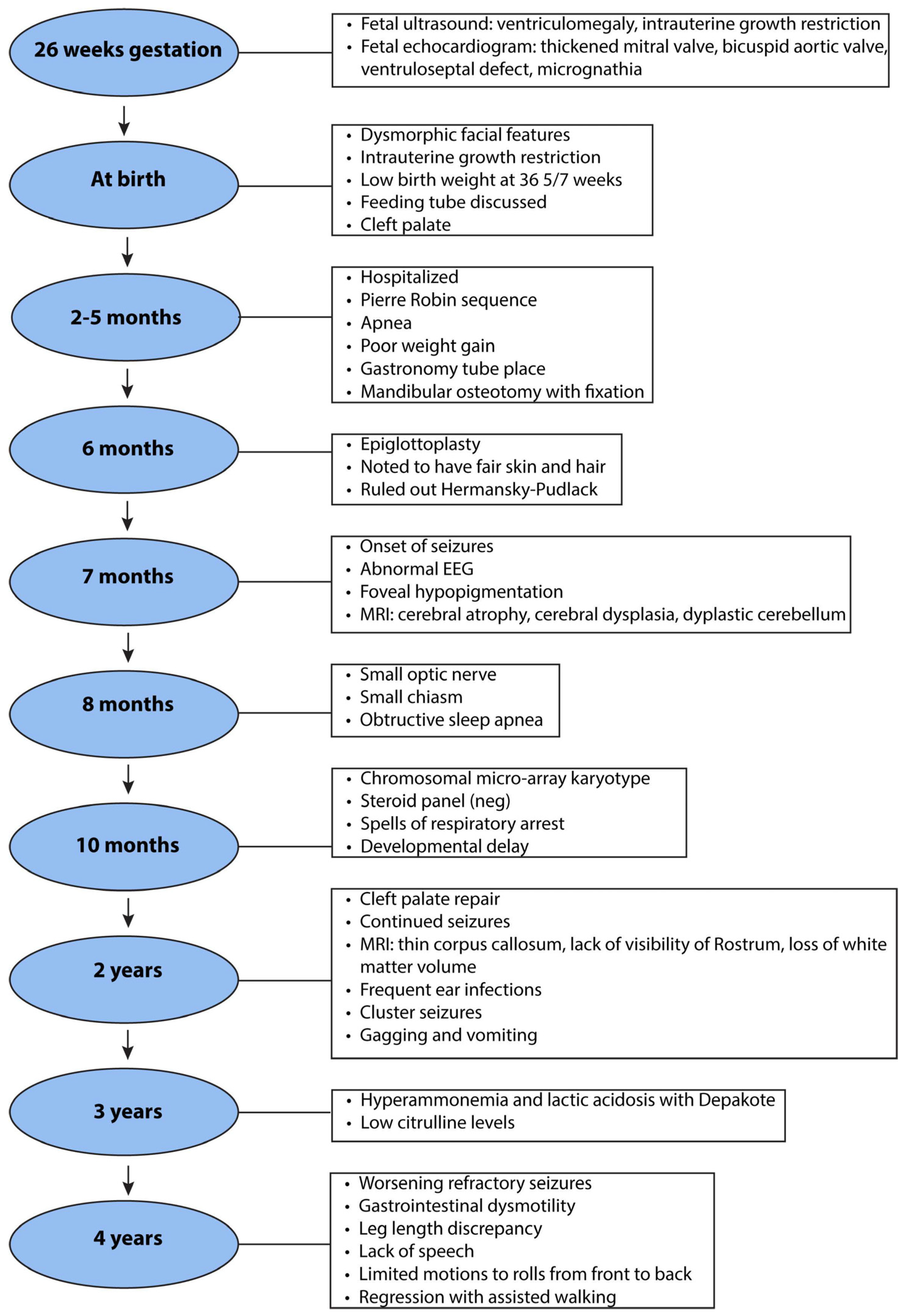{kind=link}
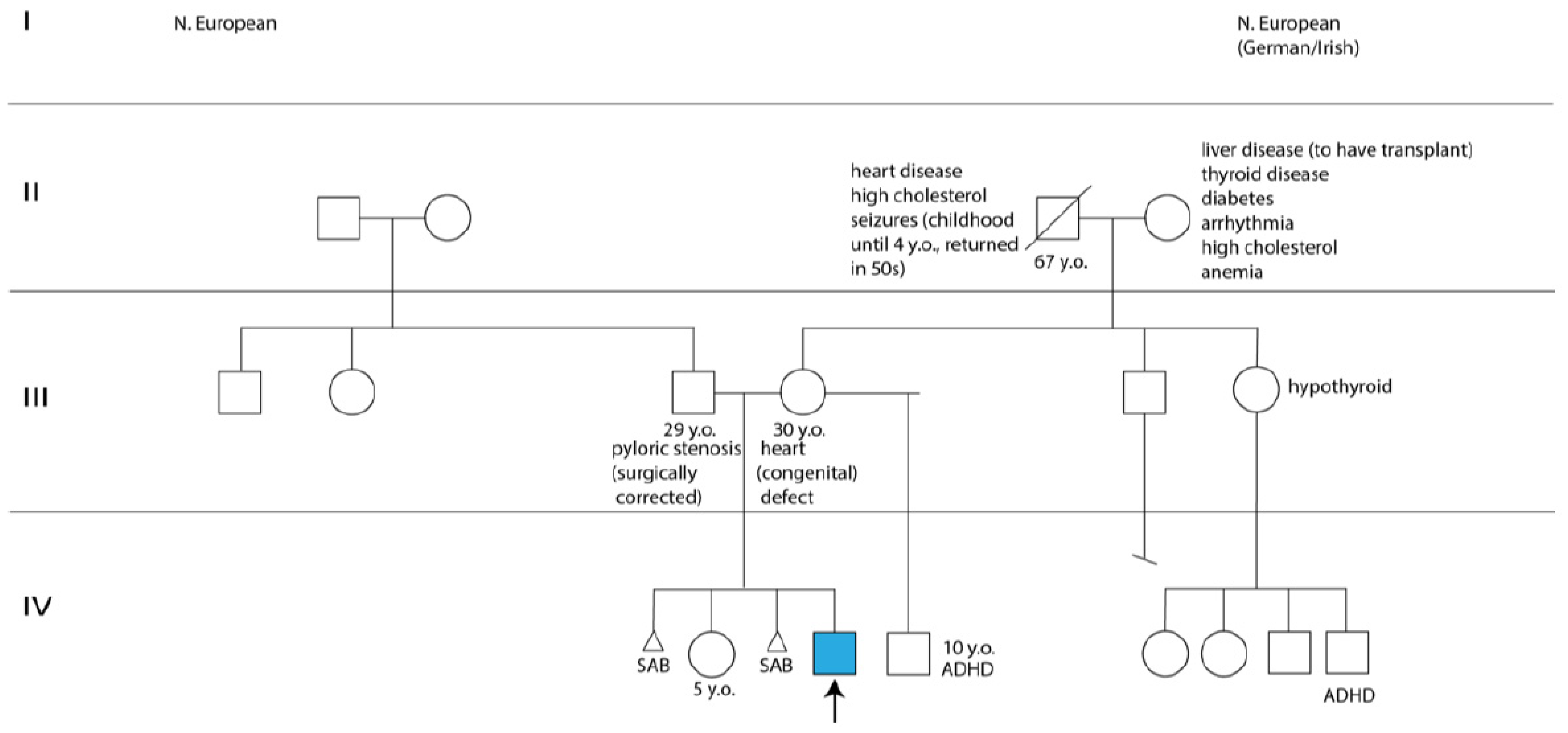{kind=link}
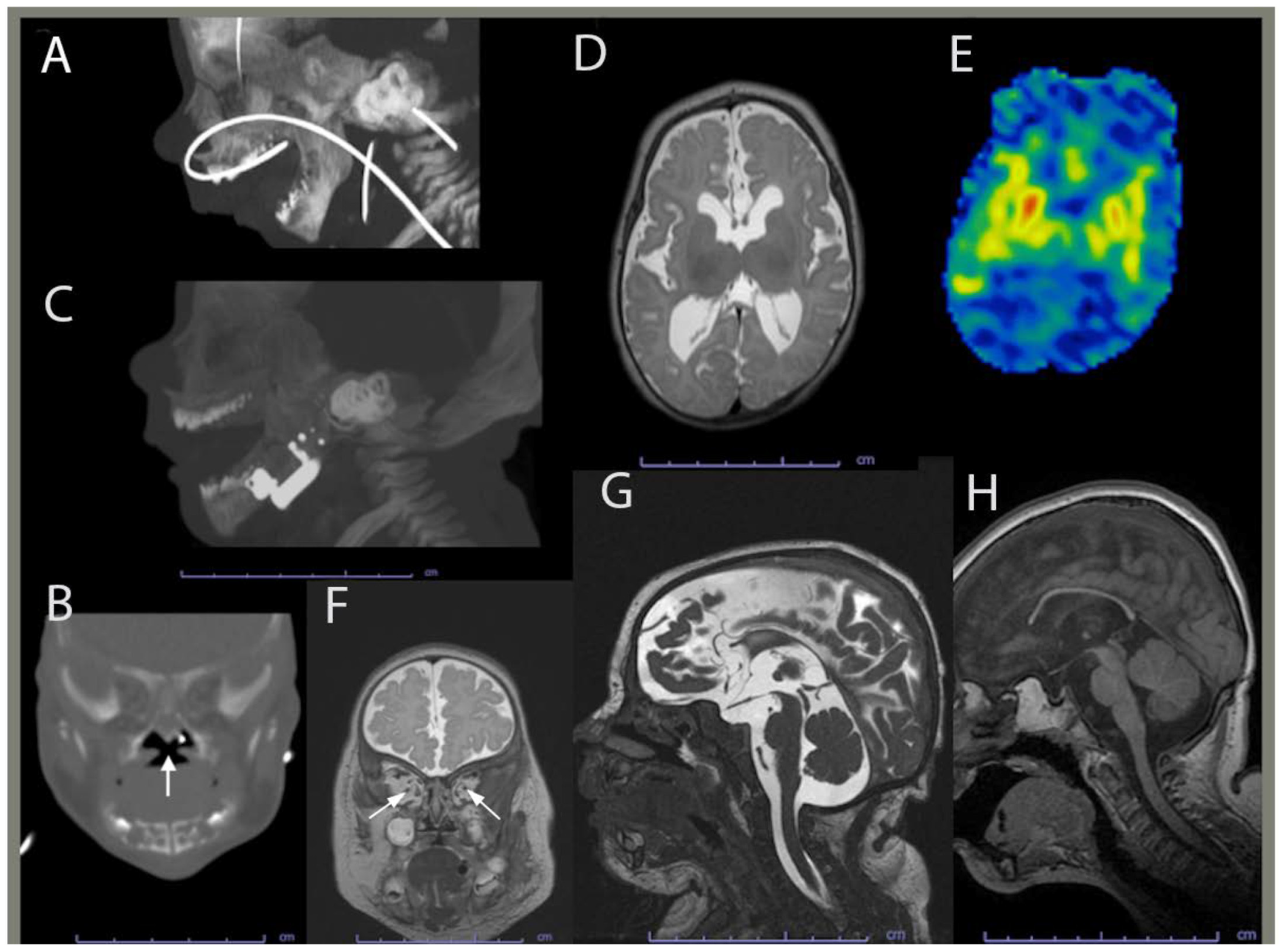{kind=link}
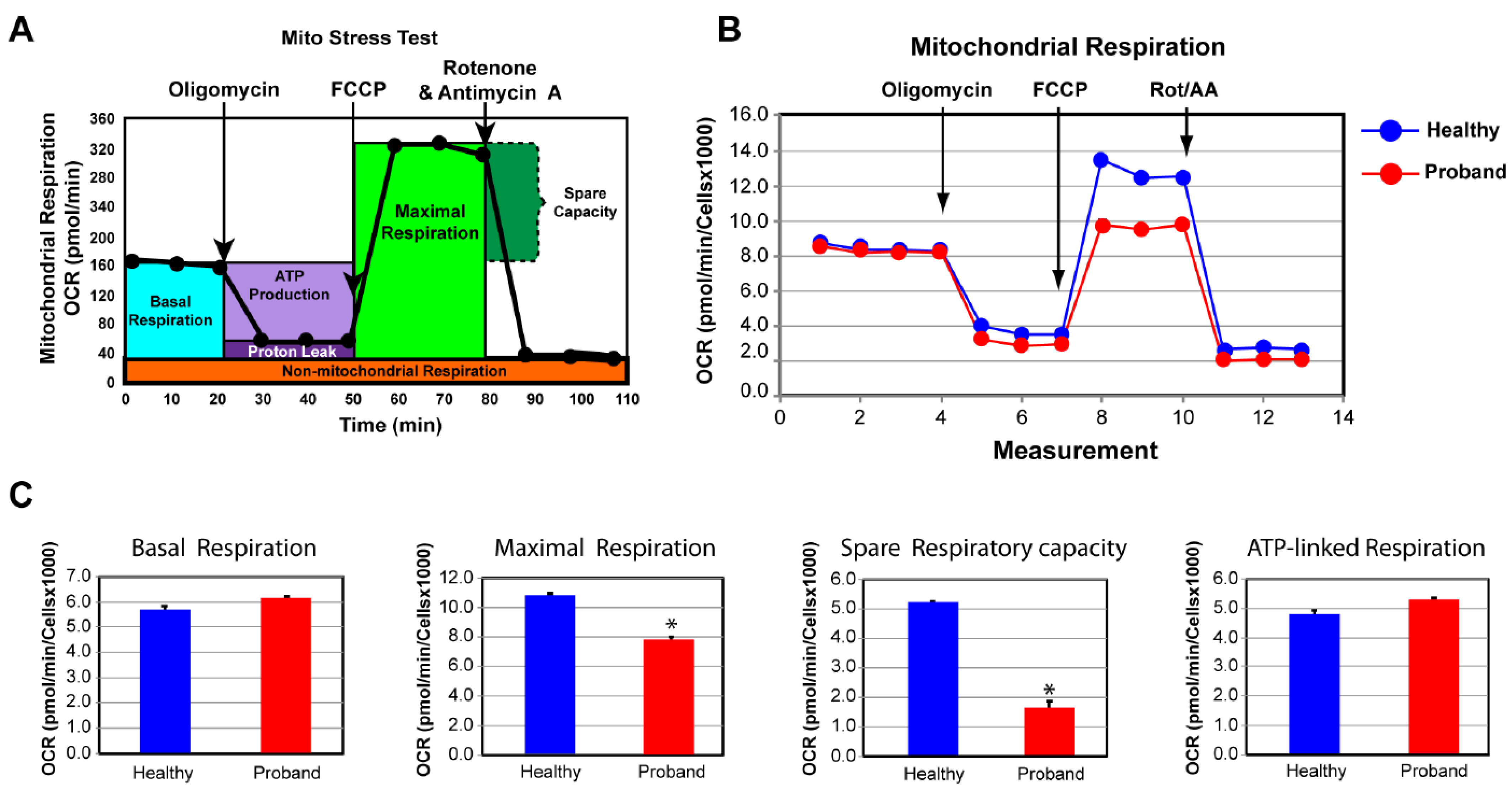{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Clinical History
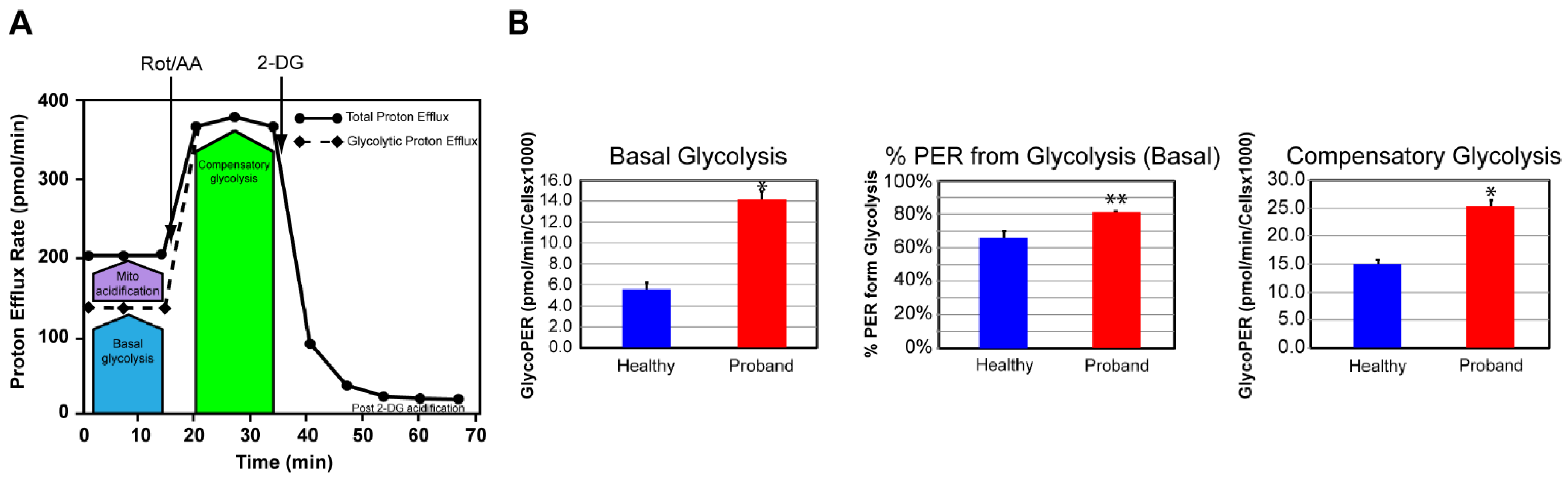
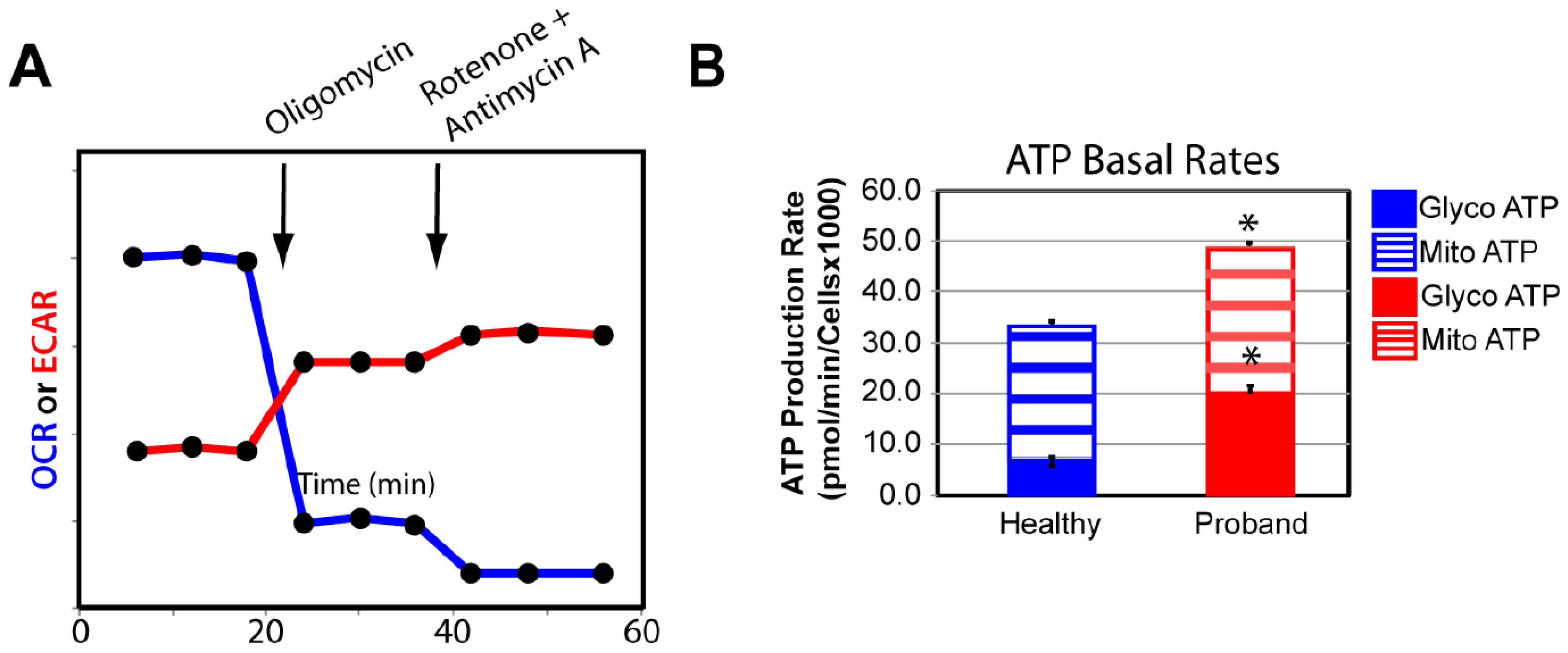
2.2. Functional Studies of the ATP Metabolism
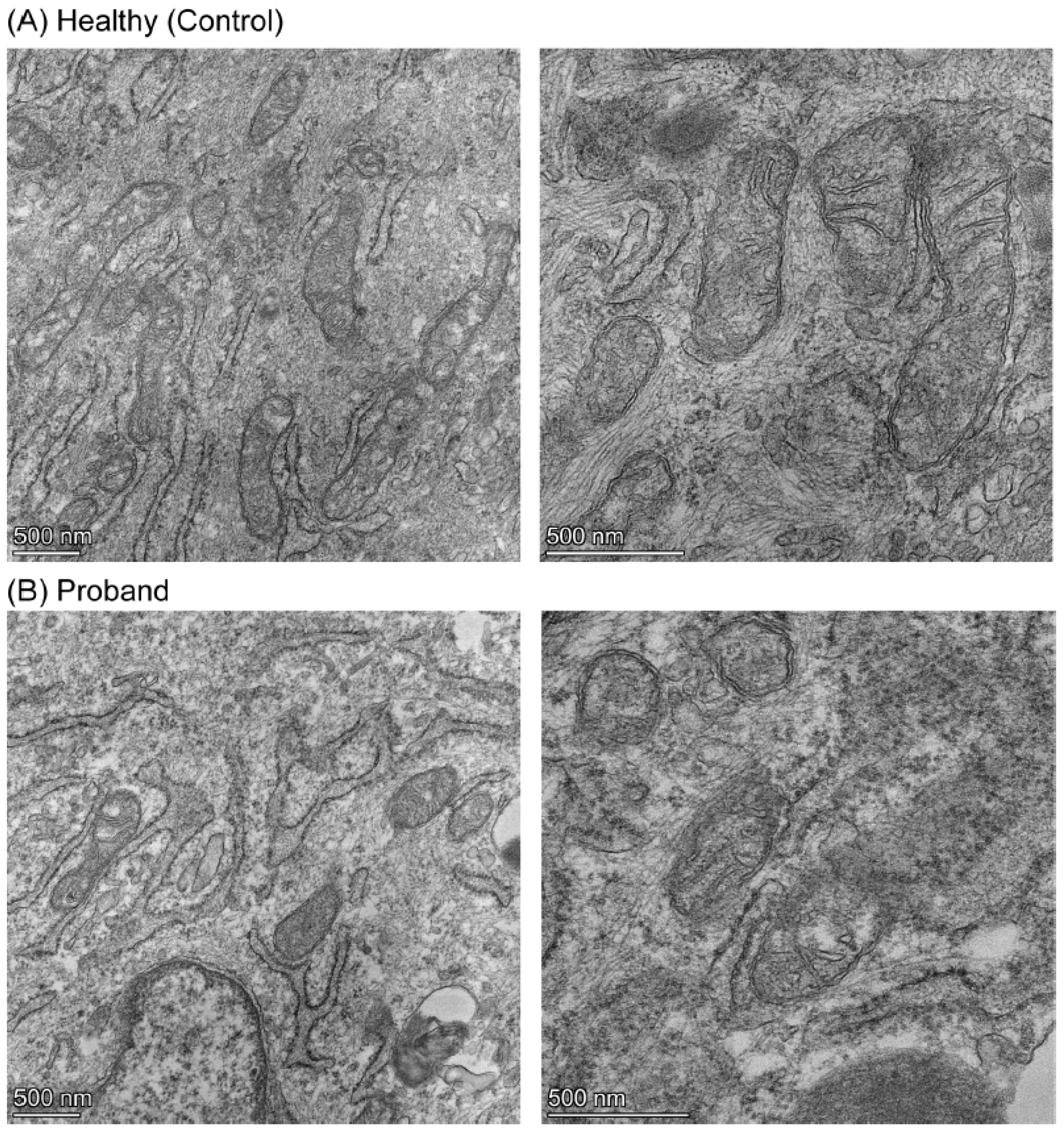
2.3. Mitochondrial Morphometric Examination of the Proband’s Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Editorial Policies and Ethical Considerations
4.2. Skin Biopsy and Fibroblast Culture
4.3. Clinical Genetic Diagnosis
4.4. Transmission Electron Microscopy
4.5. Live-Cell Measurements of Mitochondrial Respiratory and Glycolytic Activity
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Gordijn, S.J.; Beune, I.M.; Thilaganathan, B.; Papageorghiou, A.; Baschat, A.A.; Baker, P.N.; Silver, R.M.; Wynia, K.; Ganzevoort, W. Consensus definition of fetal growth restriction: A Delphi procedure. Ultrasound Obstet. Gynecol. 2016, 48, 333–339. [Google Scholar] [CrossRef] [PubMed]
- McCowan, L.M.; Figueras, F.; Anderson, N.H. Evidence-based national guidelines for the management of suspected fetal growth restriction: Comparison, consensus, and controversy. Am. J. Obstet. Gynecol. 2018, 218, S855–S868. [Google Scholar] [CrossRef] [PubMed]
- Angelica, D.; Giovanni, O.; Vassilios, F. Physiopathology of intrauterine growth retardation: From classic data to metabolomics. J. Maternal-Fetal Neonatal. Med. 2012, 25, 13–18. [Google Scholar]
