
Comparative Evaluation of STEAP1 Targeting Chimeric Antigen Receptors with Different Costimulatory Domains and Spacers
 , and
, and 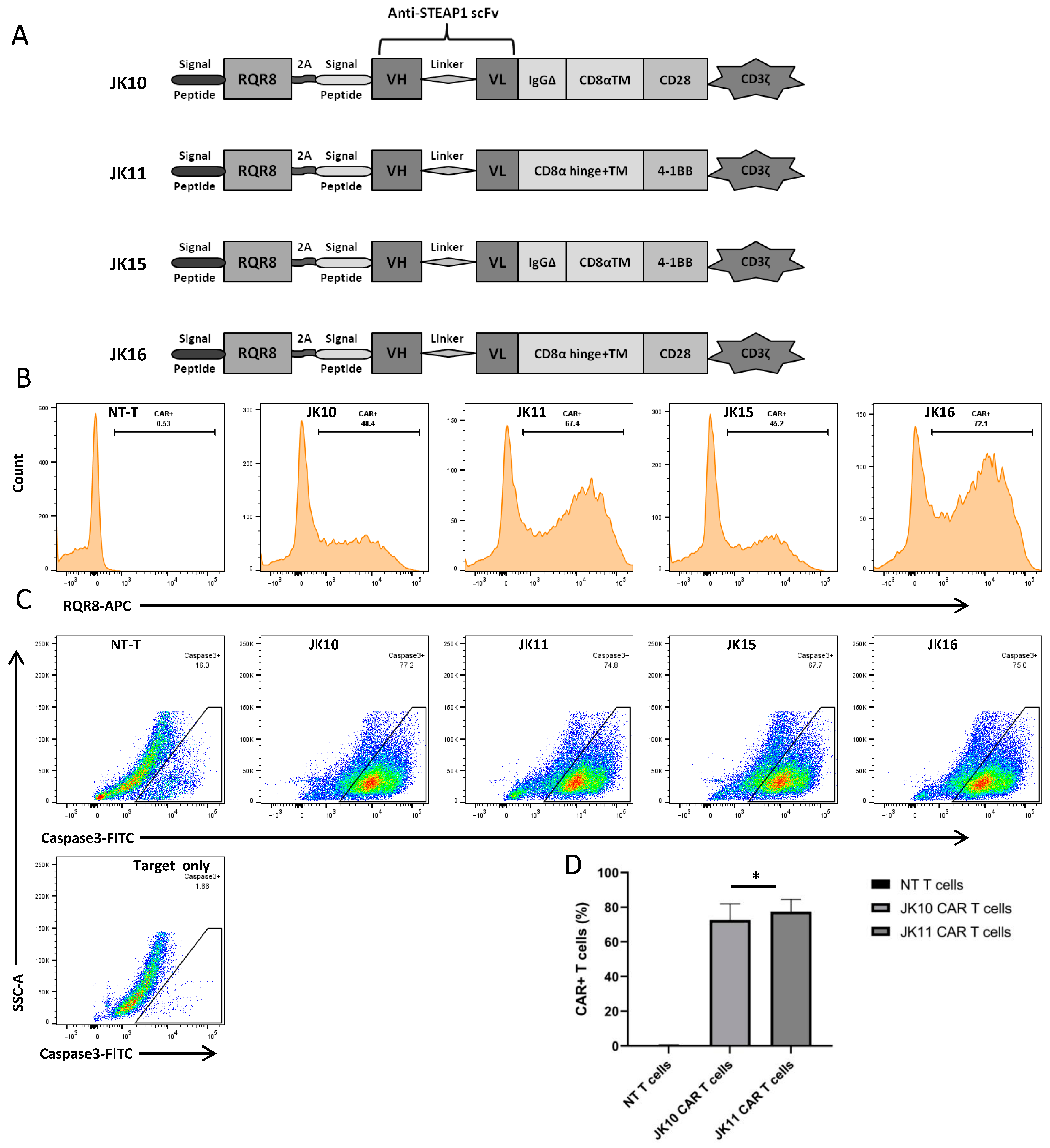{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Cloning and Expression of Alternative STEAP1 CAR Constructs
2.2. T Cells Expressing Four Alternative CAR T Variants Specifically Kill STEAP1+ Target Cells
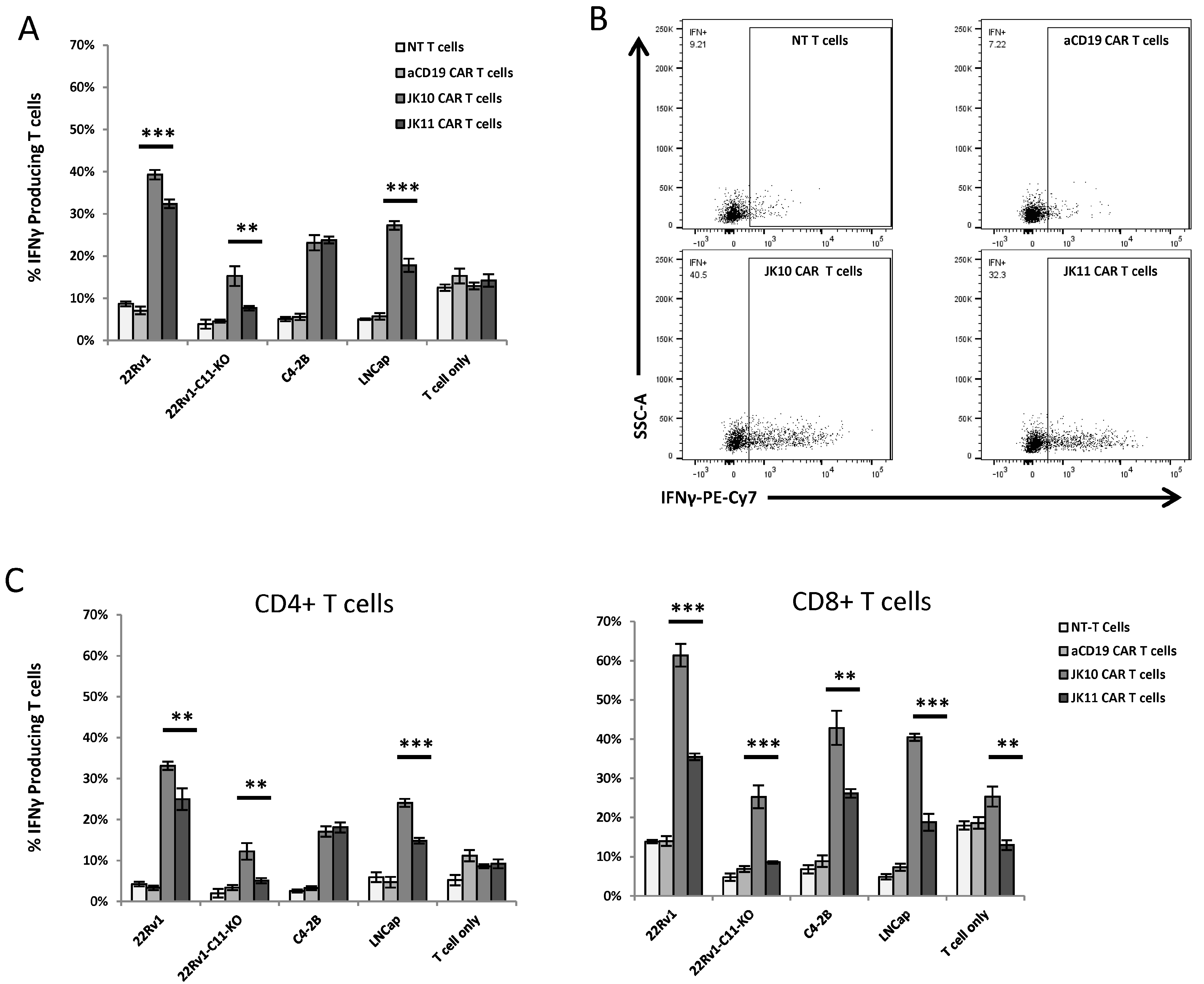
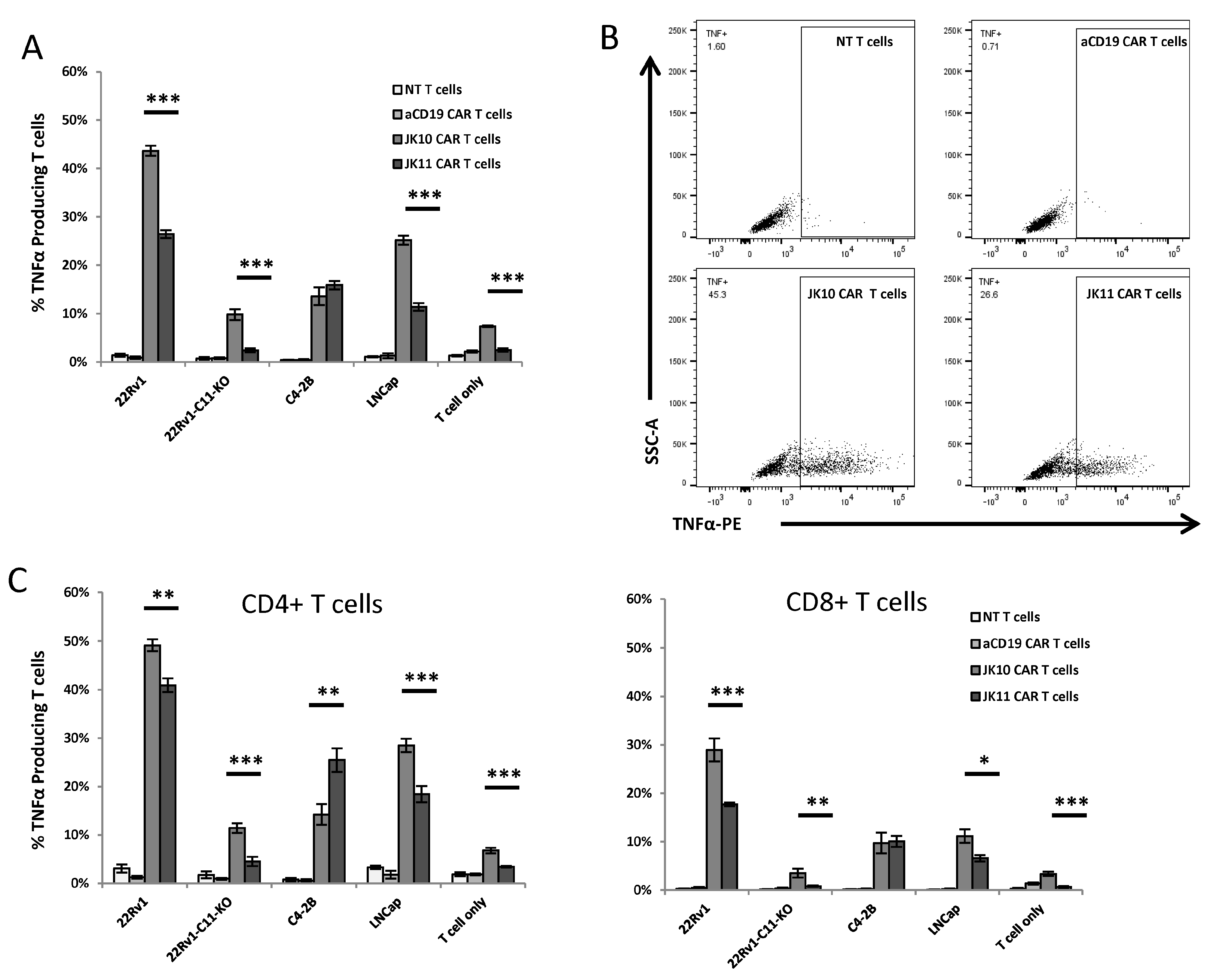
2.3. CD28-CAR T Cells Produce More IFNγ and TNFα Than 41BB-CAR T Cells
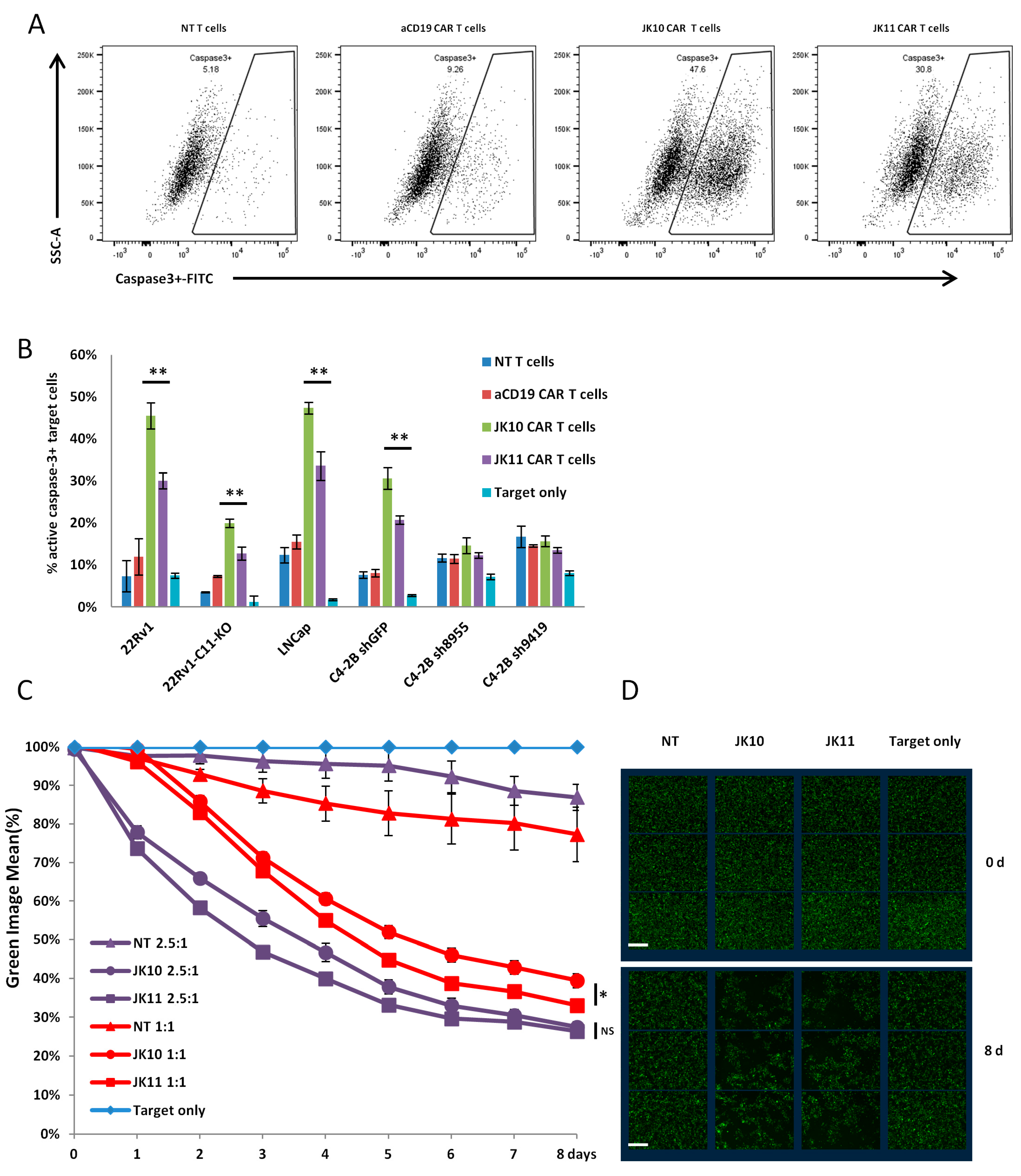
2.4. CD28-CAR T Cells Kill More STEAP1+ Targets Than 41BB-CAR T Cells in Short Term Assays
2.5. Efficacy of 41BB-CAR T Cells Is Not Inferior to CD28-CAR T Cells in Long-Term Killing Assays
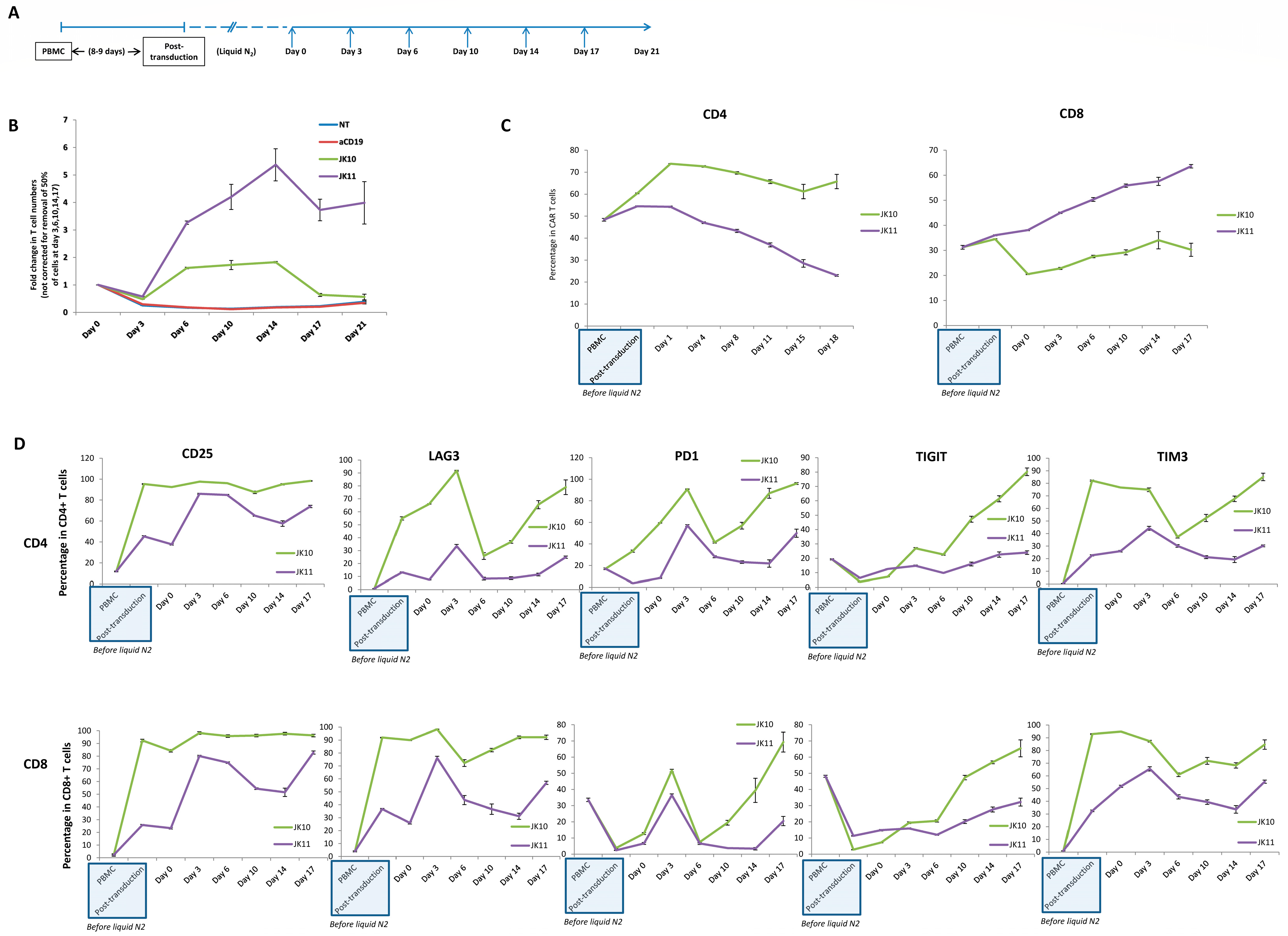
2.6. 41BB-CAR T Cells Exhibit Superior Expansion in Long-Term Assay with Repeated Stimulations
2.7. 41BB-CAR T Cells Express Lower Levels of Exhaustion Markers after Repeated Stimulations
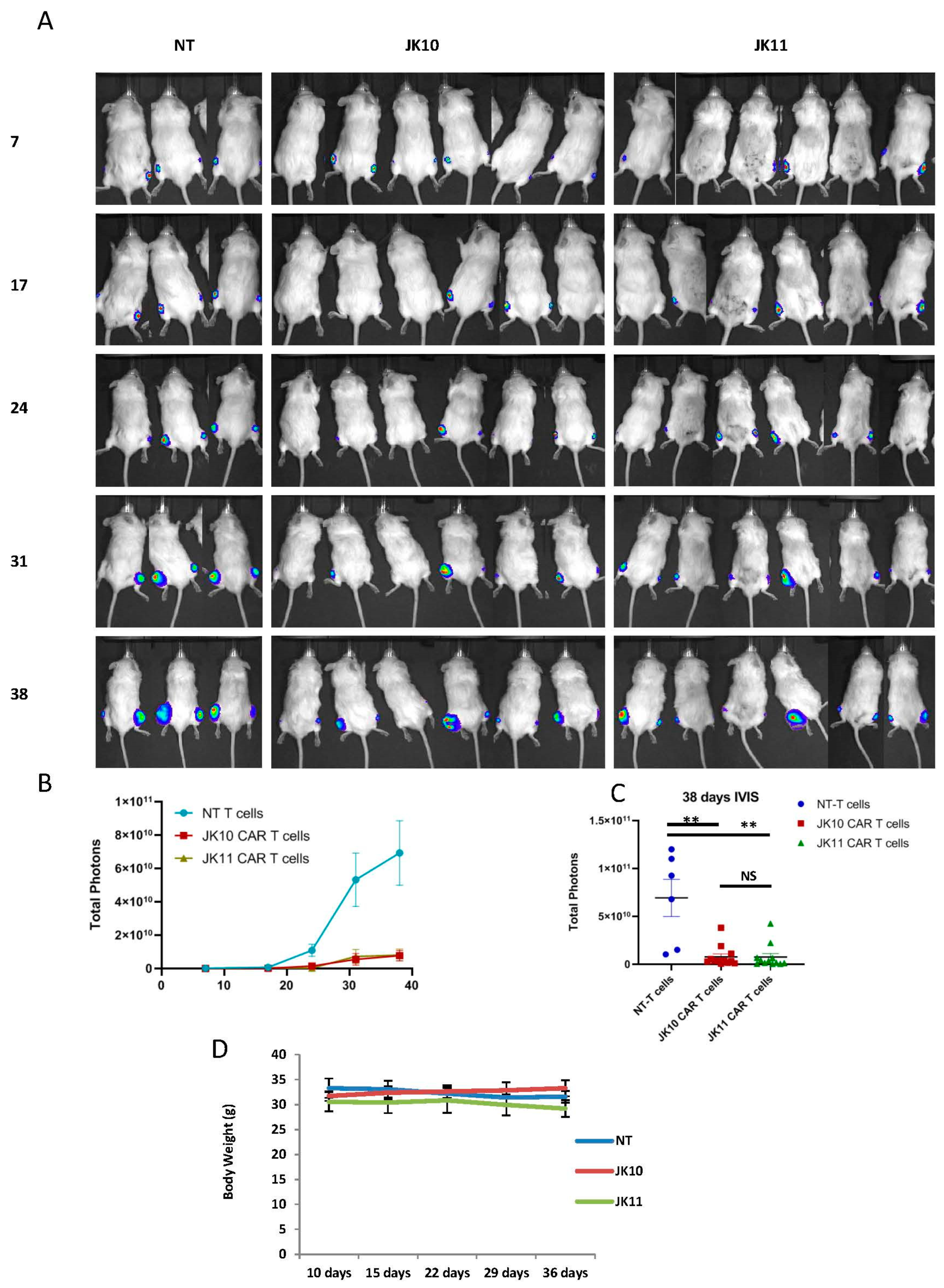
2.8. Both 41BB-CAR T Cells and CD28-CAR T Cells Have Robust Therapeutic Efficacy In Vivo
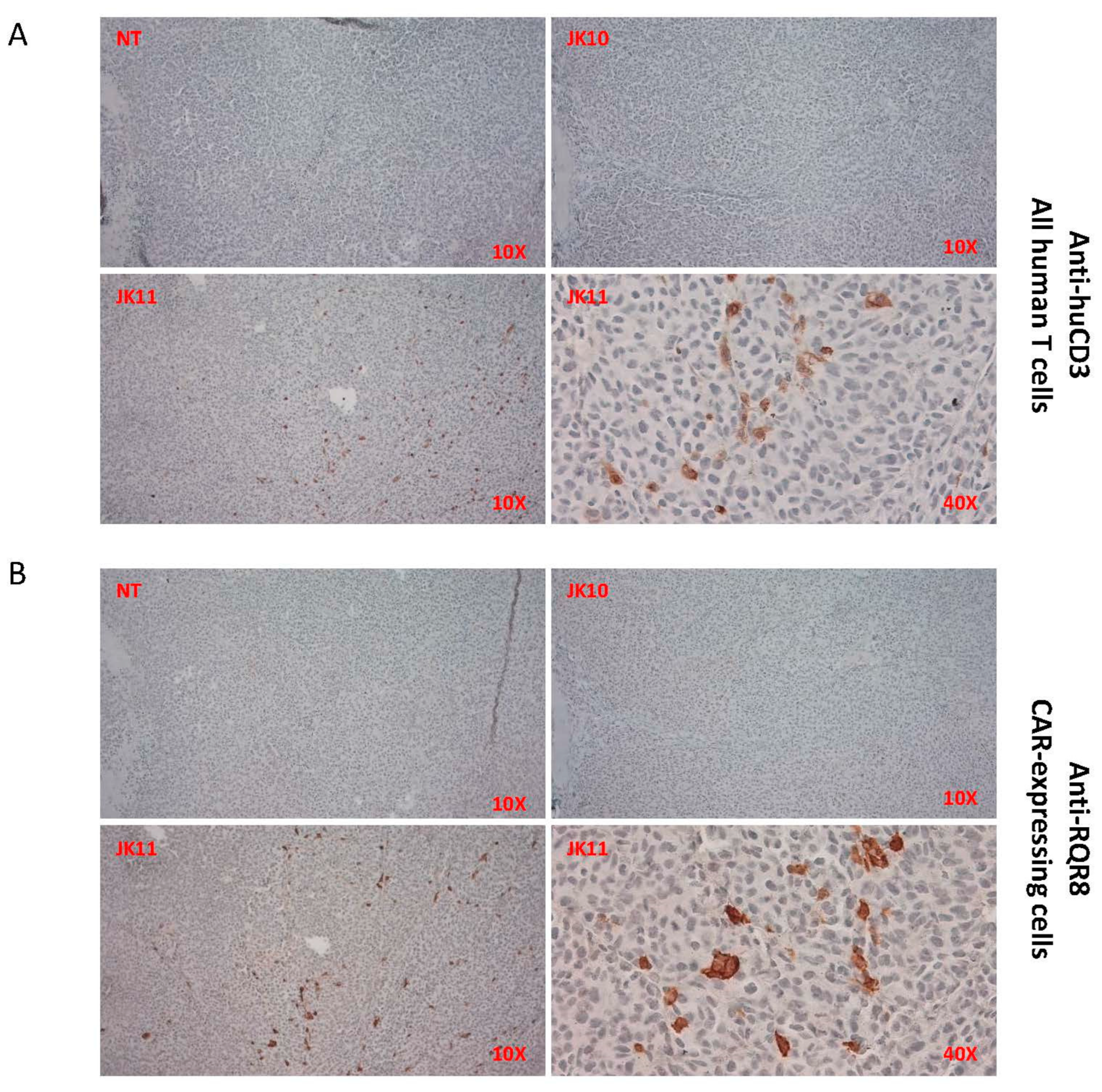
2.9. Tumor Infiltration by T Cells In Vivo Was Only Detected for 41BB-CAR T Cells
3. Discussion
4. Materials and Methods
4.1. CAR Retroviral Vector Construction
