
Bromodomain-Containing Protein 9 Regulates Signaling Pathways and Reprograms the Epigenome in Immortalized Human Uterine Fibroid Cells
 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results
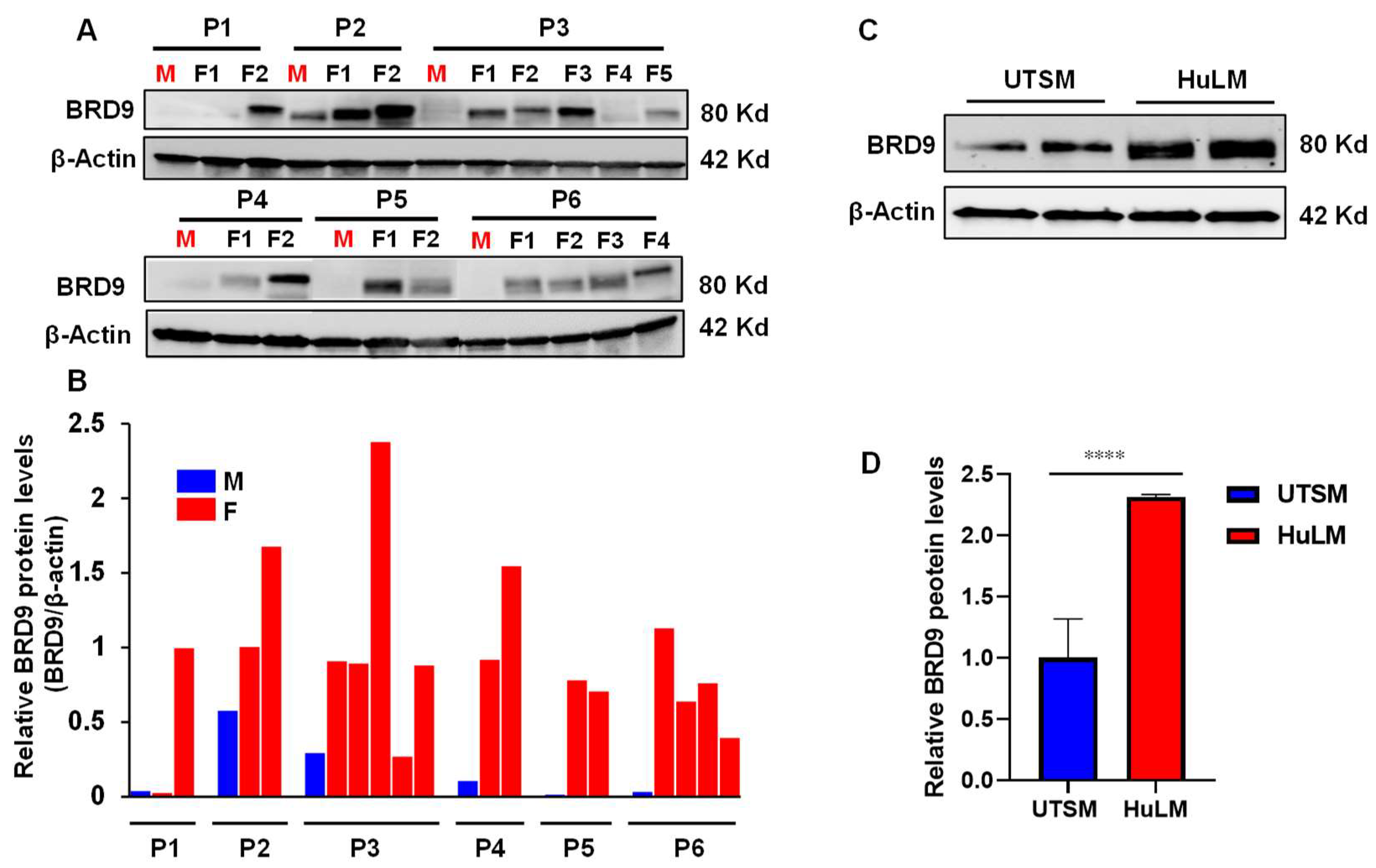
2.1. The Level of BRD9 Protein Is Aberrantly Upregulated in Uterine Fibroids
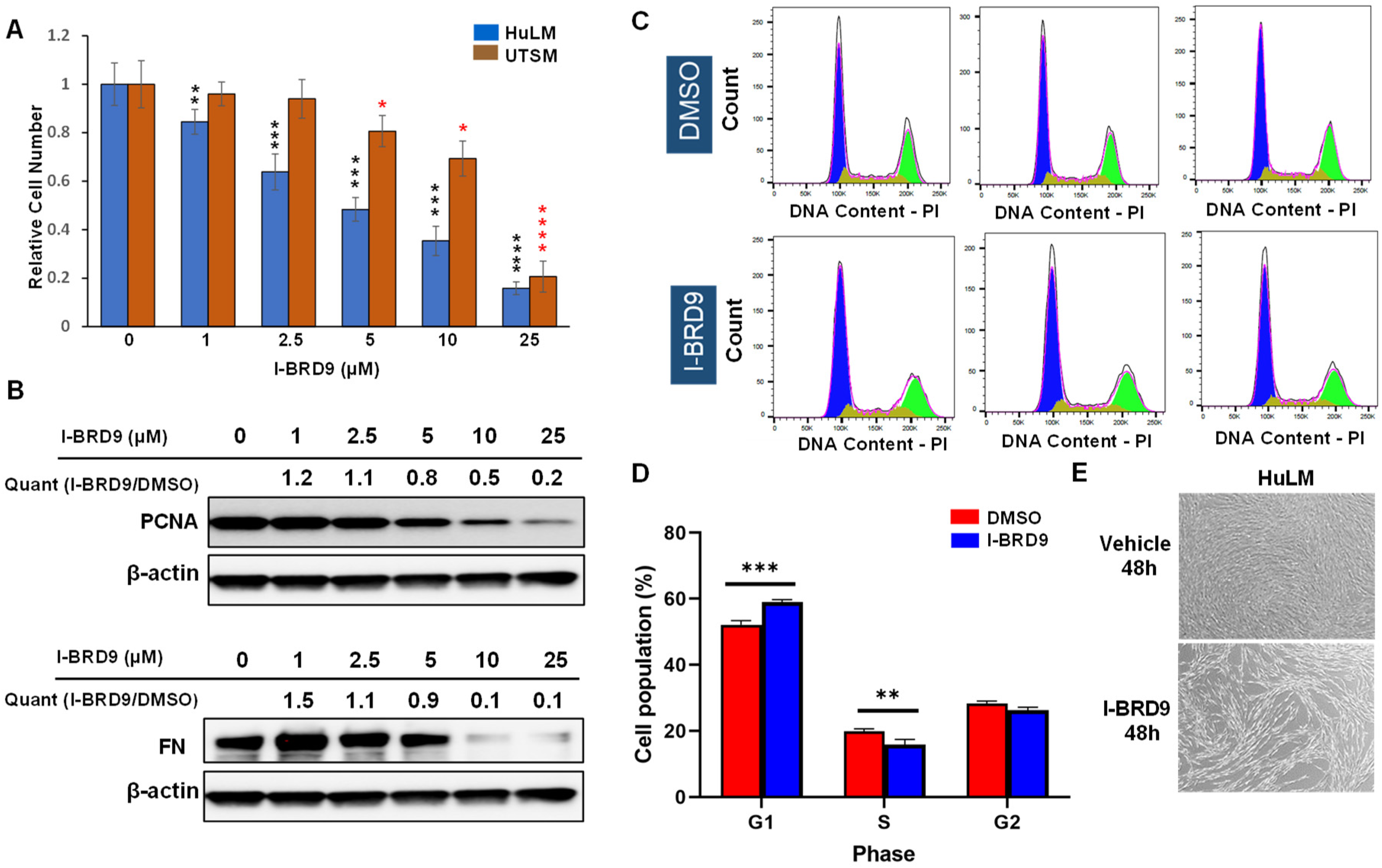
2.2. Inhibition of BRD9 Showed Decreased Cell Proliferation and Anti-Fibrotic Characteristics in Uterine Fibroid Cells
2.3. Inhibition of BRD9 Induced Apoptosis, Necrosis, and Cell Cycle Arrest in Uterine Fibroid Cells
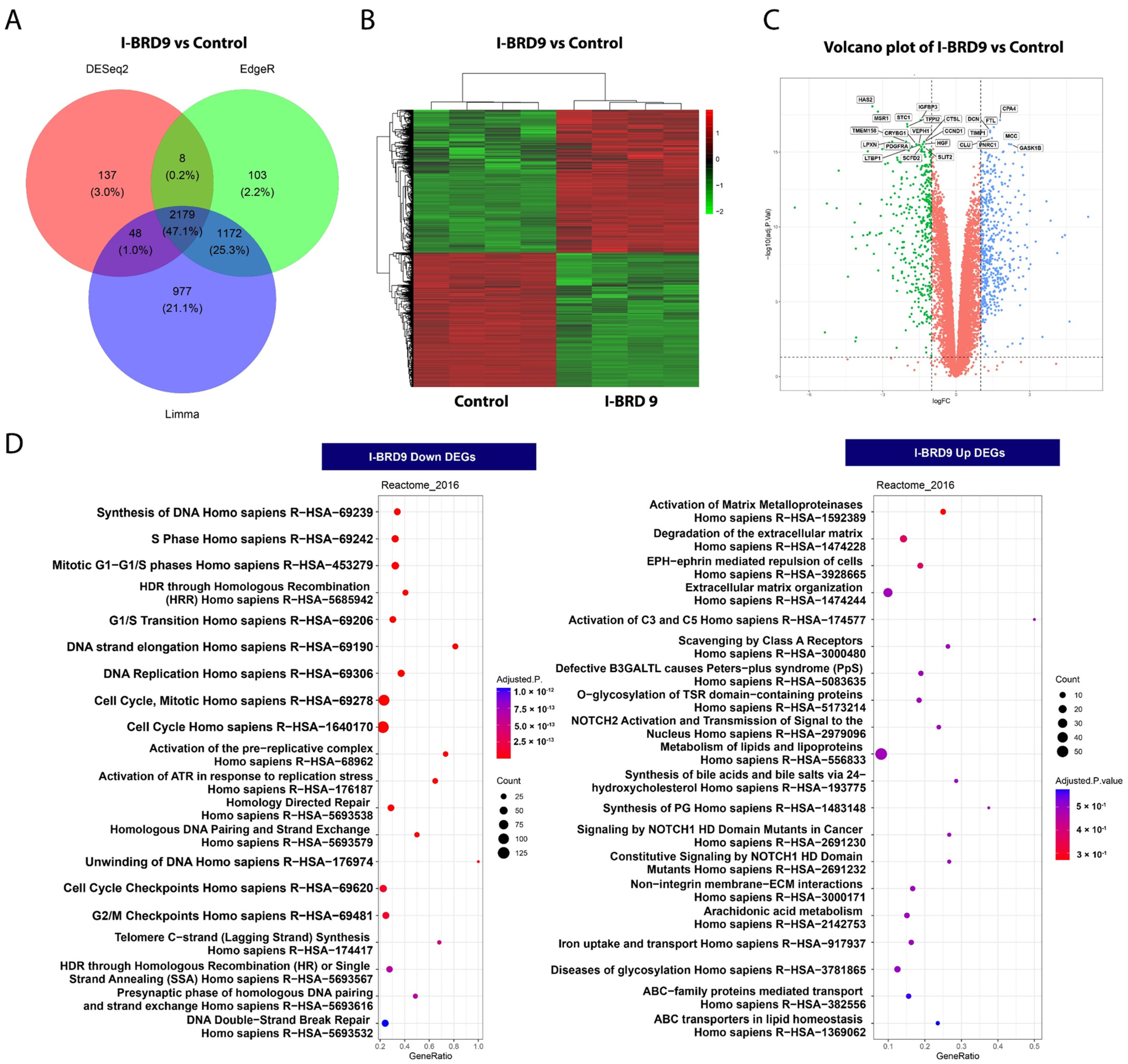
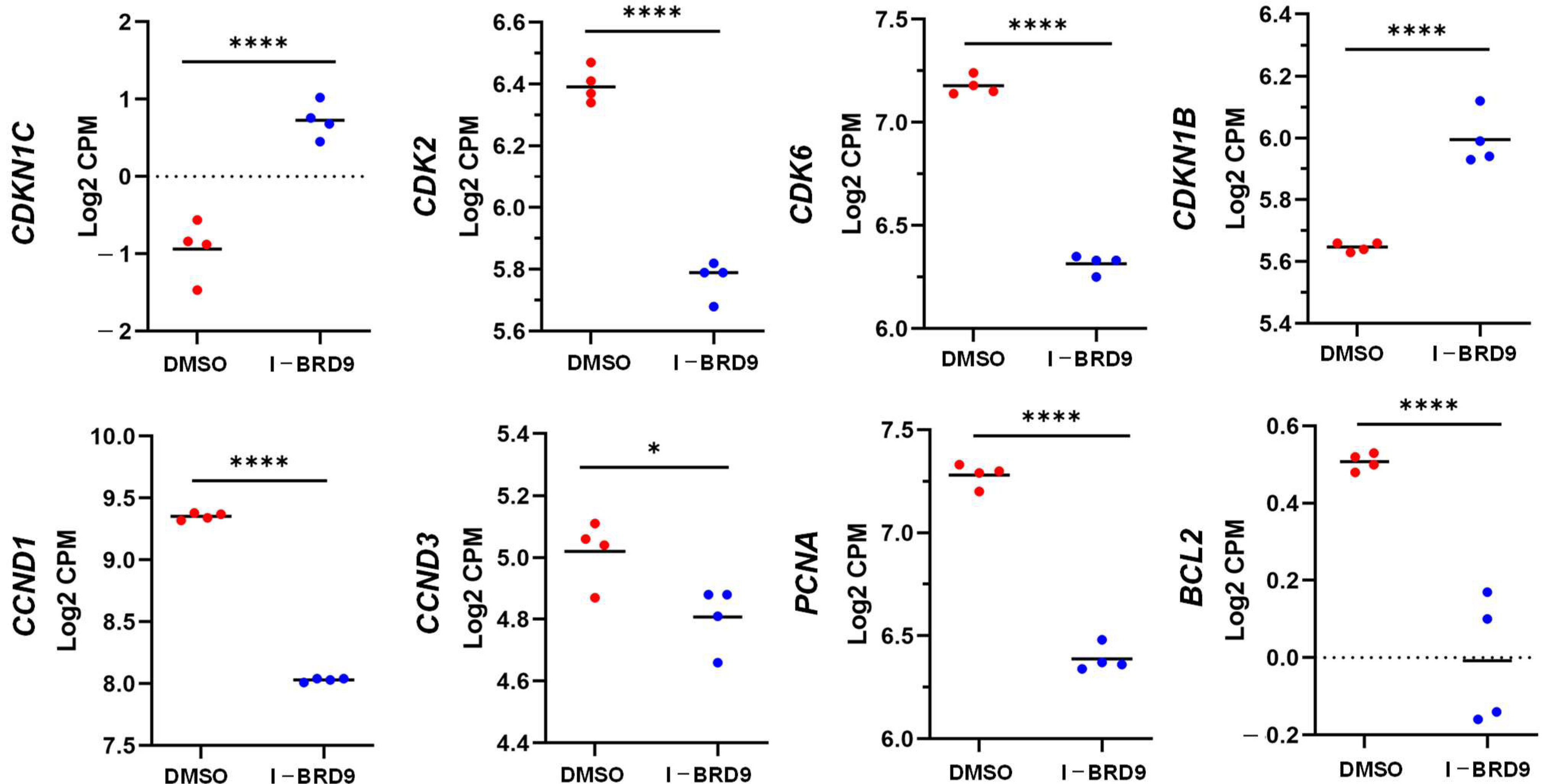
2.4. BRD9 Inhibition Causes Extensive Changes in the UF Cell Transcriptome
2.4.1. Differentially Expressed Genes upon I-BRD9 Treatment
2.4.2. Enrichment Pathway Analysis
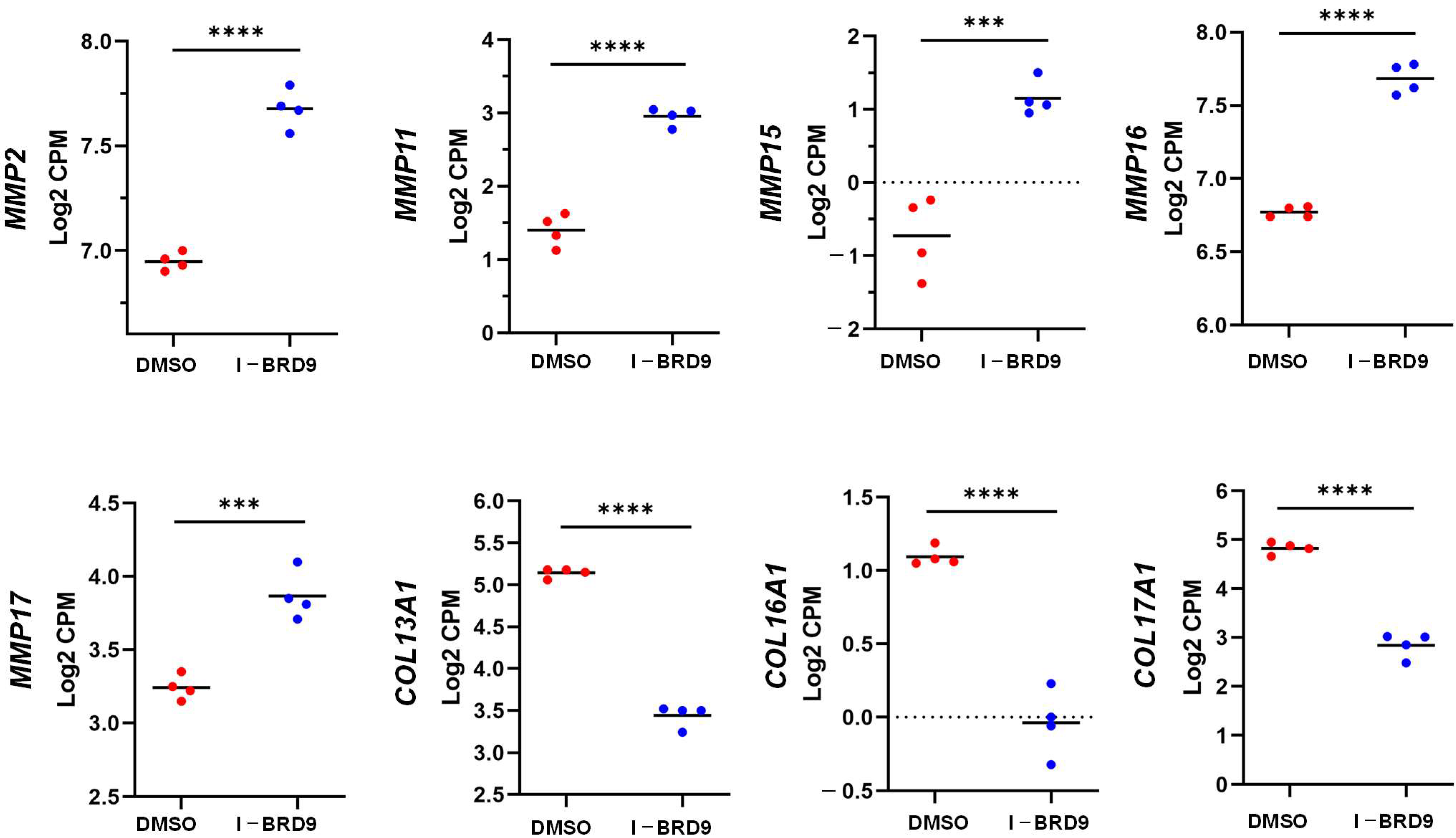
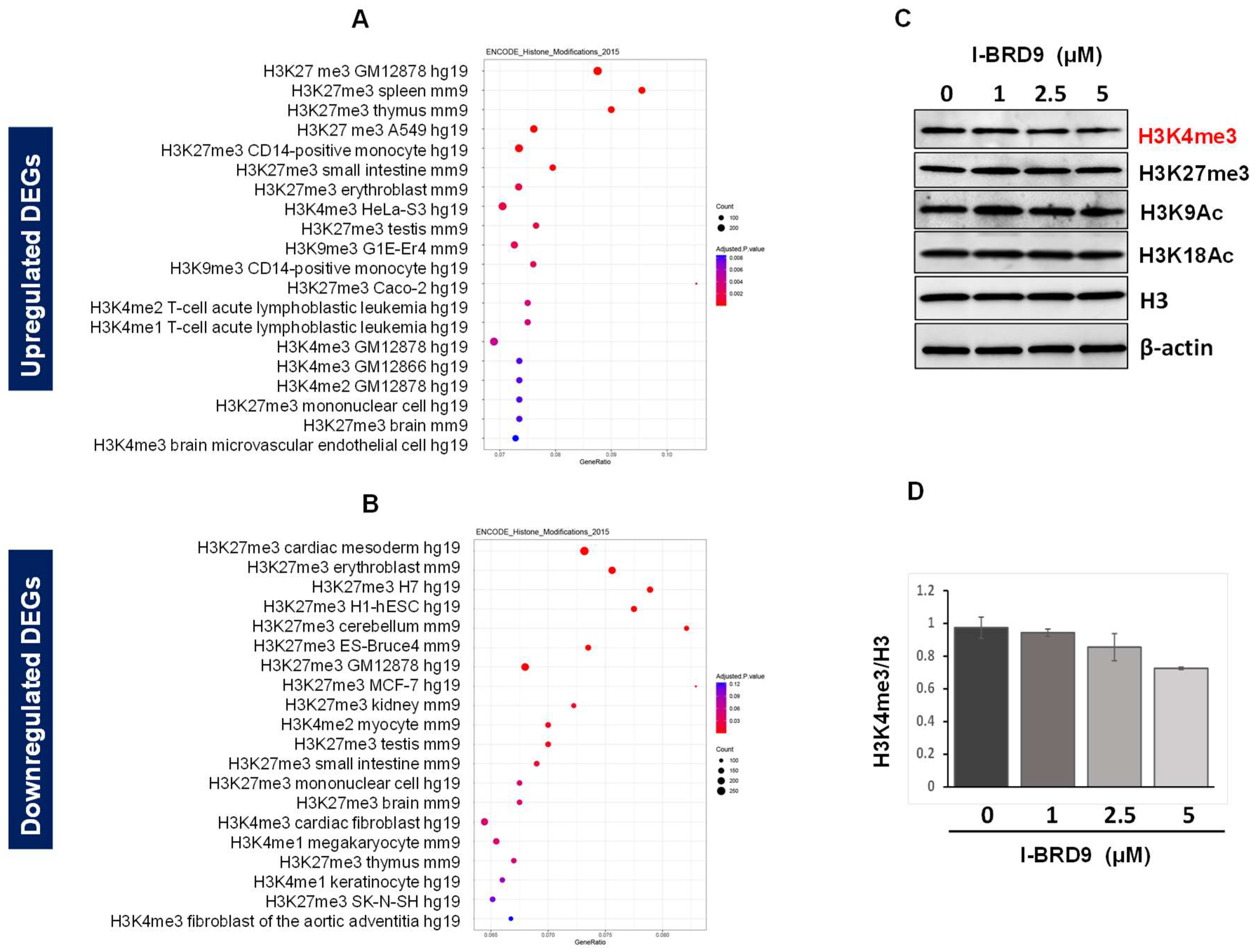
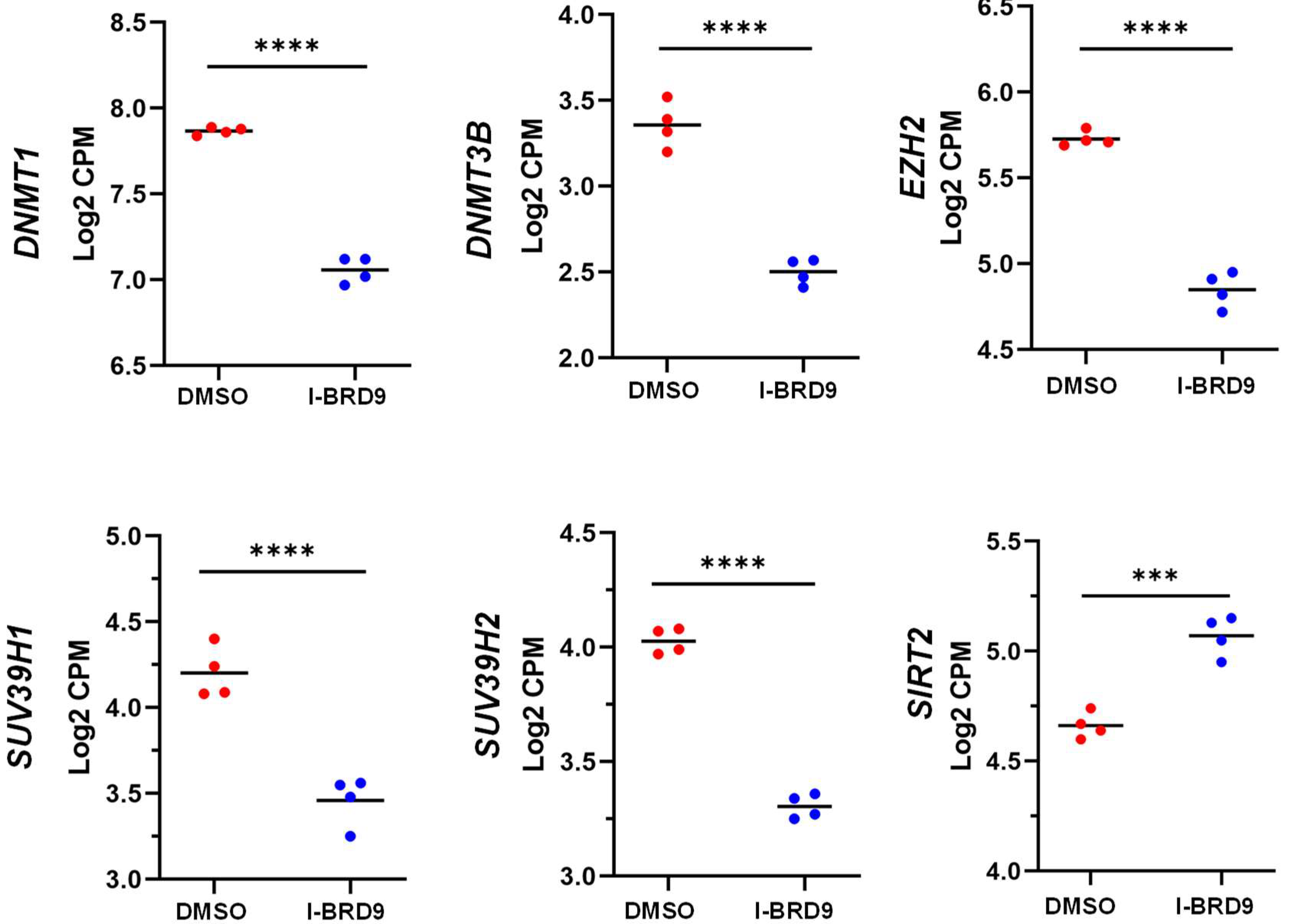
2.4.3. Inhibition of BRD9 Altered the Gene Expression Correlating to Epigenetic Modifications
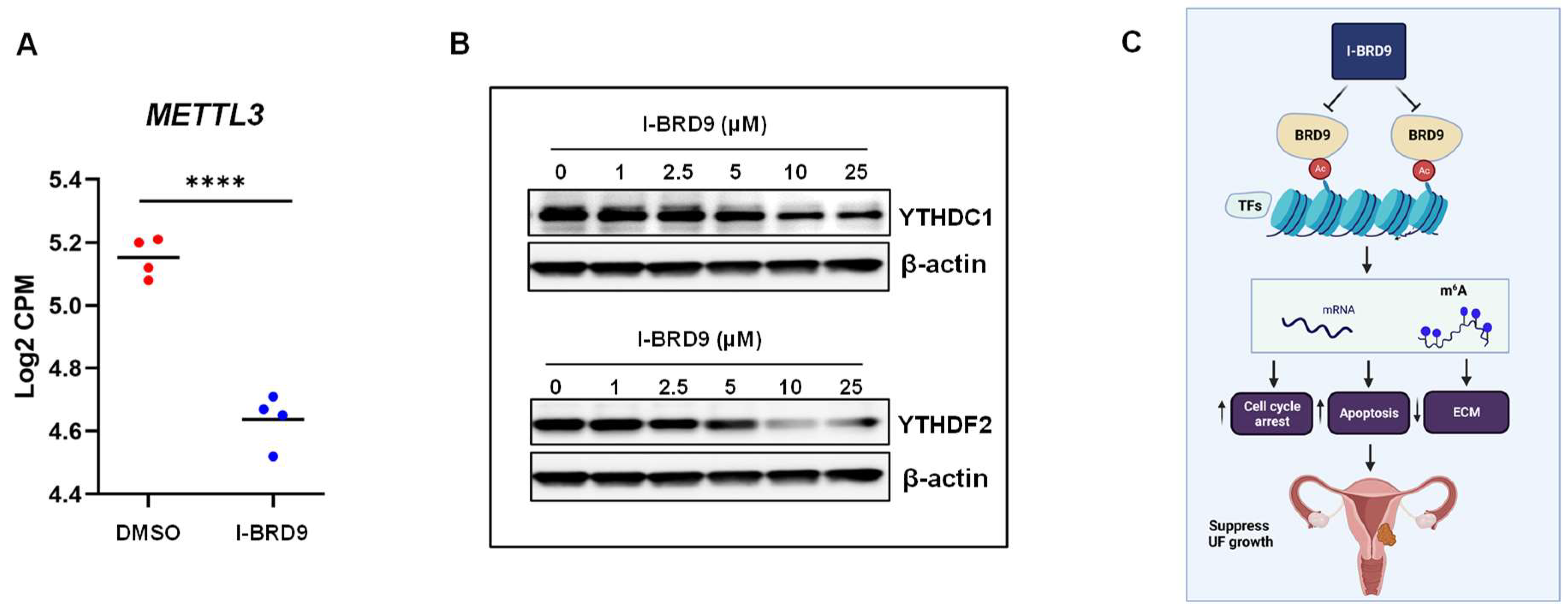
2.4.4. Inhibition of BRD9 Altered the Levels of m6A Regulators in UF Cells
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Experimental Design
4.2. Cells and Reagents
4.3. Protein Extraction and Western Blot Analysis
4.4. Proliferation Assay
4.5. Measurement of Cell Cycle Phase Distribution
4.6. Assessment of Apoptosis
4.7. RNA Sequencing
4.8. Transcriptome Profiles Analysis
4.8.1. Transcriptome Data Analysis
4.8.2. Differential Gene Expression Analysis
4.8.3. Pathway Analysis of DEGs
4.8.4. Functional and Regulatory Enrichment Analysis
4.9. RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, Q.; Ciebiera, M.; Bariani, M.V.; Ali, M.; Elkafas, H.; Boyer, T.G.; Al-Hendy, A. Comprehensive Review of Uterine Fibroids: Developmental Origin, Pathogenesis, and Treatment. Endocr. Rev. 2022, 43, 678–719. [Google Scholar] [CrossRef] [PubMed]
- Bulun, S.E. Uterine fibroids. N. Engl. J. Med. 2013, 369, 1344–1355. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.A.; Laughlin-Tommaso, S.K.; Catherino, W.H.; Lalitkumar, S.; Gupta, D.; Vollenhoven, B. Uterine fibroids. Nat. Rev. Dis. Primers 2016, 2, 16043. [Google Scholar] [CrossRef] [PubMed]
- Whynott, R.M.; Vaught, K.C.C.; Segars, J.H. The Effect of Uterine Fibroids on Infertility: A Systematic Review. Semin. Reprod. Med. 2017, 35, 523–532. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Mas, A.; Diamond, M.P.; Al-Hendy, A. The Mechanism and Function of Epigenetics in Uterine Leiomyoma Development. Reprod. Sci. 2016, 23, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Berta, D.G.; Kuisma, H.; Välimäki, N.; Räisänen, M.; Jäntti, M.; Pasanen, A.; Karhu, A.; Kaukomaa, J.; Taira, A.; Cajuso, T.; et al. Deficient H2A.Z deposition is associated with genesis of uterine leiomyoma. Nature 2021, 596, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Chuang, T.D.; Quintanilla, D.; Boos, D.; Khorram, O. Differential Expression of Super-Enhancer-Associated Long Non-coding RNAs in Uterine Leiomyomas. Reprod. Sci. 2022, 29, 2960–2976. [Google Scholar] [CrossRef]
- Chuang, T.D.; Rehan, A.; Khorram, O. Functional role of the long noncoding RNA X-inactive specific transcript in leiomyoma pathogenesis. Fertil. Steril. 2021, 115, 238–247. [Google Scholar] [CrossRef]
- Carbajo-Garcia, M.C.; Juarez-Barber, E.; Segura-Benitez, M.; Faus, A.; Trelis, A.; Monleon, J.; Carmona-Antonanzas, G.; Pellicer, A.; Flanagan, J.M.; Ferrero, H. H3K4me3 mediates uterine leiomyoma pathogenesis via neuronal processes, synapsis components, proliferation, and Wnt/beta-catenin and TGF-beta pathways. Reprod. Biol. Endocrinol. 2023, 21, 9. [Google Scholar] [CrossRef]