- Barker, J.P. Adult consequences of fetal growth restriction. Clin. Obstet. Gynecol. 2006, 49, 270–283. [Google Scholar] [CrossRef]
- Von Beckerath, A.K.; Kollman, M.; Rotky-Fast, C.; Karpf, E.; Lang, U.; Klaritsch, P. Perinatal complications and long-term neurodevelopmental outcome of infants with intrauterine growth restriction. Am. J. Obstet. Gynecol. 2013, 208, 130.e1–130.e6. [Google Scholar] [CrossRef]
- Sharma, D.; Shastri, S.; Sharma, P. Intrauterine growth restriction: Antenatal and postnatal aspects. Clin. Med. Insights Pediatr. 2016, 10, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Shastri, S.; Farahbakhsh, N.; Sharma, P. Intrauterine growth restriction—Part 1. J. Matern. Fetal Neonatal Med. 2016, 29, 3977–3987. [Google Scholar] [CrossRef]
- Suhag, A.; Berghella, V. Intrauterine growth restriction (IUGR): Etiology and diagnosis. Curr. Obstet. Gynecol. Rep. 2013, 2, 102–111. [Google Scholar] [CrossRef]
- Murthi, P. Plaacental homeobox genes and their role in regulating human fetal growth. Placenta 2014, 35, S46–S50. [Google Scholar] [CrossRef]
- Arslan, M.; Yazici, G.; Erdem, M.; Arslan, O.; Himmetoglu, O. Endothelin 1 and leptin in the pathophysiology of intrauterine growth restriction. Int. J. Gynaecol. Obstet. 2004, 84, 120–126. [Google Scholar] [CrossRef]
- Caliebe, J.; Broekman, S.; Boogaard, M.; Bosch, C.A.J.; Ruivenkamp, C.A.L.; Oostdijk, W.; Kant, S.G.; Binder, G.; Ranke, M.B.; Wit, J.M.; et al. IGF1, IGFR1R and SHOX mutation analysis in short children born small for gestational age and short children with normal birth size (idiopathic short stature). Horm. Res. Paediatr. 