

A Review of Numerical Models of Radiation Injury and Repair Considering Subcellular Targets and the Extracellular Microenvironment
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Background
2.1. Space Radiation Environment
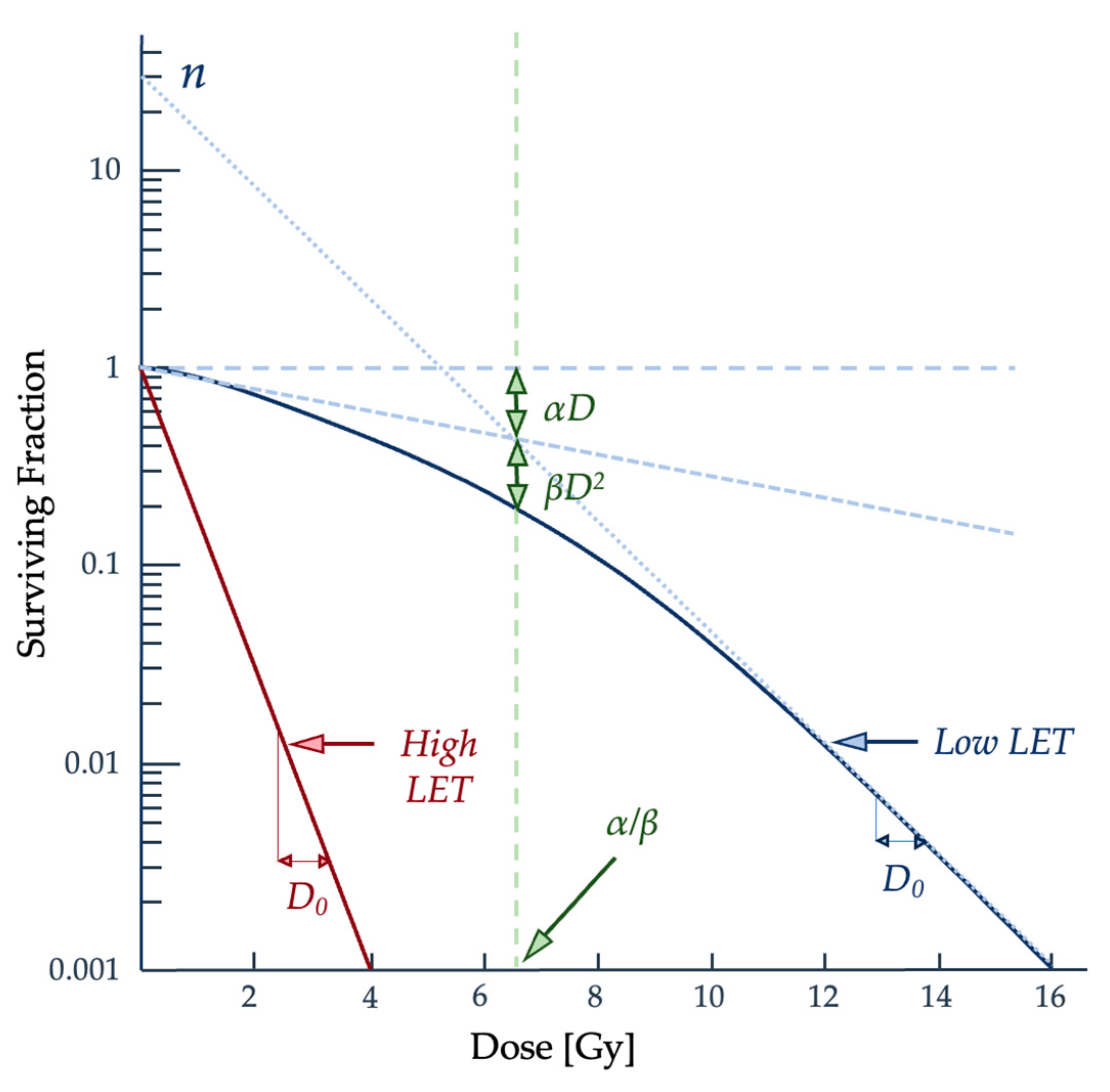
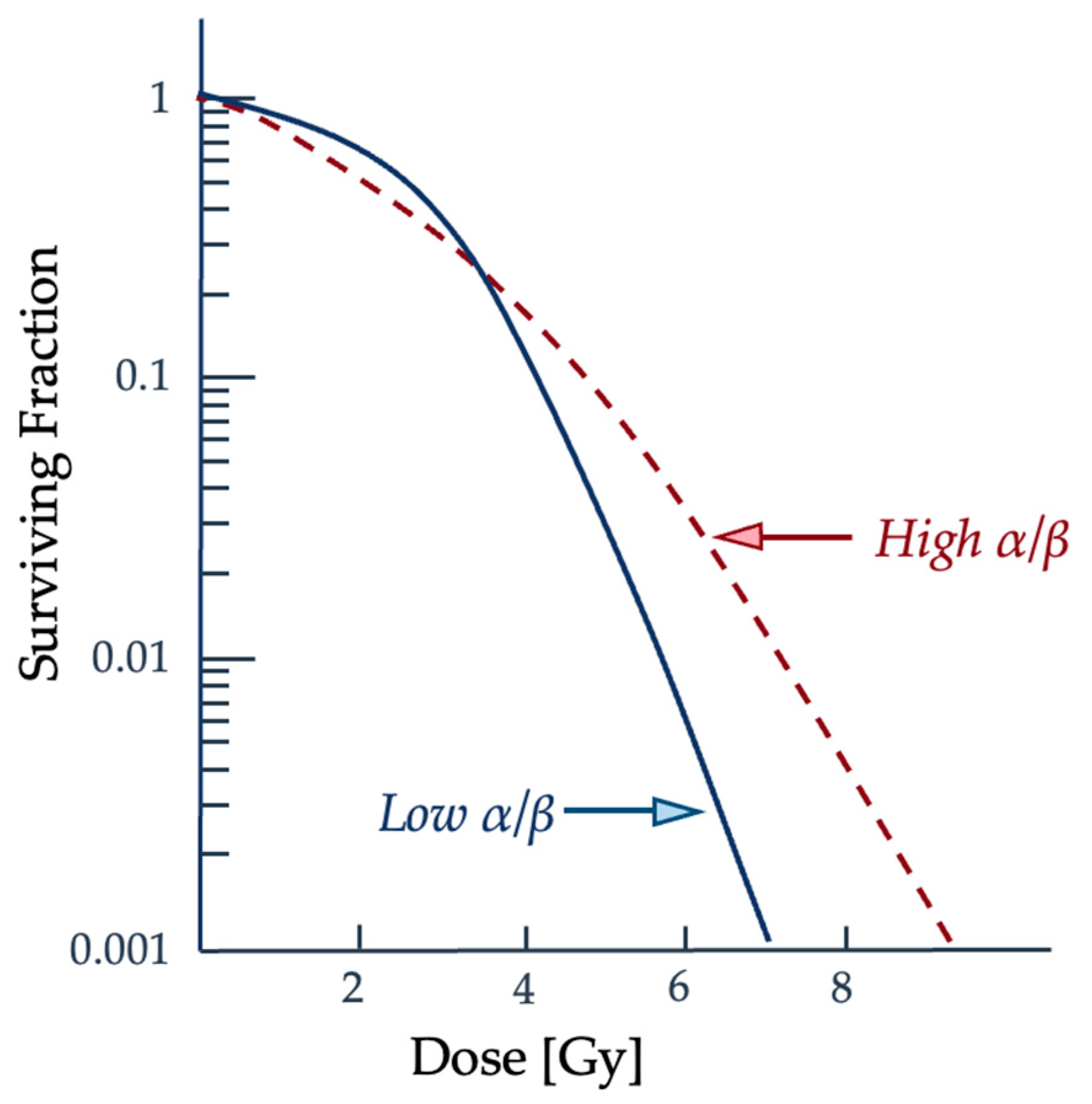
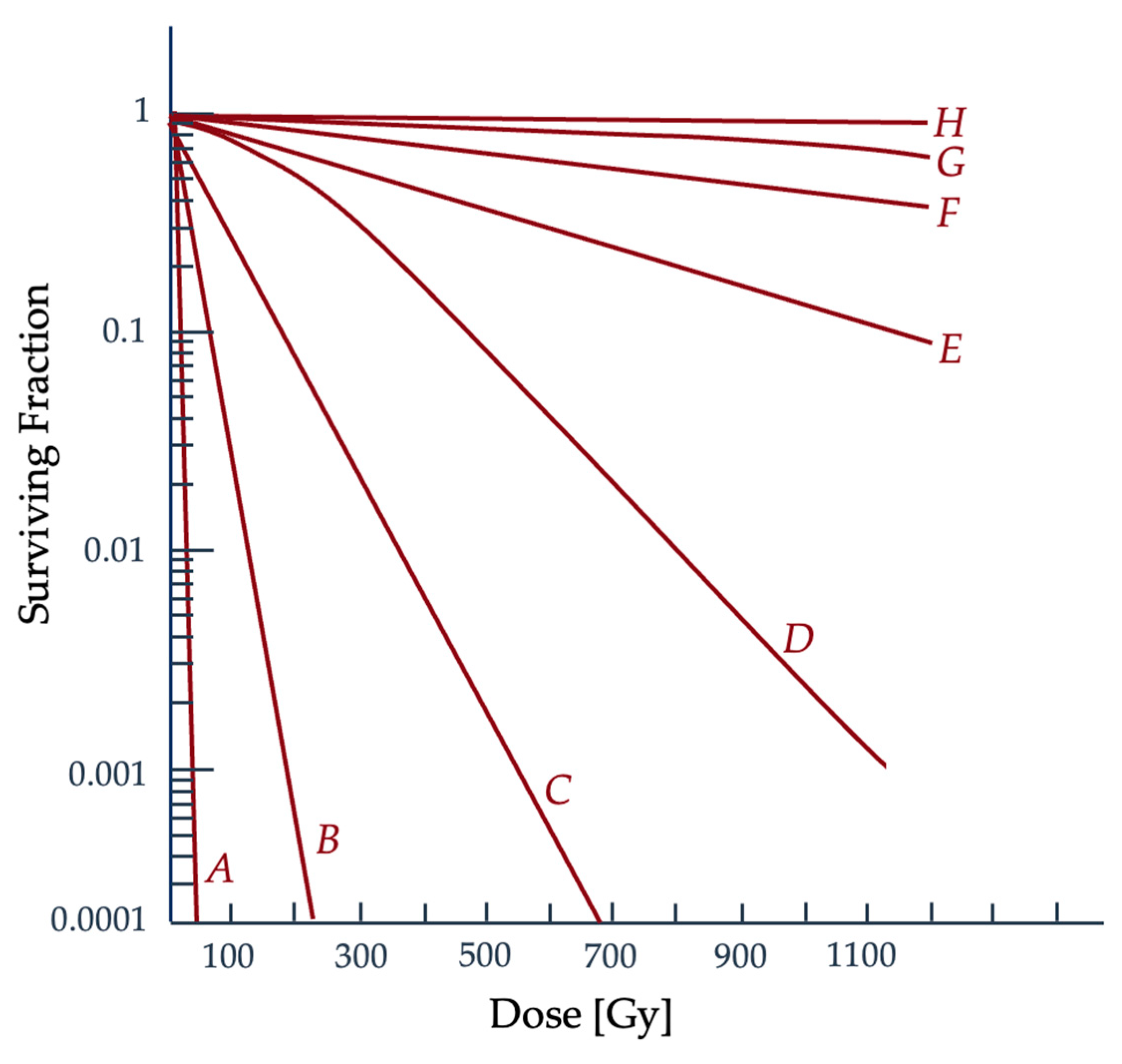
2.2. Quantifying Radiation Damage
2.3. Radiobiological Numerical Models
2.4. DNA Repair Mechanisms
2.5. Mitochondrial DNA
2.6. Epigenetics
3. Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chancellor, J.C.; Blue, R.S.; Cengel, K.A.; Auñón-Chancellor, S.M.; Rubins, K.H.; Katzgraber, H.G.; Kennedy, A.R. Limitations in predicting the space radiation health risk for exploration astronauts. Npj Microgravity 2018, 4, 8. [Google Scholar] [CrossRef]
- Hall, E.; Giaccia, A. Radiobiology for the Radiologist, 7th ed.; Wolters Kluwer Health: Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012; Volume 2, pp. 432–447. [Google Scholar]
- Warters, R.L.; Hofer, K.G.; Harris, C.R.; Smith, J.M. Radionuclide toxicity in cultured mammalian cells: Elucidation of the primary site of radiation damage. Curr. Top. Radiat. Res. Q. 1978, 12, 389–407. [Google Scholar] [PubMed]
- Okada, S.; Murakami, K.; Incerti, S.; Amako, K.; Sasaki, T. MPEXS-DNA, a new GPU-based Monte Carlo simulator for track structures and radiation chemistry at sub-cellular scale. Med. Phys. 2019, 46, 1483–1500. [Google Scholar] [CrossRef]
- Nomiya, T. Discussions on target theory: Past and present. J. Radiat. Res. 2013, 54, 1161–1163. [Google Scholar] [CrossRef]
- Williams, J.P.; Newhauser, W. Normal tissue damage: Its importance, history and challenges for the future. Br. J. Radiol. 2018, 92, 20180048. [Google Scholar] [CrossRef] [PubMed]
- Townsend, L.W.; Badhwar, G.D.; Braby, L.A.; Blakely, E.A.; Cucinotta, F.A.; Curtis, D.S.B.; Fry, R.J.M.; Land, C.E.; Smart, D.F. Report No. 153—Information Needed to Make Radiation Protection Recommendations for Space Missions beyond Low-Earth Orbit; US National Council for Radiation Protection and Measurements: Bethesda, MD, USA, 2006. [Google Scholar]
- National Council on Radiation Protection and Measurements. NCRP Report 141–142; National Council on Radiation Protection and Measurements: Bethesda, MD, USA, 2002. [Google Scholar]
- Chancellor, J.; Nowadly, C.; Williams, J.; Aunon-Chancellor, S.; Chesal, M.; Looper, J.; Newhauser, W. Everything you wanted to know about space radiation but were afraid to ask. J. Environ. Sci. Health C 2021, 39, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.R. Biological effects of space radiation and development of effective countermeasures. Life Sci. Space Res. 2014, 1, 10–43. [Google Scholar] [CrossRef]
- Rackwitz, T.; Debus, J. Clinical applications of proton and carbon ion therapy. Semin. Oncol. 2019, 46, 226–232. [Google Scholar] [CrossRef]
- Joiner, M.C.; van der Kogel, A.J. Basic Clinical Radiobiology; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Brown, A.; Suit, H. The centenary of the discovery of the Bragg peak. Radiother. Oncol. 2004, 73, 265–268. [Google Scholar] [CrossRef]
- Wilson, R.R. Radiological use of fast protons. Radiology 1946, 47, 487–491. [Google Scholar]