4.2. Retrovirus Production and CAR T Cell Transduction
4.3. Cell Cultures and Flow Cytometry T Cell Assays
4.4. CAR T Cell Proliferation and Phenotype under Longitudinal Repeated Target Exposure
4.5. Real-Time Killing Assay
4.6. In Vivo Xenograft Model Tumor Killing Assay
4.7. Immunohistochemistry (IHC)
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Weber, E.W.; Maus, M.V.; Mackall, C.L. The Emerging Landscape of Immune Cell Therapies. Cell 2020, 181, 46–62. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef]
- Abken, H. Adoptive therapy with CAR redirected T cells: The challenges in targeting solid tumors. Immunotherapy 2015, 7, 535–544. [Google Scholar] [CrossRef]
- Kyte, J.A. Strategies for Improving the Efficacy of CAR T Cells in Solid Cancers. Cancers 2022, 147, 571. [Google Scholar] [CrossRef]
- Del Bufalo, F.; De Angelis, B.; Caruana, I.; Del Baldo, G.; De Ioris, M.A.; Serra, A.; Mastronuzzi, A.; Cefalo, M.G.; Pagliara, D.; Amicucci, M.; et al. GD2-CART01 for Relapsed or Refractory High-Risk Neuroblastoma. N. Engl. J. Med. 2023, 388, 1284–1295. [Google Scholar] [CrossRef]
- Narayan, V.; Barber-Rotenberg, J.S.; Jung, I.Y.; Lacey, S.F.; Rech, A.J.; Davis, M.M.; Hwang, W.T.; Lal, P.; Carpenter, E.L.; Maude, S.L.; et al. PSMA-targeting TGFbeta-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: A phase 1 trial. Nat. Med. 2022, 28, 724–734. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef]