2012, 77, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, N.; Berghella, V. Non-placental causes of intrauterine growth restriction. Semin. Perinatol. 2008, 32, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Villar, J.; Belizán, J.M.; Spalding, J.; Klein, R.E. Postnatal growth of intrauterine growth retarded infants. Early Hum. Dev. 1982, 6, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Cosmi, E.; Fanelli, T.; Visentin, S.; Trevisanuto, D.; Zanardo, V. Consequences in infants that were intrauterine growth restricted. J. Pregnancy 2011, 2011, 364381. [Google Scholar] [CrossRef] [PubMed]
- Chernausek, S.D. Update: Consequences of abnormal fetal growth. J. Clin. Endocrinol. Metab. 2012, 97, 689–695. [Google Scholar] [CrossRef] [PubMed]
- De Bie, H.M.A.; Oostrom, K.J.; Delemarre-van de Waal, H.A. Brain development, intelligence and cognitive outcome in children born small for gestational age. Horm. Res. Paediatr. 2010, 73, 6–14. [Google Scholar] [CrossRef]
- Geva, R.; Eshel, R.; Leitner, Y.; Valevski, A.F.; Harel, S. Neuropsychological outcome of children with intrauterine growth restriction: A 9-year prospective study. Pediatrics 2006, 118, 91–101. [Google Scholar] [CrossRef]
- Samuelsen, G.B.; Packenberg, B.; Bogdanović, N.; Gundersen, H.J.G.; Larsen, J.F.; Graem, N.; Laursen, H. Severe cell reduction in the future brain cortex in human growth-restricted fetuses and infants. Am. J. Obstet. Gynecol. 2007, 197, e1–e56. [Google Scholar] [CrossRef]