- Lehnert, B.; Iyer, R. Exposure to low-level chemicals and ionizing radiation: Reactive oxygen species and cellular pathways. Hum. Exp. Toxicol. 2002, 21, 65–69. [Google Scholar] [CrossRef]
- Schardt, D.; Elsässer, T.; Schulz-Ertner, D. Heavy-ion tumor therapy: Physical and radiobiological benefits. Rev. Mod. Phys. 2010, 82, 383–425. [Google Scholar] [CrossRef]
- Nikjoo, H.; Goodhead, D.T. Track structure analysis illustrating the prominent role of low-energy electrons in radiobiological effects of low-LET radiations. Phys. Med. Biol. 1991, 36, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Edington, C.W.; Randolph, M.L. A comparison of the relative effectiveness of radiations of different average linear energy transfer on the induction of dominant and recessive lethals in Drosophila. Genetics 1958, 43, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, K.H.; Leenhouts, H.P. A molecular theory of cell survival. Phys. Med. Biol. 1973, 18, 78–87. [Google Scholar] [CrossRef]
- Dale, R.G.; Jones, B. The assessment of RBE effects using the concept of biologically effective dose. Int. J. Radiat. Oncol. 1999, 43, 639–645. [Google Scholar] [CrossRef]
- Hunter, N.; Muirhead, C.R. Review of relative biological effectiveness dependence on linear energy transfer for low-LET radia-tions. J. Radiol. Prot. 2009, 29, 5. [Google Scholar] [CrossRef]
- DeLuca, P.M.; Wambersie, A.; Seltzer, S.M. International Commission on Radiation Units & Measurements, Prescribing. Recording, and Reporting Proton-Beam Therapy (ICRU Report 78). J. ICRU 2007, 7, 29–48. [Google Scholar]
- Okayasu, R. Repair of DNA damage induced by accelerated heavy ions—A mini review. Int. J. Cancer 2011, 130, 991–1000. [Google Scholar] [CrossRef]
- Hawkins, R.B. A microdosimetric-kinetic theory of the dependence of the RBE for cell death on LET. Med. Phys. 1998, 25, 1157–1170. [Google Scholar] [CrossRef]
- Sørensen, B.S.; Overgaard, J.; Bassler, N. In vitro RBE-LET dependence for multiple particle types. Acta Oncol. 2011, 50, 757–762. [Google Scholar] [CrossRef]
- Barendsen, G. The relationships between RBE and LET for different types of lethal damage in mammalian cells: Biophysical and mo-lecular mechanisms. Radiat. Res. 1994, 139, 257–270. [Google Scholar] [CrossRef]
- Kellerer, A.M.; Rossi, H.H. RBE and the primary mechanism of radiation action. Radiat. Res. 1971, 47, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, T.; Weyrather, W.; Elsässer, T.; Durante, M.; Scholz, M. Accuracy of RBE: Experimental and theoretical considerations. Radiat. Environ. Biophys. 2010, 49, 345–349. [Google Scholar] [CrossRef]
- Jones, B. Why RBE must be a variable and not a constant in proton therapy. Br. J. Radiol. 2016, 89, 20160116. [Google Scholar] [CrossRef] [PubMed]
- Cucinotta, F.A.; Kim, M.-H.Y.; Willingham, V.; George, K.A. Physical and Biological Organ Dosimetry Analysis for International Space Station Astronauts. Radiat. Res. 2008, 170, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Karger, C.P.; Glowa, C.; Peschke, P.; Kraft-Weyrather, W. The RBE in ion beam radiotherapy: In vivo studies and clinical application. Z. Für Med. Phys. 2021, 31, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Bodgi, L.; Canet, A.; Pujo-Menjouet, L.; Lesne, A.; Victor, J.-M.; Foray, N. Mathematical models of radiation action on living cells: From the target theory to the modern approaches. A historical and critical review. J. Theor. Biol. 2016, 394, 93–101. [Google Scholar] [CrossRef]
