
The p66Shc Redox Protein and the Emerging Complications of Diabetes

 , ,
, ,  , , and
, , and 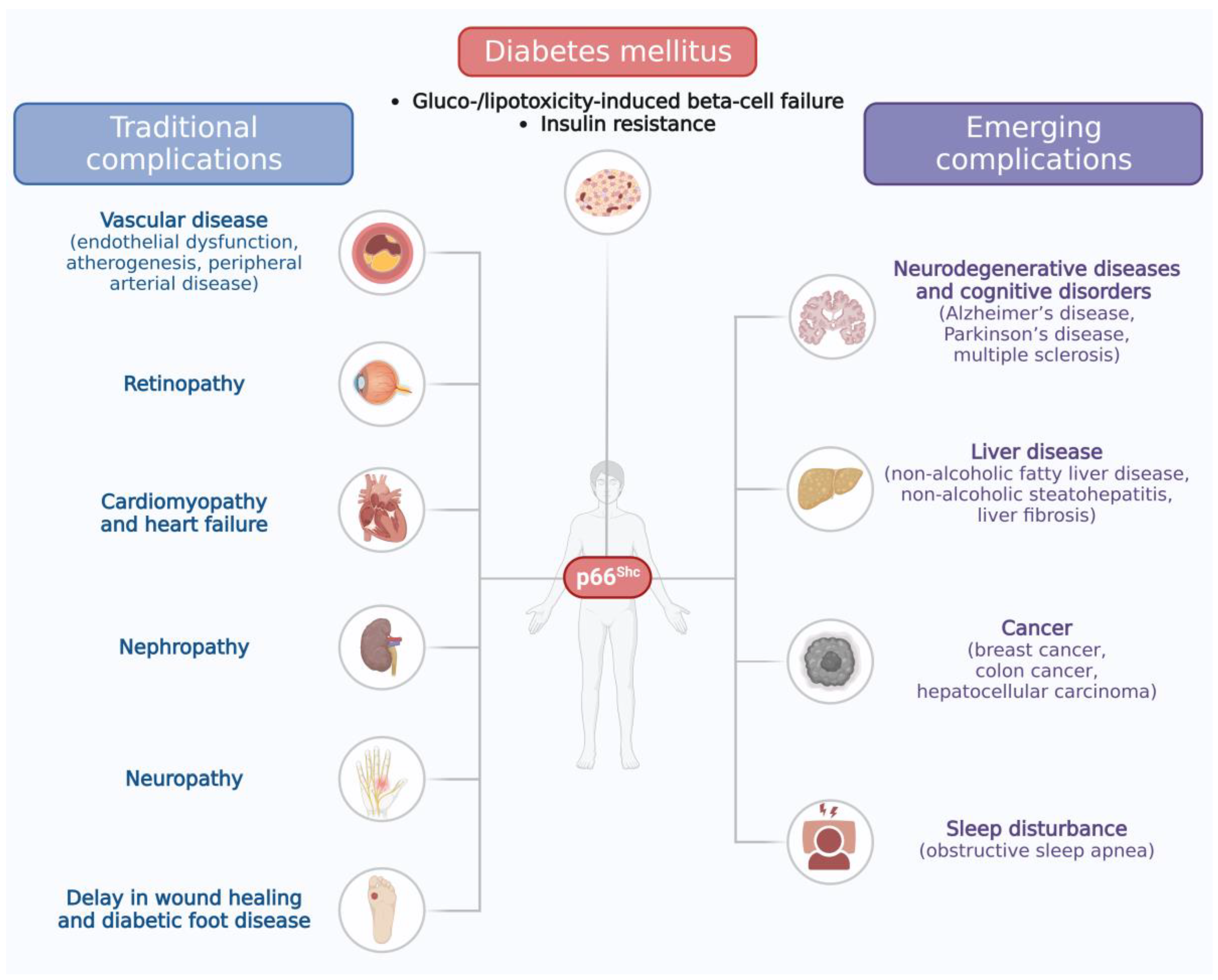{kind=link}
Abstract
:1. Introduction
2. The Role of p66Shc in Diabetes and Its Traditional Complications
2.1. The Role of p66Shc in Pancreatic Beta-Cell Failure and Insulin Resistance
2.2. The Role of p66Shc in Traditional Complications of Diabetes
3. The Role of the p66Shc Protein in Emerging Complications of Diabetes
3.1. Liver Disease
3.2. Cancer
3.3. Neurodegenerative Diseases and Cognitive Disorders
3.4. Sleep Disturbance
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- IDF DIABETES ATLAS [Internet]. 10th Edition. Books—NCBI. Available online: https://www.ncbi.nlm.nih.gov/books/NBK581934/?term=IDF%20DIABETES%20ATLAS%20%5BInternet%5D.%2010th%20edition (accessed on 23 August 2023).