- Philipson, B.I.; O’Connor, R.S.; May, M.J.; June, C.H.; Albelda, S.M.; Milone, M.C. 4-1BB costimulation promotes CAR T cell survival through noncanonical NF-kappaB signaling. Sci. Signal 2020, 13, eaay8248. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Lorvik, K.B.; Jin, Y.; Beck, C.; Sike, A.; Persiconi, I.; Kvaloy, E.; Saatcioglu, F.; Dunn, C.; Kyte, J.A. Development of STEAP1 targeting chimeric antigen receptor for adoptive cell therapy against cancer. Mol. Ther. Oncolytics 2022, 26, 189–206. [Google Scholar] [CrossRef] [PubMed]
- Moreaux, J.; Kassambara, A.; Hose, D.; Klein, B. STEAP1 is overexpressed in cancers: A promising therapeutic target. Biochem. Biophys. Res. Commun. 2012, 429, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Gomes, I.M.; Maia, C.J.; Santos, C.R. STEAP proteins: From structure to applications in cancer therapy. Mol. Cancer Res. 2012, 10, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, T.G.; Diebold, I.; Esposito, I.; Plehm, S.; Hauer, K.; Thiel, U.; da Silva-Buttkus, P.; Neff, F.; Unland, R.; Muller-Tidow, C.; et al. STEAP1 is associated with the invasive and oxidative stress phenotype of Ewing tumors. Mol. Cancer Res. 2012, 10, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ji, G.; Xie, J.; Jiao, Z.; Zhang, H.; Chen, J. Six-transmembrane epithelial antigen of the prostate 1 is associated with tumor invasion and migration in endometrial carcinomas. J. Cell Biochem. 