- Paul, E.N.; Grey, J.A.; Carpenter, T.J.; Madaj, Z.B.; Lau, K.H.; Givan, S.A.; Burns, G.W.; Chandler, R.L.; Wegienka, G.R.; Shen, H.; et al. Transcriptome and DNA methylome analyses reveal underlying mechanisms for the racial disparity in uterine fibroids. JCI Insight 2022, 7, e160274. [Google Scholar] [CrossRef] [PubMed]
- Carbajo-Garcia, M.C.; Corachan, A.; Juarez-Barber, E.; Monleon, J.; Paya, V.; Trelis, A.; Quinonero, A.; Pellicer, A.; Ferrero, H. Integrative analysis of the DNA methylome and transcriptome in uterine leiomyoma shows altered regulation of genes involved in metabolism, proliferation, extracellular matrix, and vesicles. J. Pathol. 2022, 257, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Ali, M.; Trevino, L.S.; Mas, A.; Al-Hendy, A. Developmental reprogramming of myometrial stem cells by endocrine disruptor linking to risk of uterine fibroids. Cell Mol. Life Sci. 2023, 80, 274. [Google Scholar] [CrossRef]
- Yang, Q.; Ali, M.; Trevino, L.S.; Mas, A.; Ismail, N.; Al-Hendy, A. Epigenetic Modulation of Inflammatory Pathways in Myometrial Stem Cells and Risk of Uterine Fibroids. Int. J. Mol. Sci. 2023, 24, 11641. [Google Scholar] [CrossRef] [PubMed]
- Yun, M.; Wu, J.; Workman, J.L.; Li, B. Readers of histone modifications. Cell Res. 2011, 21, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Barton, M.C. Bromodomain Histone Readers and Cancer. J. Mol. Biol. 2017, 429, 2003–2010. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.M.; Huidobro, C.; Urdinguio, R.G.; Mangas, C.; Soldevilla, B.; Dominguez, G.; Bonilla, F.; Fernandez, A.F.; Fraga, M.F. Aberrant epigenetic regulation of bromodomain BRD4 in human colon cancer. J. Mol. Med. 2012, 90, 587–595. [Google Scholar] [CrossRef]
- Sahni, J.M.; Keri, R.A. Targeting bromodomain and extraterminal proteins in breast cancer. Pharmacol. Res. 2018, 129, 156–176. [Google Scholar] [CrossRef]
- Yang, Q.; Al-Hendy, A. Non-coding RNAs: An important regulatory mechanism in pathogenesis of uterine fibroids. Fertil. Steril. 2018, 109, 802–803. [Google Scholar] [CrossRef]
- George, J.W.; Fan, H.; Johnson, B.; Carpenter, T.J.; Foy, K.K.; Chatterjee, A.; Patterson, A.L.; Koeman, J.; Adams, M.; Madaj, Z.B.; et al. Integrated Epigenome, Exome, and Transcriptome Analyses Reveal Molecular Subtypes and Homeotic Transformation in Uterine Fibroids. Cell Rep. 2019, 29, 4069–4085.e6. [Google Scholar] [CrossRef]
- Moyo, M.B.; Parker, J.B.; Chakravarti, D. Altered chromatin landscape and enhancer engagement underlie transcriptional dysregulation in MED12 mutant uterine leiomyomas. Nat. Commun. 2020, 11, 1019. [Google Scholar] [CrossRef]
- Yang, Q.; Falahati, A.; Khosh, A.; Mohammed, H.; Kang, W.; Corachan, A.; Bariani, M.V.; Boyer, T.G.; Al-Hendy, A. Targeting Class I Histone Deacetylases in Human Uterine Leiomyosarcoma. Cells 2022, 11, 3801. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Filippakopoulos, P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 246–262. [Google Scholar] [CrossRef] [PubMed]
- Ferri, E.; Petosa, C.; McKenna, C.E. Bromodomains: Structure, function and pharmacology of inhibition. Biochem. Pharmacol. 2016, 106, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hugle, M.; Lucas, X.; Ostrovskyi, D.; Regenass, P.; Gerhardt, S.; Einsle, O.; Hau, M.; Jung, M.; Breit, B.; Gunther, S.; et al. Beyond the BET Family: Targeting CBP/p300 with 4-Acyl Pyrroles. Angew. Chem. Int. Ed. Engl. 2017, 56, 12476–12480. [Google Scholar] [CrossRef]
- Clegg, M.A.; Tomkinson, N.C.O.; Prinjha, R.K.; Humphreys, P.G. Advancements in the Development of non-BET Bromodomain Chemical Probes. ChemMedChem 2019, 14, 362–385. [Google Scholar] [CrossRef]
- Vichaikul, S.; Gurrea-Rubio, M.; Amin, M.A.; Campbell, P.L.; Wu, Q.; Mattichak, M.N.; Brodie, W.D.; Palisoc, P.J.; Ali, M.; Muraoka, S.; et al. Inhibition of bromodomain extraterminal histone readers alleviates skin fibrosis in experimental models of scleroderma. JCI Insight 2022, 7, e150871. [Google Scholar] [CrossRef]