- Marrano, N.; Biondi, G.; Cignarelli, A.; Perrini, S.; Laviola, L.; Giorgino, F.; Natalicchio, A. Functional Loss of Pancreatic Islets in Type 2 Diabetes: How Can We Halt It? Metabolism 2020, 110, 154304. [Google Scholar] [CrossRef] [PubMed]
- Lytrivi, M.; Castell, A.L.; Poitout, V.; Cnop, M. Recent Insights Into Mechanisms of β-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [Google Scholar] [CrossRef] [PubMed]
- Halban, P.A.; Polonsky, K.S.; Bowden, D.W.; Hawkins, M.A.; Ling, C.; Mather, K.J.; Powers, A.C.; Rhodes, C.J.; Sussel, L.; Weir, G.C. β-Cell Failure in Type 2 Diabetes: Postulated Mechanisms and Prospects for Prevention and Treatment. J. Clin. Endocrinol. Metab. 2014, 99, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Biondi, G.; Marrano, N.; Borrelli, A.; Rella, M.; Palma, G.; Calderoni, I.; Siciliano, E.; Lops, P.; Giorgino, F.; Natalicchio, A. Adipose Tissue Secretion Pattern Influences β-Cell Wellness in the Transition from Obesity to Type 2 Diabetes. Int. J. Mol. Sci. 2022, 23, 105522. [Google Scholar] [CrossRef] [PubMed]
- Fowler, M.J. Microvascular and Macrovascular Complications of Diabetes. CliniCal Diabetes 2008, 26, 77–82. [Google Scholar] [CrossRef]
- Shah, A.D.; Langenberg, C.; Rapsomaniki, E.; Denaxas, S.; Pujades-Rodriguez, M.; Gale, C.P.; Deanfield, J.; Smeeth, L.; Timmis, A.; Hemingway, H. Type 2 Diabetes and Incidence of Cardiovascular Diseases: A Cohort Study in 1.9 Million People. Lancet Diabetes Endocrinol. 2015, 3, 105–113. [Google Scholar] [CrossRef]
- Constantino, M.I.; Molyneaux, L.; Limacher-Gisler, F.; Al-Saeed, A.; Luo, C.; Wu, T.; Twigg, S.M.; Yue, D.K.; Wong, J. Long-Term Complications and Mortality in Young-Onset Diabetes: Type 2 Diabetes Is More Hazardous and Lethal than Type 1 Diabetes. Diabetes Care 2013, 36, 3863–3869. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Lau, E.S.H.; Wu, H.; Yang, A.; Chow, E.; Kong, A.P.S.; Ma, R.C.W.; Chan, J.C.N.; Luk, A.O.Y. Higher Incidence of Cardiovascular-Kidney Complications in Chinese with Youth-Onset Type 2 Diabetes versus Youth-Onset Type 1 Diabetes Attenuated by Control of Cardio-Metabolic Risk Factors: A Population-Based Prospective Cohort Study in Hong Kong. Diabetes Res. Clin. Pract. 2023, 202, 110728. [Google Scholar] [CrossRef]
- Amutha, A.; Anjana, R.M.; Venkatesan, U.; Ranjani, H.; Unnikrishnan, R.; Venkat Narayan, K.M.; Mohan, V.; Ali, M.K. Incidence of Complications in Young-Onset Diabetes: Comparing Type 2 with Type 1 (the Young Diab Study). Diabetes Res. Clin. Pract. 2017, 123, 1–8. [Google Scholar] [CrossRef]
- Wong, J.; Constantino, M.; Yue, D.K. Morbidity and Mortality in Young-Onset Type 2 Diabetes in Comparison to Type 1 Diabetes: Where Are We Now? Curr. Diab Rep. 2015, 15, 566. [Google Scholar] [CrossRef]
- Dabelea, D.; Stafford, J.M.; Mayer-Davis, E.J.; D’Agostino, R.; Dolan, L.; Imperatore, G.; Linder, B.; Lawrence, J.M.; Marcovina, S.M.; Mottl, A.K.; et al. Association of Type 1 Diabetes vs Type 2 Diabetes Diagnosed During Childhood and Adolescence with Complications during Teenage Years and Young Adulthood. JAMA 2017, 317, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Gregg, E.W.; Li, Y.; Wang, J.; Rios Burrows, N.; Ali, M.K.; Rolka, D.; Williams, D.E.; Geiss, L. Changes in Diabetes-Related Complications in the United States, 1990–2010. N. Engl. J. Med. 2014, 370, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Gregg, E.W.; Sattar, N.; Ali, M.K. The Changing Face of Diabetes Complications. Lancet Diabetes Endocrinol. 2016, 4, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Pearson-Stuttard, J.; Buckley, J.; Cicek, M.; Gregg, E.W. The Changing Nature of Mortality and Morbidity in Patients with Diabetes. Endocrinol. Metab. Clin. North. Am. 2021, 50, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Einarson, T.R.; Acs, A.; Ludwig, C.; Panton, U.H. Prevalence of Cardiovascular Disease in Type 2 Diabetes: A Systematic Literature Review of Scientific Evidence from across the World in 2007–2017. Cardiovasc. Diabetol. 2018, 17, 83. [Google Scholar] [CrossRef] [PubMed]