2019, 120, 11172–11189. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.Y.; Jiang, J.N.; Fang, X.D.; Ji, F.J. STEAP1 Regulates Tumorigenesis and Chemoresistance During Peritoneal Metastasis of Gastric Cancer. Front. Physiol. 2018, 9, 1132. [Google Scholar] [CrossRef]
- Jiang, J.N.; Wu, Y.Y.; Fang, X.D.; Ji, F.J. EIF4E regulates STEAP1 expression in peritoneal metastasis. J. Cancer 2020, 11, 990–996. [Google Scholar] [CrossRef]
- Barroca-Ferreira, J.; Pais, J.P.; Santos, M.M.; Goncalves, A.M.; Gomes, I.M.; Sousa, I.; Rocha, S.M.; Passarinha, L.A.; Maia, C.J. Targeting STEAP1 Protein in Human Cancer: Current Trends and Future Challenges. Curr. Cancer Drug Targets 2018, 18, 222–230. [Google Scholar] [CrossRef]
- Hubert, R.S.; Vivanco, I.; Chen, E.; Rastegar, S.; Leong, K.; Mitchell, S.C.; Madraswala, R.; Zhou, Y.; Kuo, J.; Raitano, A.B.; et al. STEAP: A prostate-specific cell-surface antigen highly expressed in human prostate tumors. Proc. Natl. Acad. Sci. USA 1999, 96, 14523–14528. [Google Scholar] [CrossRef] [PubMed]
- Markey, F.B.; Romero, B.; Parashar, V.; Batish, M. Identification of a New Transcriptional Co-Regulator of STEAP1 in Ewing’s Sarcoma. Cells 2021, 10, 1300. [Google Scholar] [CrossRef] [PubMed]