- Dubois, J.; Benders, M.; Borradori-Tolsa, C.; Cachia, A.; Lazeyras, F.; Ha-Vinh Leuchter, R.; Sizonenko, S.V.; Warfield, S.K.; Mangin, J.F.; Hüppi, P.S. Primary cortical folding in the human newborn: An early marker of later functional development. Brain 2008, 131, 2028–2041. [Google Scholar] [CrossRef]
- Padilla, N.; Falcón, C.; Sanz-Cortés, M.; Figueras, F.; Bargallo, N.; Crispi, F.; Eixarch, E.; Arranz, A.; Botet, F.; Gratacós, E. Differential effects of intrauterine growth restriction on brain structure and development in preterm infants: A magnetic resonance imaging study. Brain Res. 2011, 1382, 98–108. [Google Scholar] [CrossRef]
- Ramenghi, L.A.; Martinelli, A.; De Carli, A.; Brusati, V.; Mandia, L.; Fumagalli, M.; Triulzi, F.; Mosca, F.; Cetin, I. Cerebral maturation in IUGR and appropriate for gestational age preterm babies. Reprod. Sci. 2011, 18, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Cetin, A.; Alvino, G. Intrauterine growth restriction: Implications for placental metabolism and transport. A review. Placenta 2009, 30, S77–S82. [Google Scholar] [CrossRef] [PubMed]
- Naha, R.; Anees, A.; Chakrabarty, S.; Naik, P.S.; Pandove, M.; Pandey, D.; Satyamoorthy, K. Placental mitochondrial DNA mutations and copy numbers in intrauterine growth restricted (IUGR) pregnancy. Mitochondrion 2020, 55, 85–94. [Google Scholar] [CrossRef]
- Pandey, D.; Yevale, A.; Naha, R.; Kuthethur, R.; Chakrabarty, S.; Satyamoorthy, K. Mitochondrial DNA copy number variation—A potential biomarker for early onset preeclampsia. Pregnancy Hypertens. 