- Pearson-Stuttard, J.; Bennett, J.; Cheng, Y.J.; Vamos, E.P.; Cross, A.J.; Ezzati, M.; Gregg, E.W. Trends in Predominant Causes of Death in Individuals with and without Diabetes in England from 2001 to 2018: An Epidemiological Analysis of Linked Primary Care Records. Lancet Diabetes Endocrinol. 2021, 9, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.L.; Shaw, J.E.; Peeters, A.; Davidson, S.; Magliano, D.J. Age-Specific Trends from 2000–2011 in All-Cause and Cause-Specific Mortality in Type 1 and Type 2 Diabetes: A Cohort Study of More Than One Million People. Diabetes Care 2016, 39, 1018–1026. [Google Scholar] [CrossRef]
- Gregg, E.W.; Cheng, Y.J.; Srinivasan, M.; Lin, J.; Geiss, L.S.; Albright, A.L.; Imperatore, G. Trends in Cause-Specific Mortality among Adults with and without Diagnosed Diabetes in the USA: An Epidemiological Analysis of Linked National Survey and Vital Statistics Data. Lancet 2018, 391, 2430–2440. [Google Scholar] [CrossRef]
- Tomic, D.; Shaw, J.E.; Magliano, D.J. The Burden and Risks of Emerging Complications of Diabetes Mellitus. Nat. Rev. Endocrinol. 2022, 18, 525–539. [Google Scholar] [CrossRef]
- Harding, J.L.; Pavkov, M.E.; Magliano, D.J.; Shaw, J.E.; Gregg, E.W. Global Trends in Diabetes Complications: A Review of Current Evidence. Diabetologia 2019, 62, 3–16. [Google Scholar] [CrossRef]
- Tsilidis, K.K.; Kasimis, J.C.; Lopez, D.S.; Ntzani, E.E.; Ioannidis, J.P.A. Type 2 Diabetes and Cancer: Umbrella Review of Meta-Analyses of Observational Studies. BMJ 2015, 350, 7607. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Peters, S.A.E.; Woodward, M.; Arango, S.M.; Batty, G.D.; Beckett, N.; Beiser, A.; Borenstein, A.R.; Crane, P.K.; Haan, M.; et al. Type 2 Diabetes as a Risk Factor for Dementia in Women Compared with Men: A Pooled Analysis of 2.3 Million People Comprising More Than 100,000 Cases of Dementia. Diabetes Care 2016, 39, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Tolman, K.G.; Fonseca, V.; Dalpiaz, A.; Tan, M.H. Spectrum of Liver Disease in Type 2 Diabetes and Management of Patients with Diabetes and Liver Disease. Diabetes Care 2007, 30, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Pearson-Stuttard, J.; Blundell, S.; Harris, T.; Cook, D.G.; Critchley, J. Diabetes and Infection: Assessing the Association with Glycaemic Control in Population-Based Studies. Lancet Diabetes Endocrinol. 2016, 4, 148–158. [Google Scholar] [CrossRef]
- Pearson-Stuttard, J.; Cheng, Y.J.; Bennett, J.; Vamos, E.P.; Zhou, B.; Valabhji, J.; Cross, A.J.; Ezzati, M.; Gregg, E.W. Trends in Leading Causes of Hospitalisation of Adults with Diabetes in England from 2003 to 2018: An Epidemiological Analysis of Linked Primary Care Records. Lancet Diabetes Endocrinol. 2022, 10, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Pelicci, G.; Lanfrancone, L.; Grignani, F.; McGlade, J.; Cavallo, F.; Forni, G.; Nicoletti, I.; Grignani, F.; Pawson, T.; Pelicci, P.G. A Novel Transforming Protein (SHC) with an SH2 Domain Is Implicated in Mitogenic Signal Transduction. Cell 1992, 70, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Perrini, S.; Tortosa, F.; Natalicchio, A.; Pacelli, C.; Cignarelli, A.; Palmieri, V.O.; Caccioppoli, C.; De Stefano, F.; Porro, S.; Leonardini, A.; et al. The P66Shc Protein Controls Redox Signaling and Oxidation-dependent DNA Damage in Human Liver Cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, 826–840. [Google Scholar] [CrossRef] [PubMed]
- Biondi, G.; Marrano, N.; Dipaola, L.; Borrelli, A.; Rella, M.; D’oria, R.; Genchi, V.A.; Caccioppoli, C.; Porreca, I.; Cignarelli, A.; et al. The P66Shc Protein Mediates Insulin Resistance and Secretory Dysfunction in Pancreatic β-Cells under Lipotoxic Conditions. Diabetes 2022, 71, 1763–1771. [Google Scholar] [CrossRef]
- Natalicchio, A.; Tortosa, F.; Labarbuta, R.; Biondi, G.; Marrano, N.; Carchia, E.; Leonardini, A.; Cignarelli, A.; Bugliani, M.; Marchetti, P.; et al. The P66(Shc) Redox Adaptor Protein Is Induced by Saturated Fatty Acids and Mediates Lipotoxicity-Induced Apoptosis in Pancreatic Beta Cells. Diabetologia 2015, 58, 1260–1271. [Google Scholar] [CrossRef]