- Schober, S.J.; Thiede, M.; Gassmann, H.; Prexler, C.; Xue, B.; Schirmer, D.; Wohlleber, D.; Stein, S.; Grunewald, T.G.P.; Busch, D.H.; et al. MHC Class I-Restricted TCR-Transgenic CD4(+) T Cells Against STEAP1 Mediate Local Tumor Control of Ewing Sarcoma In Vivo. Cells 2020, 9, 1581. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, D.; Grunewald, T.G.; Klar, R.; Schmidt, O.; Wohlleber, D.; Rubio, R.A.; Uckert, W.; Thiel, U.; Bohne, F.; Busch, D.H.; et al. Transgenic antigen-specific, HLA-A*02:01-allo-restricted cytotoxic T cells recognize tumor-associated target antigen STEAP1 with high specificity. Oncoimmunology 2016, 5, e1175795. [Google Scholar] [CrossRef] [PubMed]
- Danila, D.C.; Szmulewitz, R.Z.; Vaishampayan, U.; Higano, C.S.; Baron, A.D.; Gilbert, H.N.; Brunstein, F.; Milojic-Blair, M.; Wang, B.; Kabbarah, O.; et al. Phase I Study of DSTP3086S, an Antibody-Drug Conjugate Targeting Six-Transmembrane Epithelial Antigen of Prostate 1, in Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2019, 37, 3518–3527. [Google Scholar] [CrossRef] [PubMed]
- Kelly, W.; Danila, D.; Lin, C.C.; Lee, J.L.; Matsubara, N.; Ward, P.; Armstrong, A.J.; Pook, D.; Kim, M.; Dorff, T.; et al. 1765O Interim results from a phase I study of AMG 509 (xaluritamig), a STEAP1 x CD3 XmAb 2+1 immune therapy, in patients with metastatic castration-resistant prostate cancer (mCRPC). Ann. Oncol. 