- Osborne, J.C., Jr.; Miller, J.H.; Kempner, E. Molecular mass and volume in radiation target theory. Biophys. J. 2000, 78, 1698–1702. [Google Scholar] [CrossRef]
- Crowther, J.A. The Biological Action of X Rays—A Theoretical Review. Br. J. Radiol. 1938, 11, 132–145. [Google Scholar] [CrossRef]
- Lea, D.E. Actions of Radiaitons on Living Cells, 2nd ed.; Cambridge University Press: Cambridge, UK, 1955. [Google Scholar]
- McMahon, S.J. The linear quadratic model: Usage, interpretation and challenges. Phys. Med. Biol. 2018, 64, 01TR01. [Google Scholar] [CrossRef]
- Hall, E.J.; Gross, W.; Dvorak, R.F.; Kellerer, A.M.; Rossi, H.H. Survival curves and age response functions for Chinese hamster cells exposed to X-rays or high LET alpha-particles. Radiat. Res. 1972, 52, 88–98. [Google Scholar] [CrossRef] [PubMed]
- López-Sáez, J.F.; de la Torre, C.; Pincheira, J.; Giménez-Martín, G. Cell proliferation and cancer. Histol. Histopathol. 1998, 13, 1197–1214. [Google Scholar] [PubMed]
- Alpen, E.L. Radiation Biophysics; Academic Press: Cambridge, MA, USA, 1997. [Google Scholar]
- Curtis, S.B. Mechanistic models. In Physical and Chemical Mechanisms in Molecular Radiation Biology; Springer: Berlin/Heidelberg, Germany, 1991; pp. 367–386. [Google Scholar]
- Wheldon, T.E.; Deehan, C.; Wheldon, E.G.; Barrett, A. The linear-quadratic transformation of dose–volume histograms in fractionated radiotherapy. Radiother. Oncol. 1998, 46, 285–295. [Google Scholar] [CrossRef]
- Stewart, R.D. Two-lesion kinetic model of double-strand break rejoining and cell killing. Radiat. Res. 2001, 156, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.D.; Streitmatter, S.W.; Argento, D.C.; Kirkby, C.; Goorley, J.T.; Moffitt, G.; Jevremovic, T.; Sandison, G.A. Rapid MCNP simulation of DNA double strand break (DSB) relative biological effectiveness (RBE) for photons, neutrons, and light ions. Phys. Med. Biol. 2015, 60, 8249–8274. [Google Scholar] [CrossRef]
- Bellinzona, V.E.; Cordoni, F.; Missiaggia, M.; Tommasino, F.; Scifoni, E.; La Tessa, C.; Attili, A. Linking microdosimetric measurements to biological effectiveness in ion beam therapy: A review of theoretical aspects of mkm and other models. Front. Phys. 2021, 8, 578492. [Google Scholar] [CrossRef]
- Carlson, D.J.; Stewart, R.D.; Semenenko, V.A.; Sandison, G.A. Combined use of Monte Carlo DNA damage simulations and deterministic repair models to examine putative mechanisms of cell killing. Radiat. Res. 2008, 169, 447–459. [Google Scholar] [CrossRef]
- Stewart, R.D.; Yu, V.K.; Georgakilas, A.G.; Koumenis, C.; Park, J.H.; Carlson, D.J. Effects of radiation quality and oxygen on clustered DNA lesions and cell death. Radiat. Res. 2011, 176, 587–602. [Google Scholar] [CrossRef]
- Puck, T.T.; Morkovin, D.; Marcus, P.I.; Cieciura, S.J. Action of X-rays on mammalian cells. J. Exp. Med. 1957, 106, 485–500. [Google Scholar] [CrossRef]
- Sapp, J. The prokaryote-eukaryote dichotomy: Meanings and mythology. Microbiol. Mol. Biol. Rev. 2005, 69, 292–305. [Google Scholar] [CrossRef]