- Natalicchio, A.; Laviola, L.; De Tullio, C.; Renna, L.A.; Montrone, C.; Perrini, S.; Valenti, G.; Procino, G.; Svelto, M.; Giorgino, F. Role of the P66Shc Isoform in Insulin-like Growth Factor I Receptor Signaling through MEK/Erk and Regulation of Actin Cytoskeleton in Rat Myoblasts. J. Biol. Chem. 2004, 279, 43900–43909. [Google Scholar] [CrossRef]
- Migliaccio, E.; Giogio, M.; Mele, S.; Pelicci, G.; Reboldi, P.; Pandolfi, P.P.; Lanfrancone, L.; Pelicci, P.G. The P66shc Adaptor Protein Controls Oxidative Stress Response and Life Span in Mammals. Nature 1999, 402, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Natalicchio, A.; De Stefano, F.; Perrini, S.; Laviola, L.; Cignarelli, A.; Caccioppoli, C.; Quagliara, A.; Melchiorre, M.; Leonardini, A.; Conserva, A.; et al. Involvement of the P66Shc Protein in Glucose Transport Regulation in Skeletal Muscle Myoblasts. Am. J. Physiol. Endocrinol. Metab. 2009, 296, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Natalicchio, A.; Tortosa, F.; Perrini, S.; Laviola, L.; Giorgino, F. P66Shc, a Multifaceted Protein Linking Erk Signalling, Glucose Metabolism, and Oxidative Stress. Arch. Physiol. Biochem. 2011, 117, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Xi, G.; Shen, X.; Radhakrishnan, Y.; Maile, L.; Clemmons, D. Hyperglycemia-Induced P66shc Inhibits Insulin-like Growth Factor I-Dependent Cell Survival via Impairment of Src Kinase-Mediated Phosphoinositide-3 Kinase/AKT Activation in Vascular Smooth Muscle Cells. Endocrinology 2010, 151, 3611–3623. [Google Scholar] [CrossRef]
- Zhang, M.; Tang, J.; Shan, H.; Zhang, Q.; Yang, X.; Zhang, J.; Li, Y. P66Shc Mediates Mitochondrial Dysfunction Dependent on PKC Activation in Airway Epithelial Cells Induced by Cigarette Smoke. Oxid. Med. Cell Longev. 2018, 2018, 5837123. [Google Scholar] [CrossRef] [PubMed]
- Laviola, L.; Orlando, M.R.; Incalza, M.A.; Caccioppoli, C.; Melchiorre, M.; Leonardini, A.; Cignarelli, A.; Tortosa, F.; Labarbuta, R.; Martemucci, S.; et al. TNFα Signals via P66(Shc) to Induce E-Selectin, Promote Leukocyte Transmigration and Enhance Permeability in Human Endothelial Cells. PLoS ONE 2013, 8, 81930. [Google Scholar] [CrossRef] [PubMed]
- Yousef, H.; Khandoker, A.H.; Feng, S.F.; Helf, C.; Jelinek, H.F. Inflammation, Oxidative Stress and Mitochondrial Dysfunction in the Progression of Type II Diabetes Mellitus with Coexisting Hypertension. Front. Endocrinol. 2023, 14, 1173402. [Google Scholar] [CrossRef]
- Jelinek, H.F.; Helf, C.; Khalaf, K. Human SHC-Transforming Protein 1 and Its Isoforms P66shc: A Novel Marker for Prediabetes. J. Diabetes Investig. 2021, 12, 1881–1889. [Google Scholar] [CrossRef]
- Pagnin, E.; Fadini, G.; De Toni, R.; Tiengo, A.; Calò, L.; Avogaro, A. Diabetes Induces P66shc Gene Expression in Human Peripheral Blood Mononuclear Cells: Relationship to Oxidative Stress. J. Clin. Endocrinol. Metab. 2005, 90, 1130–1136. [Google Scholar] [CrossRef]
- Huang, T.-T.; Sun, W.-J.; Liu, H.-Y.; Ma, H.-L.; Cui, B.-X. P66Shc-Mediated Oxidative Stress Is Involved in Gestational Diabetes Mellitus. World J. Diabetes 2021, 12, 1894–1907. [Google Scholar] [CrossRef]
- Karunakaran, U.; Elumalai, S.; Moon, J.S.; Chang Won, K. CD36 Dependent Redoxosomes Promotes Ceramide-Mediated Pancreatic β-Cell Failure via P66Shc Activation. Free Radic. Biol. Med. 2019, 134, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, S.C.; Fusco, S.; Panieri, E.; Labate, V.; Mele, M.; Tesori, V.; Ferrara, A.M.; Maulucci, G.; De Spirito, M.; Martorana, G.E.; et al. Mammalian Life-Span Determinant P66shcA Mediates Obesity-Induced Insulin Resistance. Proc. Natl. Acad. Sci. USA 2010, 107, 13420–13425. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Xu, D.; Wu, X.; Xu, F.; Lin, L.; Zhou, H. Isosteviol Protects Free Fatty Acid- and High Fat Diet-Induced Hepatic Injury via Modulating PKC-β/P66Shc/ROS and Endoplasmic Reticulum Stress Pathways. Antioxid. Redox Signal. 2019, 30, 1949–1968. [Google Scholar] [CrossRef] [PubMed]
- Ciciliot, S.; Fadini, G.P. Modulation of Obesity and Insulin Resistance by the Redox Enzyme and Adaptor Protein P66Shc. Int. J. Mol. Sci. 2019, 20, 985. [Google Scholar] [CrossRef] [PubMed]