2021, 23, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Uittenbogaard, M.; Brantner, C.A.; Fang, Z.; Wong, L.J.; Gropman, A.; Chiaramello, A. Novel insights into the functional metabolic impact of an apparent de novo m.8993T>G variant in the MT-ATP6 gene associated with maternally inherited form of Leigh syndrome. Mol. Genet. Metab. 2018, 124, 71–81. [Google Scholar] [CrossRef]
- Mookerjee, S.A.; Goncalves, R.L.S.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. The contributions of respiratory and glycolysis to extracellular acid production. Biochem. Biophys. Acta 2015, 1847, 171–181. [Google Scholar] [PubMed]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial cristae: Where beauty meets functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef]
- Côté, A.; Fanous, A.; Almajed, A.; Lacroix, Y. Pierre-Robin sequence: Review of diagnostic and treatment challenges. Int. J. Pediatr. Otorhinolaryngol. 2015, 79, 451–464. [Google Scholar] [CrossRef]
- Gagnon, R. Placental insufficiency and its consequences. Eur. J. Obstet. Gynecol. 2003, 110, S99–S107. [Google Scholar] [CrossRef]
- Miller, S.L.; Huppi, P.S.; Mallard, C. The consequences of fetal growth restriction on brain structure and neurodevelopmental outcome. J. Physiol. 2016, 594, 807–823. [Google Scholar] [CrossRef]
- Pendleton, A.L.; Wesolowski, S.R.; Regnault, T.R.H.; Lynch, R.M.; Limesand, S.W. Dimming the powerhouse: Mitochondrial dysfunction in the liver and skeletal muscle of intrauterine growth restricted fetuses. Front. Endocrinol. 2021, 12, 612888. [Google Scholar] [CrossRef] [PubMed]
- Pardi, G.; Marconi, A.M.; Cetin, I. Placental-fetal interrelationships in IUGR fetuses—A review. Placenta 2002, 23, S136–S141. [Google Scholar] [CrossRef] [PubMed]