- Wang, Q.; Shao, X.; Leung, E.L.H.; Chen, Y.; Yao, X. Selectively targeting individual bromodomain: Drug discovery and molecular mechanisms. Pharmacol. Res. 2021, 172, 105804. [Google Scholar] [CrossRef]
- Zaware, N.; Zhou, M.M. Bromodomain biology and drug discovery. Nat. Struct. Mol. Biol. 2019, 26, 870–879. [Google Scholar] [CrossRef]
- Ali, M.M.; Naz, S.; Ashraf, S.; Knapp, S.; Ul-Haq, Z. Epigenetic modulation by targeting bromodomain containing protein 9 (BRD9): Its therapeutic potential and selective inhibition. Int. J. Biol. Macromol. 2023, 230, 123428. [Google Scholar] [CrossRef]
- Wang, X.; Song, C.; Ye, Y.; Gu, Y.; Li, X.; Chen, P.; Leng, D.; Xiao, J.; Wu, H.; Xie, S.; et al. BRD9-mediated control of the TGF-beta/Activin/Nodal pathway regulates self-renewal and differentiation of human embryonic stem cells and progression of cancer cells. Nucleic Acids Res. 2023, 51, 11634–11651. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Kondo, S.; Nomura, M.; Kato, S.; Nishimura, K.; Zang, W.; Zhang, Y.; Akashi, T.; Viny, A.; Shigehiro, T.; et al. BRD9 determines the cell fate of hematopoietic stem cells by regulating chromatin state. Nat. Commun. 2023, 14, 8372. [Google Scholar] [CrossRef] [PubMed]
- Michel, B.C.; D’Avino, A.R.; Cassel, S.H.; Mashtalir, N.; McKenzie, Z.M.; McBride, M.J.; Valencia, A.M.; Zhou, Q.; Bocker, M.; Soares, L.M.M.; et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat. Cell Biol. 2018, 20, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, S.; Troisi, E.C.; Howard, T.P.; Haswell, J.R.; Wolf, B.K.; Hawk, W.H.; Ramos, P.; Oberlick, E.M.; Tzvetkov, E.P.; et al. BRD9 defines a SWI/SNF sub-complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat. Commun. 2019, 10, 1881. [Google Scholar] [CrossRef] [PubMed]
- Sima, X.; He, J.; Peng, J.; Xu, Y.; Zhang, F.; Deng, L. The genetic alteration spectrum of the SWI/SNF complex: The oncogenic roles of BRD9 and ACTL6A. PLoS ONE 2019, 14, e0222305. [Google Scholar] [CrossRef]
- Yang, Q.; Bariani, M.V.; Falahati, A.; Khosh, A.; Lastra, R.R.; Siblini, H.; Boyer, T.G.; Al-Hendy, A. The Functional Role and Regulatory Mechanism of Bromodomain-Containing Protein 9 in Human Uterine Leiomyosarcoma. Cells 2022, 11, 2160. [Google Scholar] [CrossRef]
- Park, S.G.; Lee, D.; Seo, H.R.; Lee, S.A.; Kwon, J. Cytotoxic activity of bromodomain inhibitor NVS-CECR2-1 on human cancer cells. Sci. Rep. 2020, 10, 16330. [Google Scholar] [CrossRef]
- Ali, M.M.; Ashraf, S.; Nure, E.A.M.; Qureshi, U.; Khan, K.M.; Ul-Haq, Z. Identification of Selective BRD9 Inhibitor via Integrated Computational Approach. Int. J. Mol. Sci. 2022, 23, 13513. [Google Scholar] [CrossRef]
- Bevill, S.M.; Olivares-Quintero, J.F.; Sciaky, N.; Golitz, B.T.; Singh, D.; Beltran, A.S.; Rashid, N.U.; Stuhlmiller, T.J.; Hale, A.; Moorman, N.J.; et al. GSK2801, a BAZ2/BRD9 Bromodomain Inhibitor, Synergizes with BET Inhibitors to Induce Apoptosis in Triple-Negative Breast Cancer. Mol. Cancer Res. 2019, 17, 1503–1518. [Google Scholar] [CrossRef]
- Martin, L.J.; Koegl, M.; Bader, G.; Cockcroft, X.L.; Fedorov, O.; Fiegen, D.; Gerstberger, T.; Hofmann, M.H.; Hohmann, A.F.; Kessler, D.; et al. Structure-Based Design of an in Vivo Active Selective BRD9 Inhibitor. J. Med. Chem. 2016, 59, 4462–4475. [Google Scholar] [CrossRef]