- Paneni, F.; Costantino, S.; Cosentino, F. P66(Shc)-Induced Redox Changes Drive Endothelial Insulin Resistance. Atherosclerosis 2014, 236, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Tomilov, A.A.; Ramsey, J.J.; Hagopian, K.; Giorgio, M.; Kim, K.M.; Lam, A.; Migliaccio, E.; Lloyd, K.C.; Berniakovich, I.; Prolla, T.A.; et al. The Shc Locus Regulates Insulin Signaling and Adiposity in Mammals. Aging Cell 2011, 10, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Soliman, M.A.; Abdel Rahman, A.M.; Lamming, D.A.; Birsoy, K.; Pawling, J.; Frigolet, M.E.; Lu, H.; Fantus, I.G.; Pasculescu, A.; Zheng, Y.; et al. The Adaptor Protein P66Shc Inhibits MTOR-Dependent Anabolic Metabolism. Sci. Signal. 2014, 7, 17. [Google Scholar] [CrossRef]
- Ciciliot, S.; Albiero, M.; Menegazzo, L.; Poncina, N.; Scattolini, V.; Danesi, A.; Pagnin, E.; Marabita, M.; Blaauw, B.; Giorgio, M.; et al. P66Shc Deletion or Deficiency Protects from Obesity but Not Metabolic Dysfunction in Mice and Humans. Diabetologia 2015, 58, 2352–2360. [Google Scholar] [CrossRef]
- Ciciliot, S.; Albiero, M.; Campanaro, S.; Poncina, N.; Tedesco, S.; Scattolini, V.; Costa, F.D.; Cignarella, A.; Vettore, M.; Di Gangi, I.M.; et al. Interplay between Gut Microbiota and P66Shc Affects Obesity-Associated Insulin Resistance. FASEB J. 2018, 32, 4004–4015. [Google Scholar] [CrossRef]
- Menini, S.; Amadio, L.; Oddi, G.; Ricci, C.; Pesce, C.; Pugliese, F.; Giorgio, M.; Migliaccio, E.; Pelicci, P.G.; Iacobini, C.; et al. Deletion of P66Shc Longevity Gene Protects against Experimental Diabetic Glomerulopathy by Preventing Diabetes-Induced Oxidative Stress. Diabetes 2006, 55, 1642–1650. [Google Scholar] [CrossRef]
- Camici, G.G.; Schiavoni, M.; Francia, P.; Bachschmid, M.; Martin-Padura, I.; Hersberger, M.; Tanner, F.C.; Pelicci, P.G.; Volpe, M.; Anversa, P.; et al. Genetic Deletion of P66(Shc) Adaptor Protein Prevents Hyperglycemia-Induced Endothelial Dysfunction and Oxidative Stress. Proc. Natl. Acad. Sci. USA 2007, 104, 5217–5222. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Kim, Y.R.; Vikram, A.; Kumar, S.; Kassan, M.; Gabani, M.; Lee, S.K.; Jacobs, J.S.; Irani, K. P66Shc-Induced MicroRNA-34a Causes Diabetic Endothelial Dysfunction by Downregulating Sirtuin1. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2394–2403. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Chen, H.Z.; Wan, Y.Z.; Zhang, Q.J.; Wei, Y.S.; Huang, S.; Liu, J.J.; Lu, Y.B.; Zhang, Z.Q.; Yang, R.F.; et al. Repression of P66Shc Expression by SIRT1 Contributes to the Prevention of Hyperglycemia-Induced Endothelial Dysfunction. Circ. Res. 2011, 109, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Albiero, M.; Menegazzo, L.; Boscaro, E.; Pagnin, E.; Iori, E.; Cosma, C.; Lapolla, A.; Pengo, V.; Stendardo, M.; et al. The Redox Enzyme P66Shc Contributes to Diabetes and Ischemia-Induced Delay in Cutaneous Wound Healing. Diabetes 2010, 59, 2306–2314. [Google Scholar] [CrossRef] [PubMed]
- Rota, M.; LeCapitaine, N.; Hosoda, T.; Boni, A.; De Angelis, A.; Padin-Iruegas, M.E.; Esposito, G.; Vitale, S.; Urbanek, K.; Casarsa, C.; et al. Diabetes Promotes Cardiac Stem Cell Aging and Heart Failure, Which Are Prevented by Deletion of the P66shc Gene. Circ. Res. 2006, 99, 42–52. [Google Scholar] [CrossRef]
- Francia, P.; Cosentino, F.; Schiavoni, M.; Huang, Y.; Perna, E.; Camici, G.G.; Lüscher, T.F.; Volpe, M. P66(Shc) Protein, Oxidative Stress, and Cardiovascular Complications of Diabetes: The Missing Link. J. Mol. Med. 2009, 87, 885–891. [Google Scholar] [CrossRef]
- Zheng, D.; Tao, M.; Liang, X.; Li, Y.; Jin, J.; He, Q. P66Shc Regulates Podocyte Autophagy in High Glucose Environment Through the Notch-PTEN-PI3K/Akt/mTOR Pathway. Histol. Histopathol. 2020, 35, 405–415. [Google Scholar]
- Bock, F.; Shahzad, K.; Wang, H.; Stoyanov, S.; Wolter, J.; Dong, W.; Pelicci, P.G.; Kashif, M.; Ranjan, S.; Schmidt, S.; et al. Activated Protein C Ameliorates Diabetic Nephropathy by Epigenetically Inhibiting the Redox Enzyme P66Shc. Proc. Natl. Acad. Sci. USA 2013, 110, 648–653. [Google Scholar] [CrossRef]