- Erecinska, M.; Cherian, S.; Silver, I.A. Energy metabolism in mammalian brain during development. Prog. Neurobiol. 2004, 73, 397–445. [Google Scholar] [CrossRef] [PubMed]
- Uittenbogaard, M.; Chiaramello, A. Mitochondrial biogenesis: A therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. Curr. Pharm. Des. 2014, 20, 5574–5593. [Google Scholar] [CrossRef] [PubMed]
- Salmeri, N.; Carbone, I.M.; Cavoretto, P.I.; Farina, A.; Morano, D. Epigenetics beyond fetal growth restriction: A comprehensive overview. Mol. Diagn. Therapy 2022, 26, 607–626. [Google Scholar] [CrossRef]
- Uittenbogaard, M.; Brantner, C.A.; Chiaramello, A. Epigenetic modifiers promote mitochondrial biogenesis and oxidative metabolism leading to enhanced differentiation of neuroprogenitor cells. Cell Death Dis. 2018, 9, 360. [Google Scholar] [CrossRef]
- Nicholls, D.G. Spare respiratory capacity oxidative stress excitotoxicity. Biochem. Soc. Trans. 2009, 37, 1385–1388. [Google Scholar] [CrossRef]
- Gropman, A.; Uittenbogaard, M.; Brantner, C.A.; Wang, Y.; Wong, L.J.; Chiaramello, A. Molecular genetic and mitochondrial metabolic analyses confirm the suspected mitochondrial etiology in a pediatric patient with an atypical form of alternating hemiplegia of childhood. Mol. Genet. Metab. Rep. 2020, 24, 100609. [Google Scholar] [CrossRef]
- Facucho-Oliveira, J.M.; St John, J.C. The relationship between pluripotency and mitochondrial DNA proliferation during early embryo development and embryonic stem cell differentiation. Stem Cell Rev. Rep. 2009, 5, 140158. [Google Scholar] [CrossRef]