- Mason, L.D.; Chava, S.; Reddi, K.K.; Gupta, R. The BRD9/7 Inhibitor TP-472 Blocks Melanoma Tumor Growth by Suppressing ECM-Mediated Oncogenic Signaling and Inducing Apoptosis. Cancers 2021, 13, 5516. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S.; Ciavattini, A.; Petraglia, F.; Castellucci, M.; Ciarmela, P. Extracellular matrix in uterine leiomyoma pathogenesis: A potential target for future therapeutics. Hum. Reprod. Update 2018, 24, 59–85. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S.; Afrin, S.; Singh, B.; Jayes, F.L.; Brennan, J.T.; Borahay, M.A.; Leppert, P.C.; Segars, J.H. Extracellular matrix and Hippo signaling as therapeutic targets of antifibrotic compounds for uterine fibroids. Clin. Transl. Med. 2021, 11, e475. [Google Scholar] [CrossRef]
- Jamaluddin, M.F.B.; Nahar, P.; Tanwar, P.S. Proteomic Characterization of the Extracellular Matrix of Human Uterine Fibroids. Endocrinology 2018, 159, 2656–2669. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Al-Hendy, A. Update on the Role and Regulatory Mechanism of Extracellular Matrix in the Pathogenesis of Uterine Fibroids. Int. J. Mol. Sci. 2023, 24, 5778. [Google Scholar] [CrossRef] [PubMed]
- Leppert, P.C.; Jayes, F.L.; Segars, J.H. The extracellular matrix contributes to mechanotransduction in uterine fibroids. Obstet. Gynecol. Int. 2014, 2014, 783289. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.A.; Jamaluddin, M.F.B.; Adebayo, M.; Bajwa, P.; Scott, R.J.; Dharmarajan, A.M.; Nahar, P.; Tanwar, P.S. Extracellular matrix (ECM) activates beta-catenin signaling in uterine fibroids. Reproduction 2018, 155, 61–71. [Google Scholar] [CrossRef]
- Norian, J.M.; Owen, C.M.; Taboas, J.; Korecki, C.; Tuan, R.; Malik, M.; Catherino, W.H.; Segars, J.H. Characterization of tissue biomechanics and mechanical signaling in uterine leiomyoma. Matrix Biol. 2012, 31, 57–65. [Google Scholar] [CrossRef]
- Yue, Y.; Liu, J.; He, C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 2015, 29, 1343–1355. [Google Scholar] [CrossRef]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef]
- Jia, G.; Fu, Y.; He, C. Reversible RNA adenosine methylation in biological regulation. Trends Genet. 2013, 29, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Scheitl, C.P.M.; Ghaem Maghami, M.; Lenz, A.K.; Hobartner, C. Site-specific RNA methylation by a methyltransferase ribozyme. Nature 2020, 587, 663–667. [Google Scholar] [CrossRef]
- Shen, H.; Lan, Y.; Zhao, Y.; Shi, Y.; Jin, J.; Xie, W. The emerging roles of N6-methyladenosine RNA methylation in human cancers. Biomark. Res. 2020, 8, 24. [Google Scholar] [CrossRef]
- Bert, S.A.; Robinson, M.D.; Strbenac, D.; Statham, A.L.; Song, J.Z.; Hulf, T.; Sutherland, R.L.; Coolen, M.W.; Stirzaker, C.; Clark, S.J. Regional activation of the cancer genome by long-range epigenetic remodeling. Cancer Cell 2013, 23, 9–22. [Google Scholar] [CrossRef]
- Garcia, N.; Al-Hendy, A.; Baracat, E.C.; Carvalho, K.C.; Yang, Q. Targeting Hedgehog Pathway and DNA Methyltransferases in Uterine Leiomyosarcoma Cells. Cells 2020, 10, 53. [Google Scholar] [CrossRef]
- Zaib, S.; Rana, N.; Khan, I. Histone Modifications and their Role in Epigenetics of Cancer. Curr. Med. Chem. 2022, 29, 2399–2411. [Google Scholar] [CrossRef] [PubMed]
- Keyvani-Ghamsari, S.; Khorsandi, K.; Rasul, A.; Zaman, M.K. Current understanding of epigenetics mechanism as a novel target in reducing cancer stem cells resistance. Clin. Epigenet. 2021, 13, 120. [Google Scholar] [CrossRef]
- Ali, M.; Shahin, S.M.; Sabri, N.A.; Al-Hendy, A.; Yang, Q. Activation of beta-Catenin Signaling and its Crosstalk with Estrogen and Histone Deacetylases in Human Uterine Fibroids. J. Clin. Endocrinol. Metab. 2020, 105, e1517–e1535. [Google Scholar] [CrossRef]
- Schrump, D.S.; Hong, J.A.; Nguyen, D.M. Utilization of chromatin remodeling agents for lung cancer therapy. Cancer J. 2007, 13, 56–64. [Google Scholar] [CrossRef]
- Qi, J. Bromodomain and extraterminal domain inhibitors (BETi) for cancer therapy: Chemical modulation of chromatin structure. Cold Spring Harb. Perspect. Biol. 2014, 6, a018663. [Google Scholar] [CrossRef]