- Miller, B.S.; Blumenthal, S.R.; Shalygin, A.; Wright, K.D.; Staruschenko, A.; Imig, J.D.; Sorokin, A. Inactivation of P66Shc Decreases Afferent Arteriolar KATP Channel Activity and Decreases Renal Damage in Diabetic Dahl SS Rats. Diabetes 2018, 67, 2206–2212. [Google Scholar] [CrossRef]
- Mishra, M.; Duraisamy, A.J.; Bhattacharjee, S.; Kowluru, R.A. Adaptor Protein P66Shc: A Link Between Cytosolic and Mitochondrial Dysfunction in the Development of Diabetic Retinopathy. Antioxid. Redox Signal. 2019, 30, 1621–1634. [Google Scholar] [CrossRef]
- Al Sabaani, N. Exendin-4 Inhibits High Glucose-Induced Oxidative Stress in Retinal Pigment Epithelial Cells by Modulating the Expression and Activation of P66Shc. Cutan. Ocul. Toxicol. 2021, 40, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Albiero, M.; Poncina, N.; Tjwa, M.; Ciciliot, S.; Menegazzo, L.; Ceolotto, G.; Vigili De Kreutzenberg, S.; Moura, R.; Giorgio, M.; Pelicci, P.; et al. Diabetes Causes Bone Marrow Autonomic Neuropathy and Impairs Stem Cell Mobilization via Dysregulated P66Shc and Sirt1. Diabetes 2014, 63, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Albiero, M.; Ciciliot, S.; Tedesco, S.; Menegazzo, L.; D’Anna, M.; Scattolini, V.; Cappellari, R.; Zuccolotto, G.; Rosato, A.; Cignarella, A.; et al. Diabetes-Associated Myelopoiesis Drives Stem Cell Mobilopathy through an OSM-P66Shc Signaling Pathway. Diabetes 2019, 68, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Albiero, M.; D’anna, M.; Bonora, B.M.; Zuccolotto, G.; Rosato, A.; Giorgio, M.; Iori, E.; Avogaro, A.; Fadini, G.P. Hematopoietic and Nonhematopoietic P66Shc Differentially Regulates Stem Cell Traffic and Vascular Response to Ischemia in Diabetes. Antioxid. Redox Signal. 2022, 36, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Paneni, F.; Mocharla, P.; Akhmedov, A.; Costantino, S.; Osto, E.; Volpe, M.; Lüscher, T.F.; Cosentino, F. Gene Silencing of the Mitochondrial Adaptor P66(Shc) Suppresses Vascular Hyperglycemic Memory in Diabetes. Circ. Res. 2012, 111, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, K.; Gadi, I.; Nazir, S.; Al-Dabet, M.M.; Kohli, S.; Bock, F.; Breitenstein, L.; Ranjan, S.; Fuchs, T.; Halloul, Z.; et al. Activated Protein C Reverses Epigenetically Sustained P66Shc Expression in Plaque-Associated Macrophages in Diabetes. Commun. Biol. 2018, 1, 104. [Google Scholar] [CrossRef] [PubMed]
- Costantino, S.; Paneni, F.; Mitchell, K.; Mohammed, S.A.; Hussain, S.; Gkolfos, C.; Berrino, L.; Volpe, M.; Schwarzwald, C.; Lüscher, T.F.; et al. Hyperglycaemia-Induced Epigenetic Changes Drive Persistent Cardiac Dysfunction via the Adaptor P66Shc. Int. J. Cardiol. 2018, 268, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Albiero, M.; Bonora, B.M.; Poncina, N.; Vigili de Kreutzenberg, S.; Avogaro, A. P66Shc Gene Expression in Peripheral Blood Mononuclear Cells and Progression of Diabetic Complications. Cardiovasc. Diabetol. 2018, 17, 16. [Google Scholar] [CrossRef]
- Mousavi, S.; Khazeei Tabari, M.A.; Bagheri, A.; Samieefar, N.; Shaterian, N.; Kelishadi, R. The Role of P66Shc in Diabetes: A Comprehensive Review from Bench to Bedside. J. Diabetes Res. 2022, 24, 7703520. [Google Scholar] [CrossRef]
- Costantino, S.; Paneni, F.; Virdis, A.; Hussain, S.; Mohammed, S.A.; Capretti, G.; Akhmedov, A.; Dalgaard, K.; Chiandotto, S.; Pospisilik, J.A.; et al. Interplay Among H3K9-editing Enzymes SUV39H1, JMJD2C and SRC-1 Drives P66Shc Transcription and Vascular Oxidative Stress in Obesity. Eur. Heart J. 2019, 40, 383–391. [Google Scholar] [CrossRef]
- Mengozzi, A.; Costantino, S.; Paneni, F.; Duranti, E.; Nannipieri, M.; Mancini, R.; Lai, M.; La Rocca, V.; Puxeddu, I.; Antonioli, L.; et al. Targeting SIRT1 Rescues Age- and Obesity-Induced Microvascular Dysfunction in Ex Vivo Human Vessels. Circ. Res. 2022, 131, 476–491. [Google Scholar] [CrossRef] [PubMed]
- Arany, I.; Hall, S.; Reed, D.K.; Dixit, M. The Pro-oxidant Gene P66shc Increases Nicotine Exposure-induced Lipotoxic Oxidative Stress in Renal Proximal Tubule Cells. Mol. Med. Rep. 2016, 14, 2771–2777. [Google Scholar] [CrossRef] [PubMed]