2023, 34, S953–S954. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.; Riviere, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef]
- Sun, C.; Shou, P.; Du, H.; Hirabayashi, K.; Chen, Y.; Herring, L.E.; Ahn, S.; Xu, Y.; Suzuki, K.; Li, G.; et al. THEMIS-SHP1 Recruitment by 4-1BB Tunes LCK-Mediated Priming of Chimeric Antigen Receptor-Redirected T Cells. Cancer Cell 2020, 37, 216–225.e216. [Google Scholar] [CrossRef]
- Zhao, Z.; Condomines, M.; van der Stegen, S.J.C.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef]
- Almasbak, H.; Walseng, E.; Kristian, A.; Myhre, M.R.; Suso, E.M.; Munthe, L.A.; Andersen, J.T.; Wang, M.Y.; Kvalheim, G.; Gaudernack, G.; et al. Inclusion of an IgG1-Fc spacer abrogates efficacy of CD19 CAR T cells in a xenograft mouse model. Gene Ther. 2015, 22, 391–403. [Google Scholar] [CrossRef]
- Kintz, H.; Nylen, E.; Barber, A. Inclusion of Dap10 or 4-1BB costimulation domains in the chPD1 receptor enhances anti-tumor efficacy of T cells in murine models of lymphoma and melanoma. Cell Immunol. 2020, 351, 104069. [Google Scholar] [CrossRef] [PubMed]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B.; et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, S.; Picco, G.; Foster, J.; Davies, D.M.; Julien, S.; Cooper, L.; Arif, S.; Mather, S.J.; Taylor-Papadimitriou, J.; Burchell, J.M.; et