- Krisko, A.; Radman, M. Protein damage and death by radiation in Escherichia coli and Deinococcus radiodurans. Proc. Natl. Acad. Sci. USA 2010, 107, 14373–14377. [Google Scholar] [CrossRef] [PubMed]
- Pitchiaya, S.; Krishnan, Y. First blueprint, now bricks: DNA as construction material on the nanoscale. Chem. Soc. Rev. 2006, 35, 1111–1121. [Google Scholar] [CrossRef]
- Sobol, R.W.; Watson, D.E.; Nakamura, J.; Yakes, F.M.; Hou, E.; Horton, J.K.; Ladapo, J.; Van Houten, B.; Swenberg, J.A.; Tindall, K.R.; et al. Mutations associated with base excision repair deficiency and methylation-induced genotoxic stress. Proc. Natl. Acad. Sci. USA 2002, 99, 6860–6865. [Google Scholar] [CrossRef]
- Markkanen, E.; Fischer, R.; Ledentcova, M.; Dianov, G.L. Cells deficient in base-excision repair reveal cancer hallmarks originating from adjustments to genetic instability. Nucleic Acids Res. 2015, 43, 3667–3679. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Dutta, A.; Chakraborty, A. External modulators and redox homeostasis: Scenario in radiation-induced by-stander cells. Mutat. Res. Rev. Mutat. Res. 2021, 787, 108368. [Google Scholar] [CrossRef] [PubMed]
- Nikitaki, Z.; Velalopoulou, A.; Zanni, V.; Tremi, I.; Havaki, S.; Kokkoris, M.; Gorgoulis, V.G.; Koumenis, C.; Georgakilas, A.G. Key biological mechanisms involved in high-LET radiation therapies with a focus on DNA damage and repair. Expert Rev. Mol. Med. 2022, 24, e15. [Google Scholar] [CrossRef] [PubMed]
- Pouget, J.-P.; Mather, S.J. General aspects of the cellular response to low- and high-LET radiation. Eur. J. Nucl. Med. 2001, 28, 541–561. [Google Scholar] [CrossRef]
- Hegde, M.L.; Hazra, T.K.; Mitra, S. Early steps in the DNA base excision/single-strand interruption repair pathway in mam-malian cells. Cell Res. 2008, 18, 27–47. [Google Scholar] [CrossRef]
- Coppedè, F. An overview of DNA repair in amyotrophic lateral sclerosis. Sci. World J. 2011, 11, 1679–1691. [Google Scholar] [CrossRef]
- Le May, N.; Mota-Fernandes, D.; Vélez-Cruz, R.; Iltis, I.; Biard, D.; Egly, J.M. NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol. Cell 2010, 38, 54–66. [Google Scholar] [CrossRef]
- Tsao, D.; Kalogerinis, P.; Tabrizi, I.; Dingfelder, M.; Stewart, R.D.; Georgakilas, A.G. Induction and processing of oxidative clustered DNA lesions in 56Fe-ion-irradiated human monocytes. Radiat. Res. 2007, 168, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Limoli, C.; Ponnaiya, B.; Corcoran, J.; Giedzinski, E.; Kaplan, M.; Hartmann, A.; Morgan, W. Genomic instability induced by high and low let ionizing radiation. Adv. Space Res. 2000, 25, 2107–2117. [Google Scholar] [CrossRef] [PubMed]
- Mavragani, I.V.; Nikitaki, Z.; Kalospyros, S.A.; Georgakilas, A.G. Ionizing radiation and complex DNA damage: From prediction to detection challenges and biological signifi-cance. Cancers 2019, 11, 1789. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Bao, C.; Shang, Y.; He, X.; Ma, C.; Lei, X.; Mi, D.; Sun, Y. The determinant of DNA repair pathway choices in ionising radiation-induced DNA double-strand breaks. BioMed Res. Int. 2020, 2020, 4834965. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. DNA single-strand break repair and human genetic disease. Trends Cell Biol. 2022, 32, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Iyama, T.; Wilson, D.M., III. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 2013, 12, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Tian, B.; Liu, L.; Sheng, D.; Shen, B.; Hua, Y. Expression of Deinococcus radiodurans PprI enhances the radioresistance of Escherichia coli. DNA Repair 2003, 2, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.R.; Li, M.; Chen, W.; Liu, T.; de Toledo, S.M.; Pandey, B.N.; Li, H.; Rabin, B.M.; Azzam, E.I. In vivo space radiation-induced non-targeted responses: Late effects on molecular signaling in mitochondria. Curr. Mol. Pharmacol. 2011, 4, 106–114. [Google Scholar] [CrossRef]
- Atig, R.K.-B.; Hsouna, S.; Beraud-Colomb, E.; Abdelhak, S. [Mitochondrial DNA: Properties and applications]. Arch. Inst. Pasteur Tunis 2009, 86, 3–14. [Google Scholar]
- Greaves, L.C.; Reeve, A.K.; Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA and disease. J. Pathol. 2012, 226, 274–286. [Google Scholar] [CrossRef]
- Averbeck, D.; Rodriguez-Lafrasse, C. Role of Mitochondria in Radiation Responses: Epigenetic, Metabolic, and Signaling Impacts. Int. J. Mol. Sci. 2021, 22, 11047. [Google Scholar] [CrossRef] [PubMed]
- Moraes, C.T.; Kenyon, L.; Hao, H. Mechanisms of Human Mitochondrial DNA Maintenance: The Determining Role of Primary Sequence and Length over Function. Mol. Biol. Cell 1999, 10, 3345–3356. [Google Scholar] [CrossRef]
- Filograna, R.; Mennuni, M.; Alsina, D.; Larsson, N.-G. Mitochondrial DNA copy number in human disease: The more the better? FEBS Lett. 2021, 595, 976–1002. [Google Scholar] [CrossRef]
- Stein, A.; Sia, E.A. Mitochondrial DNA repair and damage tolerance. Front. Biosci. Landmark 2017, 22, 920–943. [Google Scholar]
- Dahal, S.; Dubey, S.; Raghavan, S.C. Homologous recombination-mediated repair of DNA double-strand breaks operates in mammalian mitochondria. Cell. Mol. Life Sci. 2018, 75, 1641–1655. [Google Scholar] [CrossRef] [PubMed]
- McVey, M.; Lee, S.E. MMEJ repair of double-strand breaks (director’s cut): Deleted sequences and alternative endings. Trends Genet. 2008, 24, 529–538. [Google Scholar] [CrossRef]
- Moretton, A.; Morel, F.; Macao, B.; Lachaume, P.; Ishak, L.; Lefebvre, M.; Garreau-Balandier, I.; Vernet, P.; Falkenberg, M.; Farge, G. Selective mitochondrial DNA degradation following double-strand breaks. PLoS ONE 2017, 12, e01767952017. [Google Scholar] [CrossRef]
- Delp, M.D.; Charvat, J.M.; Limoli, C.L.; Globus, R.K.; Ghosh, P. Apollo lunar astronauts show higher cardiovascular disease mortality: Possible deep space radiation effects on the vascular endothelium. Sci. Rep. 2016, 6, 29901. [Google Scholar] [CrossRef]
- Garrett-Bakelman, F.E.; Darshi, M.; Green, S.J.; Gur, R.C.; Lin, L.; Macias, B.R.; McKenna, M.J.; Meydan, C.; Mishra, T.; Nasrini, J.; et al. The NASA Twins Study: A multidimensional analysis of a year-long human spaceflight. Science 2019, 364, eaau8650. [Google Scholar] [CrossRef]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. Embo Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Zinovkina, L.A. Mechanisms of mitochondrial DNA repair in mammals. Biochemistry 2018, 83, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Maremonti, E.; Brede, D.A.; Olsen, A.-K.; Eide, D.M.; Berg, E.S. Ionizing radiation, genotoxic stress, and mitochondrial DNA copy-number variation in Caenorhabditis elegans: Droplet digital PCR analysis. Mutat. Res. Toxicol. Environ. Mutagen. 2020, 858–860, 503277. [Google Scholar] [CrossRef] [PubMed]