- Arany, I.; Clark, J.S.; Reed, D.K.; Juncos, L.A.; Dixit, M. Role of P66shc in Renal Toxicity of Oleic Acid. Am. J. Nephrol. 2013, 38, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A New Definition for Metabolic Dysfunction-Associated Fatty Liver Disease: An International Expert Consensus Statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Sanderson, S.; Lindor, K.D.; Angulo, P. The Histological Course of Nonalcoholic Fatty Liver Disease: A Longitudinal Study of 103 Patients with Sequential Liver Biopsies. J. Hepatol. 2005, 42, 132–138. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The Global Epidemiology of NAFLD and NASH in Patients with Type 2 Diabetes: A Systematic Review and Meta-Analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Larter, C.Z.; Farrell, G.C. Insulin Resistance, Adiponectin, Cytokines in NASH: Which Is the Best Target to Treat? J. Hepatol. 2006, 44, 253–261. [Google Scholar] [CrossRef]
- Stefan, N. Causes, Consequences, and Treatment of Metabolically Unhealthy Fat Distribution. Lancet Diabetes Endocrinol. 2020, 8, 616–627. [Google Scholar] [CrossRef]
- Stefan, N.; Häring, H.U.; Cusi, K. Non-Alcoholic Fatty Liver Disease: Causes, Diagnosis, Cardiometabolic Consequences, and Treatment Strategies. Lancet Diabetes Endocrinol. 2019, 7, 313–324. [Google Scholar] [CrossRef]
- Marchesini, G.; Brizi, M.; Morselli-Labate, A.M.; Bianchi, G.; Bugianesi, E.; McCullough, A.J.; Forlani, G.; Melchionda, N. Association of Nonalcoholic Fatty Liver Disease with Insulin Resistance. Am. J. Med. 1999, 107, 450–455. [Google Scholar] [CrossRef]
- Kim, H.; Lee, D.S.; An, T.H.; Park, H.J.; Kim, W.K.; Bae, K.H.; Oh, K.J. Metabolic Spectrum of Liver Failure in Type 2 Diabetes and Obesity: From NAFLD to NASH to HCC. Int. J. Mol. Sci. 2021, 22, 94495. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Wang, S.; Chen, F.; Zhang, C.; Wang, Q.; Zhao, Y.; Zhang, Z. Hepatic Knockdown of Endothelin Type A Receptor (ETAR) Ameliorates Hepatic Insulin Resistance and Hyperglycemia Through Suppressing P66Shc-Mediated Mitochondrial Fragmentation in High-Fat Diet-Fed Mice. Diabetes Metab. Syndr. Obes. 2021, 14, 963–981. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Teratani, T.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Nishiyama, K.; Mataki, N.; Irie, R.; Minamino, T.; et al. P53/P66Shc-Mediated Signaling Contributes to the Progression of Non-Alcoholic Steatohepatitis in Humans and Mice. J. Hepatol. 2012, 57, 837–843. [Google Scholar] [CrossRef]
- Shan, W.; Gao, L.; Zeng, W.; Hu, Y.; Wang, G.; Li, M.; Zhou, J.; Ma, X.; Tian, X.; Yao, J. Activation of the SIRT1/P66shc Antiapoptosis Pathway via Carnosic Acid-Induced Inhibition of MiR-34a Protects Rats against Nonalcoholic Fatty Liver Disease. Cell Death Dis. 2015, 6, 196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, Y.; Wang, B.; Luo, Y.; Shi, J.; Zhao, B. The P66shc-Mediated Regulation of Hepatocyte Senescence Influences Hepatic Steatosis in Nonalcoholic Fatty Liver Disease. Med. Sci. Monit. 2020, 26, e921887. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, Z.; Feng, D.; Zhao, H.; Lin, M.; Hu, Y.; Zhang, N.; Lv, L.; Gao, Z.; Zhai, X.; et al. P66Shc Contributes to Liver Fibrosis through the Regulation of Mitochondrial Reactive Oxygen Species. Theranostics 2019, 9, 1510–1522. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, Z.; Zhou, J.; Feng, D.; Li, Y.; Hu, Y.; Zhang, F.; Chen, Z.; Wang, G.; Ma, X.; et al. LncRNA Mical2/MiR-203a-3p Sponge Participates in Epithelial-Mesenchymal Transition by Targeting P66Shc in Liver Fibrosis. Toxicol. Appl. Pharmacol. 2020, 403, 115125. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, Y.; Zhao, H.; Zhou, J.; Feng, D.; Tang, F.; Li, Y.; Lv, L.; Chen, Z.; Ma, X.; et al. Inhibition of P66Shc Oxidative Signaling via CA-Induced Upregulation of MiR-203a-3p Alleviates Liver Fibrosis Progression. Mol. Ther. Nucleic Acids 2020, 21, 751–763. [Google Scholar] [CrossRef]
- Ling, S.; Brown, K.; Miksza, J.K.; Howells, L.; Morrison, A.; Issa, E.; Yates, T.; Khunti, K.; Davies, M.J.; Zaccardi, F. Association of Type 2 Diabetes with Cancer: A Meta-Analysis with Bias Analysis for Unmeasured Confounding in 151 Cohorts Comprising 32 Million People. Diabetes Care 2020, 43, 2313–2322. [Google Scholar] [CrossRef]