- Kaur, J.; Daoud, A.; Eblen, S.T. Targeting Chromatin Remodeling for Cancer Therapy. Curr. Mol. Pharmacol. 2019, 12, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Magnani, L.; Stoeck, A.; Zhang, X.; Lanczky, A.; Mirabella, A.C.; Wang, T.L.; Gyorffy, B.; Lupien, M. Genome-wide reprogramming of the chromatin landscape underlies endocrine therapy resistance in breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, E1490–E1499. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Qiu, Y.; Jiao, Y.; Qiu, Z.; Liu, D. Small Molecules Targeting HATs, HDACs, and BRDs in Cancer Therapy. Front. Oncol. 2020, 10, 560487. [Google Scholar] [CrossRef] [PubMed]
- Santillan, D.A.; Theisler, C.M.; Ryan, A.S.; Popovic, R.; Stuart, T.; Zhou, M.M.; Alkan, S.; Zeleznik-Le, N.J. Bromodomain and histone acetyltransferase domain specificities control mixed lineage leukemia phenotype. Cancer Res. 2006, 66, 10032–10039. [Google Scholar] [CrossRef]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Lucas, X.; Gunther, S. Targeting the BET family for the treatment of leukemia. Epigenomics 2014, 6, 153–155. [Google Scholar] [CrossRef]
- Belkina, A.C.; Nikolajczyk, B.S.; Denis, G.V. BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. J. Immunol. 2013, 190, 3670–3678. [Google Scholar] [CrossRef]
- Qiu, H.; Jackson, A.L.; Kilgore, J.E.; Zhong, Y.; Chan, L.L.; Gehrig, P.A.; Zhou, C.; Bae-Jump, V.L. JQ1 suppresses tumor growth through downregulating LDHA in ovarian cancer. Oncotarget 2015, 6, 6915–6930. [Google Scholar] [CrossRef]
- Richter, G.H.S.; Hensel, T.; Schmidt, O.; Saratov, V.; von Heyking, K.; Becker-Dettling, F.; Prexler, C.; Yen, H.Y.; Steiger, K.; Fulda, S.; et al. Combined Inhibition of Epigenetic Readers and Transcription Initiation Targets the EWS-ETS Transcriptional Program in Ewing Sarcoma. Cancers 2020, 12, 304. [Google Scholar] [CrossRef]
- Lu, J.; Qian, C.; Ji, Y.; Bao, Q.; Lu, B. Gene Signature Associated with Bromodomain Genes Predicts the Prognosis of Kidney Renal Clear Cell Carcinoma. Front. Genet. 2021, 12, 643935. [Google Scholar] [CrossRef]
- Boyson, S.P.; Gao, C.; Quinn, K.; Boyd, J.; Paculova, H.; Frietze, S.; Glass, K.C. Functional Roles of Bromodomain Proteins in Cancer. Cancers 2021, 13, 3606. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Gu, X.; Wei, W.; Wu, Z.; Gong, F.; Dong, X. BRD9 is an essential regulator of glycolysis that creates an epigenetic vulnerability in colon adenocarcinoma. Cancer Med. 2023, 12, 1572–1587. [Google Scholar] [CrossRef]
- Kurata, K.; Samur, M.K.; Liow, P.; Wen, K.; Yamamoto, L.; Liu, J.; Morelli, E.; Gulla, A.; Tai, Y.T.; Qi, J.; et al. BRD9 Degradation Disrupts Ribosome Biogenesis in Multiple Myeloma. Clin. Cancer Res. 2023, 29, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Magic, Z.; Supic, G.; Brankovic-Magic, M. Towards targeted epigenetic therapy of cancer. J. BUON 2009, 14 (Suppl. 1), S79–S88. [Google Scholar] [PubMed]
- Zhao, R.; Casson, A.G. Epigenetic aberrations and targeted epigenetic therapy of esophageal cancer. Curr. Cancer Drug Targets 2008, 8, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Cao, D.; Yang, J.; Li, M.; Zhang, Z.; Shen, K. Genetic and epigenetic heterogeneity of epithelial ovarian cancer and the clinical implications for molecular targeted therapy. J. Cell Mol. Med. 2016, 20, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Moustakim, M.; Clark, P.G.K.; Hay, D.A.; Dixon, D.J.; Brennan, P.E. Chemical probes and inhibitors of bromodomains outside the BET family. MedChemComm 2016, 7, 2246–2264. [Google Scholar] [CrossRef]
- Kramer, K.F.; Moreno, N.; Fruhwald, M.C.; Kerl, K. BRD9 Inhibition, Alone or in Combination with Cytostatic Compounds as a Therapeutic Approach in Rhabdoid Tumors. Int. J. Mol. Sci. 2017, 18, 1537. [Google Scholar] [CrossRef]