- Anh, C.S.; Metallo, C.M. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015, 3, 1. [Google Scholar] [CrossRef]
- Gardner, D.K.; Wale, P.L. Analysis of metabolism to select viable human embryos for transfer. Fertil. Steril. 2013, 99, 1062–1072. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Dzeja, P.P.; Faustino, R.S.; Perz-Terzic, C.; Behfar, A.; Terzic, A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat. Clin. Pract. Cardovasc. Med. 2007, 4, S60–S67. [Google Scholar] [CrossRef]
- Harvey, A.J. Mitochondria in early developmemt: Linking the microenvironment, metabolism and the epigenome. Reproduction 2019, 157, R159–R179. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Cano, A.; Calzada-Mendoza, C.C.; Estrada-Gutierrez, G.; Mendoza-Ortega, J.A.; Perichart-Perera, O. Nutrients, mitochondrial function, and perinatal health. Nutrients 2020, 12, 2166. [Google Scholar] [CrossRef] [PubMed]
- Hay, W.W.; Myers, S.A.; Sparks, J.W.; Wilkening, R.B.; Meschia, G.; Battaglia, F.C. Glucose and lactate oxidation rates in the fetal lamb. Proc. Soc. Exp. Biol. Med. 1983, 173, 553–563. [Google Scholar] [CrossRef]
- Thorn, S.R.; Brown, L.D.; Rozance, P.J.; Hay, W.W.; Friedman, J.E. Hepatic glucose production in fetal sheep with intrauterine growth restriction is not suppressed by insulin. Diabetis 2013, 62, 65–73. [Google Scholar] [CrossRef]