- Rong, Z.; Tu, P.; Xu, P.; Sun, Y.; Yu, F.; Tu, N.; Guo, L.; Yang, Y. The mitochondrial response to DNA damage. Front. Cell Dev. Biol. 2021, 9, 669379. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Doublié, S. Base excision repair in the mitochondria. J. Cell. Biochem. 2015, 116, 1490–1499. [Google Scholar] [CrossRef] [PubMed]
- Yatagai, F.; Honma, M.; Dohmae, N.; Ishioka, N. Biological effects of space environmental factors: A possible interaction between space radiation and microgravity. Life Sci. Space Res. 2019, 20, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Koturbash, I.; Pogribny, I.; Kovalchuk, O. Stable loss of global DNA methylation in the radiation-target tissue—A possible mechanism contributing to radiation carcinogenesis? Biochem. Biophys. Res. Commun. 2005, 337, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Kalinich, J.F.; Catravas, G.N.; Snyder, S.L. The effect of γ radiation on DNA methylation. Radiat. Res. 1989, 117, 185–197. [Google Scholar] [CrossRef]
- Giotopoulos, G.; McCormick, C.; Cole, C.; Zanker, A.; Jawad, M.; Brown, R.; Plumb, M. DNA methylation during mouse hemopoietic differentiation and radiation-induced leukemia. Exp. Hematol. 2006, 34, 1462–1470. [Google Scholar] [CrossRef]
- Loree, J.; Koturbash, I.; Kutanzi, K.; Baker, M.; Pogribny, I.; Kovalchuk, O. Radiation-induced molecular changes in rat mammary tissue: Possible implications for radiation-induced carcinogenesis. Int. J. Radiat. Biol. 2006, 82, 805–815. [Google Scholar] [CrossRef]
- Kovalchuk, O.; Burke, P.; Besplug, J.; Slovack, M.; Filkowski, J.; Pogribny, I. Methylation changes in muscle and liver tissues of male and female mice exposed to acute and chronic low-dose X-ray-irradiation. Mutat. Res. Mol. Mech. Mutagen. 2004, 548, 75–84. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed]
- de Koning, A.P.J.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011, 7, e1002384. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Liang, G.; Molloy, P.L.; Jones, P.A. DNA methylation enables transposable element-driven genome expansion. Proc. Natl. Acad. Sci. USA 2020, 117, 19359–19366. [Google Scholar] [CrossRef] [PubMed]
- Miousse, I.R.; Chalbot, M.-C.G.; Lumen, A.; Ferguson, A.; Kavouras, I.G.; Koturbash, I. Response of transposable elements to environmental stressors. Mutat. Res. Mol. Mech. Mutagen. 2015, 765, 19–39. [Google Scholar] [CrossRef] [PubMed]
- Miousse, I.R.; Koturbash, I. The fine LINE: Methylation drawing the cancer landscape. BioMed Res. Int. 2015, 2015, 131547. [Google Scholar] [CrossRef]
- Xu, B.; Li, X.; Zhang, S.; Lian, M.; Huang, W.; Zhang, Y.; Wang, Y.; Huang, Z. Pan cancer characterization of genes whose expression has been associated with LINE-1 antisense promoter activity. Mob. DNA 2023, 14, 13. [Google Scholar] [CrossRef]
- Belinsky, S.A. Plutonium targets the p16 gene for inactivation by promoter hypermethylation in human lung adenocarcinoma. Carcinogenesis 2004, 25, 1063–1067. [Google Scholar] [CrossRef]