- Zhou, Q.; Huang, J.; Zhang, C.; Zhao, F.; Kim, W.; Tu, X.; Zhang, Y.; Nowsheen, S.; Zhu, Q.; Deng, M.; et al. The bromodomain containing protein BRD-9 orchestrates RAD51-RAD54 complex formation and regulates homologous recombination-mediated repair. Nat. Commun. 2020, 11, 2639. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, L.; Lou, W.; Su, J.; Huang, J.; Liu, A.; Xu, Y.; He, H.; Gao, Y.; Xu, D.; et al. Aberrant activation of m6A demethylase FTO renders HIF2alpha(low/-) clear cell renal cell carcinoma sensitive to BRD9 inhibitors. Sci. Transl. Med. 2021, 13, eabf6045. [Google Scholar] [CrossRef]
- Zhou, L.; Yao, Q.; Li, H.; Chen, J. Targeting BRD9 by I-BRD9 efficiently inhibits growth of acute myeloid leukemia cells. Transl. Cancer Res. 2021, 10, 3364–3372. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y. Epigenetic cross-talk between DNA methylation and histone modifications in human cancers. Yonsei Med. J. 2009, 50, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.; Fischle, W. Epigenetic markers and their cross-talk. Essays Biochem. 2010, 48, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Szulwach, K.E.; Li, X.; Smrt, R.D.; Li, Y.; Luo, Y.; Lin, L.; Santistevan, N.J.; Li, W.; Zhao, X.; Jin, P. Cross talk between microRNA and epigenetic regulation in adult neurogenesis. J. Cell Biol. 2010, 189, 127–141. [Google Scholar] [CrossRef]
- Lopez, G.; Song, Y.; Lam, R.; Ruder, D.; Creighton, C.J.; Bid, H.K.; Bill, K.L.; Bolshakov, S.; Zhang, X.; Lev, D.; et al. HDAC Inhibition for the Treatment of Epithelioid Sarcoma: Novel Cross Talk Between Epigenetic Components. Mol. Cancer Res. 2016, 14, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Xu, Y.; Wang, Y.; Gao, D.; King, J.; Xu, Y.; Liang, F.S. Investigating crosstalk between H3K27 acetylation and H3K4 trimethylation in CRISPR/dCas-based epigenome editing and gene activation. Sci. Rep. 2021, 11, 15912. [Google Scholar] [CrossRef] [PubMed]
- Koranda, J.L.; Dore, L.; Shi, H.; Patel, M.J.; Vaasjo, L.O.; Rao, M.N.; Chen, K.; Lu, Z.; Yi, Y.; Chi, W.; et al. Mettl14 Is Essential for Epitranscriptomic Regulation of Striatal Function and Learning. Neuron 2018, 99, 283–292.e5. [Google Scholar] [CrossRef]
- Weng, Y.L.; Wang, X.; An, R.; Cassin, J.; Vissers, C.; Liu, Y.; Liu, Y.; Xu, T.; Wang, X.; Wong, S.Z.H.; et al. Epitranscriptomic m6A Regulation of Axon Regeneration in the Adult Mammalian Nervous System. Neuron 2018, 97, 313–325.e6. [Google Scholar] [CrossRef]
- Hsu, P.J.; Shi, H.; He, C. Epitranscriptomic influences on development and disease. Genome Biol. 2017, 18, 197. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, W.; Shen, F.; Yang, X.; Liu, H.; Dai, S.; Sun, X.; Huang, J.; Guo, Q. YTH Domain Proteins: A Family of m6A Readers in Cancer Progression. Front. Oncol. 2021, 11, 629560. [Google Scholar] [CrossRef]
- Kontur, C.; Jeong, M.; Cifuentes, D.; Giraldez, A.J. Ythdf m6A Readers Function Redundantly during Zebrafish Development. Cell Rep. 2020, 33, 108598. [Google Scholar] [CrossRef]
- Zhen, D.; Wu, Y.; Zhang, Y.; Chen, K.; Song, B.; Xu, H.; Tang, Y.; Wei, Z.; Meng, J. m6A Reader: Epitranscriptome Target Prediction and Functional Characterization of N6-Methyladenosine (m6A) Readers. Front. Cell Dev. Biol. 2020, 8, 741. [Google Scholar] [CrossRef] [PubMed]
- Berlivet, S.; Scutenaire, J.; Deragon, J.M.; Bousquet-Antonelli, C. Readers of the m6A epitranscriptomic code. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 329–342. [Google Scholar] [CrossRef]
- Li, F.; He, C.; Yao, H.; Zhao, Y.; Ye, X.; Zhou, S.; Zou, J.; Li, Y.; Li, J.; Chen, S.; et al. Glutamate from nerve cells promotes perineural invasion in pancreatic cancer by regulating tumor glycolysis through HK2 mRNA-m6A modification. Pharmacol. Res. 2023, 187, 106555. [Google Scholar] [CrossRef] [PubMed]