- Uittenbogaard, M.; Wang, H.; Zhang, V.W.; Wong, L.J.; Brantner, C.A.; Gropman, A.; Chiaramello, A. The nuclear background influences the penetrance of the near-homoplasmic m.1630A>G MELAS variant in a symptomatic proband and asymptomatic mother. Mol. Genet. Metab. 2019, 126, 429–438. [Google Scholar] [CrossRef]
- Uittenbogaard, M.; Gropman, A.; Brantner, C.A.; Chiaramello, A. Novel metabolic signatures of compound heterozygous Szt2 variants in a case of early-onset of epileptic encephalopathy. Clin. Case Rep. 2018, 6, 2376. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uittenbogaard, M.; Gropman, A.L.; Whitehead, M.T.; Brantner, C.A.; Gropman, E.; Chiaramello, A. Dysfunctional Postnatal Mitochondrial Energy Metabolism in a Patient with Neurodevelopmental Defects Caused by Intrauterine Growth Restriction Due to Idiopathic Placental Insufficiency. Int. J. Mol. Sci. 2024, 25, 1386. https://doi.org/10.3390/ijms25031386
Uittenbogaard M, Gropman AL, Whitehead MT, Brantner CA, Gropman E, Chiaramello A. Dysfunctional Postnatal Mitochondrial Energy Metabolism in a Patient with Neurodevelopmental Defects Caused by Intrauterine Growth Restriction Due to Idiopathic Placental Insufficiency. International Journal of Molecular Sciences. 2024; 25(3):1386. https://doi.org/10.3390/ijms25031386
Chicago/Turabian StyleUittenbogaard, Martine, Andrea L. Gropman, Matthew T. Whitehead, Christine A. Brantner, Eliana Gropman, and Anne Chiaramello. 2024. "Dysfunctional Postnatal Mitochondrial Energy Metabolism in a Patient with Neurodevelopmental Defects Caused by Intrauterine Growth Restriction Due to Idiopathic Placental Insufficiency" International Journal of Molecular Sciences 25, no. 3: 1386. https://doi.org/10.3390/ijms25031386