- Lyon, C.M.; Klinge, D.M.; Liechty, K.C.; Gentry, F.D.; March, T.H.; Kang, T.; Gilliland, F.D.; Adamova, G.; Rusinova, G.; Telnov, V.; et al. Radiation-induced lung adenocarcinoma is associated with increased frequency of genes inactivated by promoter hypermethylation. Radiat. Res. 2007, 168, 409–414. [Google Scholar] [CrossRef]
- Aypar, U.; Morgan, W.F.; Baulch, J.E. Radiation-induced epigenetic alterations after low and high LET irradiations. Mutat. Res. Mol. Mech. Mutagen. 2011, 707, 24–33. [Google Scholar] [CrossRef]
- Goetz, W.; Morgan, M.N.M.; Baulch, J.E. The effect of radiation quality on genomic DNA methylation profiles in irradiated human cell lines. Radiat. Res. 2011, 175, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Koturbash, I.; Miousse, I.R.; Sridharan, V.; Nzabarushimana, E.; Skinner, C.M.; Melnyk, S.B.; Pavliv, O.; Hauer-Jensen, M.; Nelson, G.A.; Boerma, M. Radiation-induced changes in DNA methylation of repetitive elements in the mouse heart. Mutat. Res. Mol. Mech. Mutagen. 2016, 787, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Lima, F.; Ding, D.; Goetz, W.; Yang, A.J.; Baulch, J.E. High LET 56Fe ion irradiation induces tissue-specific changes in DNA methylation in the mouse. Environ. Mol. Mutagen. 2014, 55, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Prior, S.; Miousse, I.R.; Nzabarushimana, E.; Pathak, R.; Skinner, C.; Kutanzi, K.R.; Allen, A.R.; Raber, J.; Tackett, A.J.; Hauer-Jensen, M.; et al. Densely ionizing radiation affects DNA methylation of selective LINE-1 elements. Environ. Res. 2016, 150, 470–481. [Google Scholar] [CrossRef]
- Miousse, I.R.; Shao, L.; Chang, J.; Feng, W.; Wang, Y.; Allen, A.R.; Turner, J.; Stewart, B.; Raber, J.; Zhou, D.; et al. Exposure to low-dose 56Fe-ion radiation induces long-term epigenetic alterations in mouse bone marrow hematopoietic progenitor and stem cells. Radiat. Res. 2014, 182, 92–101. [Google Scholar] [CrossRef]
- Nzabarushimana, E.; Miousse, I.R.; Shao, L.; Chang, J.; Allen, A.R.; Turner, J.; Stewart, B.; Raber, J.; Koturbash, I. Long-term epigenetic effects of exposure to low doses of 56Fe in the mouse lung. J. Radiat. Res. 2014, 55, 823–828. [Google Scholar] [CrossRef]
- Miousse, I.R.; Skinner, C.M.; Sridharan, V.; Seawright, J.W.; Singh, P.; Landes, R.D.; Cheema, A.K.; Hauer-Jensen, M.; Boerma, M.; Koturbash, I. Changes in one-carbon metabolism and DNA methylation in the hearts of mice exposed to space environ-ment-relevant doses of oxygen ions (16O). Life Sci. Space Res. 2019, 22, 8–15. [Google Scholar] [CrossRef]
- Koturbash, I. LINE-1 in response to exposure to ionizing radiation. Mob. Genet. Elem. 2017, 7, 470–481. [Google Scholar] [CrossRef]
- Ohtani, H.; Liu, M.; Zhou, W.; Liang, G.; Jones, P.A. Switching roles for DNA and histone methylation depend on evolutionary ages of human endogenous retroviruses. Genome Res. 2018, 28, 1147–1157. [Google Scholar] [CrossRef]
- Ewing, L.E.; Pathak, R.; Landes, R.D.; Skinner, C.M.; Binz, R.; Young, S.G.; Riklon, S.; Stahr, S.; Su, J.; Boerma, M.; et al. Cytogenetic and epigenetic aberrations in peripheral lymphocytes of northwest Arkansas Marshallese. Int. J. Radiat. Biol. 2023, 99, 644–655. [Google Scholar] [CrossRef]