- Shimura, T.; Kandimalla, R.; Okugawa, Y.; Ohi, M.; Toiyama, Y.; He, C.; Goel, A. Novel evidence for m6A methylation regulators as prognostic biomarkers and FTO as a potential therapeutic target in gastric cancer. Br. J. Cancer 2022, 126, 228–237. [Google Scholar] [CrossRef]
- Wu, C.; Chen, W.; He, J.; Jin, S.; Liu, Y.; Yi, Y.; Gao, Z.; Yang, J.; Yang, J.; Cui, J.; et al. Interplay of m6A and H3K27 trimethylation restrains inflammation during bacterial infection. Sci. Adv. 2020, 6, eaba0647. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; He, C. Chromatin and transcriptional regulation by reversible RNA methylation. Curr. Opin. Cell Biol. 2021, 70, 109–115. [Google Scholar] [CrossRef]
- Omar, M.; Laknaur, A.; Al-Hendy, A.; Yang, Q. Myometrial progesterone hyper-responsiveness associated with increased risk of human uterine fibroids. BMC Women’s Health 2019, 19, 92. [Google Scholar] [CrossRef]
- Paul, E.N.; Burns, G.W.; Carpenter, T.J.; Grey, J.A.; Fazleabas, A.T.; Teixeira, J.M. Transcriptome Analyses of Myometrium from Fibroid Patients Reveals Phenotypic Differences Compared to Non-Diseased Myometrium. Int. J. Mol. Sci. 2021, 22, 3618. [Google Scholar] [CrossRef]
- Meng, J.; Lu, Z.; Liu, H.; Zhang, L.; Zhang, S.; Chen, Y.; Rao, M.K.; Huang, Y. A protocol for RNA methylation differential analysis with MeRIP-Seq data and exomePeak R/Bioconductor package. Methods 2014, 69, 274–281. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, Y.; Sun, B.; Wang, L.; Yang, Y.; Ma, D.; Lv, J.; Heng, J.; Ding, Y.; Xue, Y.; et al. m6A modulates haematopoietic stem and progenitor cell specification. Nature 2017, 549, 273–276. [Google Scholar] [CrossRef]
- Hu, L.; Liu, S.; Peng, Y.; Ge, R.; Su, R.; Senevirathne, C.; Harada, B.T.; Dai, Q.; Wei, J.; Zhang, L.; et al. m6A RNA modifications are measured at single-base resolution across the mammalian transcriptome. Nat. Biotechnol. 2022, 40, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Meng, H.; Ge, R.; Liu, S.; Chen, M.; He, C.; Hu, L. Detection of m6A RNA modifications at single-nucleotide resolution using m6A-selective allyl chemical labeling and sequencing. STAR Protoc. 2022, 3, 101677. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Carney, S.A.; Tahara, H.; Swartz, C.D.; Risinger, J.I.; He, H.; Moore, A.B.; Haseman, J.K.; Barrett, J.C.; Dixon, D. Immortalization of human uterine leiomyoma and myometrial cell lines after induction of telomerase activity: Molecular and phenotypic characteristics. Lab. Investig. 2002, 82, 719–728. [Google Scholar] [CrossRef]
- Yang, Q.; Nair, S.; Laknaur, A.; Ismail, N.; Diamond, M.P.; Al-Hendy, A. The Polycomb Group Protein EZH2 Impairs DNA Damage Repair Gene Expression in Human Uterine Fibroids. Biol. Reprod. 2016, 94, 69. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Dahl, M.J.; Albertine, K.H.; Ramchandran, R.; Sun, M.; Raj, J.U. Role of histone deacetylases in regulation of phenotype of ovine newborn pulmonary arterial smooth muscle cells. Cell Prolif. 2013, 46, 654–664. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Elam, L.; Laknaur, A.; Gavrilova-Jordan, L.; Lue, J.; Diamond, M.P.; Al-Hendy, A. Altered DNA repair genes in human uterine fibroids are epigenetically regulated via EZH2 histone methyltransferase. Fertil. Steril. 2015, 104, e72. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies | Company | Catalog# | Source | Application | Dilution | Size (KDa) |
|---|---|---|---|---|---|---|
| BRD9 | Cell signaling | 58906 | Rabbit | WB | 1:1000 | 80 |
| PCNA | GeneTex | GTX100539 | Rabbit | WB | 1:1000 | 29 |
| FN | cell signaling | 26836 | Rabbit | WB | 1:1000 | 300 |
| YTHDC1 | Abcam | ab122340 | Rabbit | WB | 1:1000 | 85 |
| YTHDF2 | Abcam | ab220163 | Rabbit | WB | 1:1000 | 62 |