al. Retargeting of human T cells to tumor-associated MUC1: The evolution of a chimeric antigen receptor. J. Immunol. 2008, 180, 4901–4909. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.; Hombach, A.A.; Abken, H. Adoptive immunotherapy with genetically engineered T cells: Modification of the IgG1 Fc ‘spacer’ domain in the extracellular moiety of chimeric antigen receptors avoids ‘off-target’ activation and unintended initiation of an innate immune response. Gene Ther. 2010, 17, 1206–1213. [Google Scholar] [CrossRef] [PubMed]
- Philip, B.; Kokalaki, E.; Mekkaoui, L.; Thomas, S.; Straathof, K.; Flutter, B.; Marin, V.; Marafioti, T.; Chakraverty, R.; Linch, D.; et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood 2014, 124, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.; Casey, N.P.; Persiconi, I.; Moharrami, N.N.; Sike, A.; Jin, Y.; Kyte, J.A. Development of a TGFbeta-IL-2/15 Switch Receptor for Use in Adoptive Cell Therapy. Biomedicines 2023, 11, 459. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef]
- Kyte, J.A.; Fane, A.; Pule, M.; Gaudernack, G. Transient redirection of T cells for adoptive cell therapy with telomerase-specific T helper cell receptors isolated from long term survivors after cancer