- Redon, C.E.; Dickey, J.S.; Bonner, W.M.; Sedelnikova, O.A. γ-H2AX as a biomarker of DNA damage induced by ionizing radiation in human peripheral blood lymphocytes and artificial skin. Adv. Space Res. 2009, 43, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Pogribny, I.; Koturbash, I.; Tryndyak, V.; Hudson, D.; Stevenson, S.M.; Sedelnikova, O.; Bonner, W.; Kovalchuk, O. Fractionated low-dose radiation exposure leads to accumulation of DNA damage and profound alterations in DNA and histone meth-ylation in the murine thymus. Mol. Cancer Res. 2005, 3, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Höckel, M.; Vaupel, P. Tumor Hypoxia: Definitions and Current Clinical, Biologic, and Molecular Aspects. JNCI J. Natl. Cancer Inst. 2001, 93, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Somosy, Z. Radiation response of cell organelles. Micron 2000, 31, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Budinger, G.S. The cellular basis for diverse responses to oxygen. Free. Radic. Biol. Med. 2007, 42, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Kouthouridis, S.; Goepp, J.; Martini, C.; Matthes, E.; Hanrahan, J.W.; Moraes, C. Oxygenation as a driving factor in epithelial differentiation at the air–liquid interface. Integr. Biol. 2021, 13, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Weissman, L.; Jo, D.-G.; Sørensen, M.M.; de Souza-Pinto, N.C.; Markesbery, W.R.; Mattson, M.P.; Bohr, V.A. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007, 35, 5545–5555. [Google Scholar] [CrossRef] [PubMed]
- Cucinotta, F.A. Once we know all the radiobiology we need to know, how can we use it to predict space radiation risks and achieve fame and fortune? Phys. Medica 2001, 17 (Suppl. S1), 5–12. [Google Scholar]
- Chancellor, J.C.; Scott, G.B.I.; Sutton, J.P. Space radiation: The number one risk to astronaut health beyond low earth orbit. Life 2014, 4, 491–510. [Google Scholar] [CrossRef]
- Moreno-Villanueva, M.; Wong, M.; Lu, T.; Zhang, Y.; Wu, H. Interplay of space radiation and microgravity in DNA damage and DNA damage response. npj Microgravity 2017, 3, 14. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afshari, N.; Koturbash, I.; Boerma, M.; Newhauser, W.; Kratz, M.; Willey, J.; Williams, J.; Chancellor, J. A Review of Numerical Models of Radiation Injury and Repair Considering Subcellular Targets and the Extracellular Microenvironment. Int. J. Mol. Sci. 2024, 25, 1015. https://doi.org/10.3390/ijms25021015
Afshari N, Koturbash I, Boerma M, Newhauser W, Kratz M, Willey J, Williams J, Chancellor J. A Review of Numerical Models of Radiation Injury and Repair Considering Subcellular Targets and the Extracellular Microenvironment. International Journal of Molecular Sciences. 2024; 25(2):1015. https://doi.org/10.3390/ijms25021015
Chicago/Turabian StyleAfshari, Nousha, Igor Koturbash, Marjan Boerma, Wayne Newhauser, Maria Kratz, Jeffrey Willey, Jacqueline Williams, and Jeffery Chancellor. 2024. "A Review of Numerical Models of Radiation Injury and Repair Considering Subcellular Targets and the Extracellular Microenvironment" International Journal of Molecular Sciences 25, no. 2: 1015. https://doi.org/10.3390/ijms25021015