| H3K4me3 | Active Motif | 39160 | Rabbit | WB | 1:1000 | 17 |
| H3K27me3 | Active Motif | 39157 | Rabbit | WB | 1:1000 | 17 |
| H3K9Ac | Active Motif | 39918 | Rabbit | WB | 1:1000 | 17 |
| H3K18Ac | Active Motif | 39756 | Rabbit | WB | 1:1000 | 17 |
| H3 | Cell Signaling | 4499 | Rabbit | WB | 1:2000 | 17 |
| β-actin | Sigma | A5316 | Mouse | WB | 1:5000 | 42 |
| Gene Symbol | Primer Sequences | F or R | Assay | Species | Size (bp) | Accession |
|---|---|---|---|---|---|---|
| CDK2 | AGATGGACGGAGCTTGTTATC | F | q-PCR | Human | 103 | X62071 |
| CDK2 | CTTGGTCACATCCTGGAAGAA | R | q-PCR | Human | 103 | |
| CCND1 | GGGTTGTGCTACAGATGATAGAG | F | q-PCR | Human | 112 | NM-053056.3 |
| CCND1 | AGACGCCTCCTTTGTGTTAAT | R | q-PCR | Human | 112 | |
| CCND3 | GTGTTGTCCCTTCTAGGGTTATT | F | q-PCR | Human | 102 | M92287.1 |
| CCND3 | TGAGAGGAGCCATCTAGACTATT | R | q-PCR | Human | 102 | |
| PCNA | GGACACTGCTGGTGGTATTT | F | q-PCR | Human | 105 | J04718 |
| PCNA | CAGAACTGGTGGAGGGTAAAC | R | q-PCR | Human | 105 | |
| Col3A1 | CTGGGCTTCCTGGTTTACAT | F | q-PCR | Human | 106 | NM_001130103.2 |
| Col3A1 | GCTCCTTGGTCTCCCTTATC | R | q-PCR | Human | 106 | |
| Col17A1 | TTGTCCGTAGGCCCATACTA | F | q-PCR | Human | 113 | NM_000494.4 |
| Col17A1 | CCTCTTCTCCCTTTATTCCTTCC | R | q-PCR | Human | 113 | |
| MMP11 | TCCTGACTTCTTTGGCTGTG | F | q-PCR | Human | 114 | NM_005940.5 |
| MMP11 | CATGGGTCTCTAGCCTGATATTC | R | q-PCR | Human | 114 | |
| MMP15 | CTGCTCCAGACAGGGAATTAG | F | q-PCR | Human | 139 | NM_002428.4 |
| MMP15 | CAAAGAGAGCCTGGCAGTTA | R | q-PCR | Human | 139 | |
| MMP16 | GACATACATCCCAACCTCTCTC | F | q-PCR | Human | 97 | NM_005941.5 |
| MMP16 | ACAGGCAATACCCATCATACTC | R | q-PCR | Human | 97 | |
| DNMT1 | CGGCCTCATCGAGAAGAATATC | F | q-PCR | Human | 95 | NM_001130823.3 |
| DNMT1 | TGCCATTAACACCACCTTCA | R | q-PCR | Human | 95 | |
| DNMT3B | GGAGCCACGACGTAACAAATA | F | q-PCR | Human | 98 | NM_006892.4 |
| DNMT3B | GTAAACTCTAGGCATCCGTCATC | R | q-PCR | Human | 98 | |
| SIRT2 | GGACAACAGAGAGGGAGAAAC | F | q-PCR | Human | 120 | AY030277.1 |
| SIRT2 | AGACAAGAACTGCTGGTTAAGA | R | q-PCR | Human | 120 | |
| SUV39H1 | CGAGGAGCTCACCTTTGATTAC | F | q-PCR | Human | 122 | NM_001282166.2 |
| SUV39H1 | CAATACGGACCCGCTTCTTAG | R | q-PCR | Human | 122 | |
| METTL3 | CACTGATGCTGTGTCCATCT | F | q-PCR | Human | 131 | NM_019852.5 |
| METTL3 | CTTGTAGGAGACCTCGCTTTAC | R | q-PCR | Human | 131 | |
| METTL14 | CCTGGGAATGAAGTCAGGATAG | F | q-PCR | Human | 119 | NM_020961.4 |
| METTL14 | CCCAGGGTATGGAACGTAATAG | R | q-PCR | Human | 119 | |
| 18S | CACGGACAGGATTGACAGATT | F | q-PCR | Human | 119 | NR_145820 |
| 18S | GCCAGAGTCTCGTTCGTTATC | R | q-PCR | Human | 119 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Q.; Vafaei, S.; Falahati, A.; Khosh, A.; Bariani, M.V.; Omran, M.M.; Bai, T.; Siblini, H.; Ali, M.; He, C.; et al. Bromodomain-Containing Protein 9 Regulates Signaling Pathways and Reprograms the Epigenome in Immortalized Human Uterine Fibroid Cells. Int. J. Mol. Sci. 2024, 25, 905. https://doi.org/10.3390/ijms25020905
Yang Q, Vafaei S, Falahati A, Khosh A, Bariani MV, Omran MM, Bai T, Siblini H, Ali M, He C, et al. Bromodomain-Containing Protein 9 Regulates Signaling Pathways and Reprograms the Epigenome in Immortalized Human Uterine Fibroid Cells. International Journal of Molecular Sciences. 2024; 25(2):905. https://doi.org/10.3390/ijms25020905
Chicago/Turabian StyleYang, Qiwei, Somayeh Vafaei, Ali Falahati, Azad Khosh, Maria Victoria Bariani, Mervat M. Omran, Tao Bai, Hiba Siblini, Mohamed Ali, Chuan He, and et al. 2024. "Bromodomain-Containing Protein 9 Regulates Signaling Pathways and Reprograms the Epigenome in Immortalized Human Uterine Fibroid Cells" International Journal of Molecular Sciences 25, no. 2: 905. https://doi.org/10.3390/ijms25020905