vaccination. Oncoimmunology 2019, 8, e1565236. [Google Scholar] [CrossRef]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Bhatia, V.; Kamat, N.V.; Pariva, T.E.; Wu, L.T.; Tsao, A.; Sasaki, K.; Sun, H.; Javier, G.; Nutt, S.; Coleman, I.; et al. Targeting advanced prostate cancer with STEAP1 chimeric antigen receptor T cell and tumor-localized IL-12 immunotherapy. Nat. Commun. 2023, 14, 2041. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, Y.; Dunn, C.; Persiconi, I.; Sike, A.; Skorstad, G.; Beck, C.; Kyte, J.A. Comparative Evaluation of STEAP1 Targeting Chimeric Antigen Receptors with Different Costimulatory Domains and Spacers. Int. J. Mol. Sci. 2024, 25, 586. https://doi.org/10.3390/ijms25010586
Jin Y, Dunn C, Persiconi I, Sike A, Skorstad G, Beck C, Kyte JA. Comparative Evaluation of STEAP1 Targeting Chimeric Antigen Receptors with Different Costimulatory Domains and Spacers. International Journal of Molecular Sciences. 2024; 25(1):586. https://doi.org/10.3390/ijms25010586
Chicago/Turabian StyleJin, Yixin, Claire Dunn, Irene Persiconi, Adam Sike, Gjertrud Skorstad, Carole Beck, and Jon Amund Kyte. 2024. "Comparative Evaluation of STEAP1 Targeting Chimeric Antigen Receptors with Different Costimulatory Domains and Spacers" International Journal of Molecular Sciences 25, no. 1: 586. https://doi.org/10.3390/ijms25010586