- Pearson-Stuttard, J.; Papadimitriou, N.; Markozannes, G.; Cividini, S.; Kakourou, A.; Gill, D.; Rizos, E.C.; Monori, G.; Ward, H.A.; Kyrgiou, M.; et al. Type 2 Diabetes and Cancer: An Umbrella Review of Observational and Mendelian Randomization Studies. Cancer Epidemiol. Biomark. Prev. 2021, 30, 1218–1228. [Google Scholar] [CrossRef]
- Natalicchio, A.; Faggiano, A.; Zatelli, M.C.; Argentiero, A.; D’Oronzo, S.; Marrano, N.; Beretta, G.D.; Acquati, S.; Adinolfi, V.; Di Bartolo, P.; et al. Metabolic Disorders and Gastroenteropancreatic-Neuroendocrine Tumors (GEP-NETs): How Do They Influence Each Other? An Italian Association of Medical Oncology (AIOM)/Italian Association of Medical Diabetologists (AMD)/Italian Society of Endocrinology (SIE)/Italian Society of Pharmacology (SIF) Multidisciplinary Consensus Position Paper. Crit. Rev. Oncol. Hematol. 2022, 169, 103572. [Google Scholar] [PubMed]
- Cignarelli, A.; Genchi, V.A.; Caruso, I.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Diabetes and Cancer: Pathophysiological Fundamentals of a “Dangerous Affair”. Diabetes Res. Clin. Pract. 2018, 143, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Natalicchio, A.; Montagnani, M.; Gallo, M.; Marrano, N.; Faggiano, A.; Zatelli, M.C.; Mazzilli, R.; Argentiero, A.; Danesi, R.; D’Oronzo, S.; et al. MiRNA Dysregulation Underlying Common Pathways in Type 2 Diabetes and Cancer Development: An Italian Association of Medical Oncology (AIOM)/Italian Association of Medical Diabetologists (AMD)/Italian Society of Diabetology (SID)/Italian Society of Endocrinology (SIE)/Italian Society of Pharmacology (SIF) Multidisciplinary Critical View. ESMO Open 2023, 8, 101573. [Google Scholar] [PubMed]
- Matsuda, M.; Shimomura, I. Increased Oxidative Stress in Obesity: Implications for Metabolic Syndrome, Diabetes, Hypertension, Dyslipidemia, Atherosclerosis, and Cancer. Obes. Res. Clin. Pract. 2013, 7, e330–e341. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Chan, J.C.N. Evidence for DNA Damage as a Biological Link between Diabetes and Cancer. Chin. Med. J. 2015, 128, 1543–1548. [Google Scholar] [CrossRef] [PubMed]
- Lebiedzinska-Arciszewska, M.; Oparka, M.; Vega-Naredo, I.; Karkucinska-Wieckowska, A.; Pinton, P.; Duszynski, J.; Wieckowski, M.R. The Interplay between P66Shc, Reactive Oxygen Species and Cancer Cell Metabolism. Eur. J. Clin. Investig. 2015, 45 (Suppl. S1), 25–31. [Google Scholar] [CrossRef]
- Mir, H.A.; Ali, R.; Mushtaq, U.; Khanday, F.A. Structure-Functional Implications of Longevity Protein P66Shc in Health and Disease. Ageing Res. Rev. 2020, 63, 101139. [Google Scholar] [CrossRef]
- Beltrami, E.; Valtorta, S.; Moresco, R.; Marcu, R.; Belloli, S.; Fassina, A.; Fazio, F.; Pelicci, P.; Giorgio, M. The P53-P66Shc Apoptotic Pathway Is Dispensable for Tumor Suppression Whereas the P66Shc-Generated Oxidative Stress Initiates Tumorigenesis. Curr. Pharm. Des. 2013, 19, 2708–2714. [Google Scholar] [CrossRef]
- Bhat, S.S.; Anand, D.; Khanday, F.A. P66Shc as a Switch in Bringing about Contrasting Responses in Cell Growth: Implications on Cell Proliferation and Apoptosis. Mol. Cancer 2015, 14, 76. [Google Scholar] [CrossRef]
- Lewis, K.; Kiepas, A.; Hudson, J.; Senecal, J.; Ha, J.R.; Voorand, E.; Annis, M.G.; Sabourin, V.; Ahn, R.; La Selva, R.; et al. P66ShcA Functions as a Contextual Promoter of Breast Cancer Metastasis. Breast Cancer Res. 2020, 22, 7. [Google Scholar] [CrossRef]
- Haines, E.; Saucier, C.; Claing, A. The Adaptor Proteins P66Shc and Grb2 Regulate the Activation of the GTPases ARF1 and ARF6 in Invasive Breast Cancer Cells. J. Biol. Chem. 2014, 289, 5687–5703. [Google Scholar] [CrossRef] [PubMed]
- Ursini-Siegel, J.; Muller, W.J. The ShcA Adaptor Protein Is a Critical Regulator of Breast Cancer Progression. Cell Cycle 2008, 7, 1936–1943. [Google Scholar] [CrossRef] [PubMed]
- Ursini-Siegel, J.; Hardy, W.R.; Zheng, Y.; Ling, C.; Zuo, D.; Zhang, C.; Podmore, L.; Pawson, T.; Muller, W.J. The ShcA SH2 Domain Engages a 14-3-3/PI3’K Signaling Complex and Promotes Breast Cancer Cell Survival. Oncogene 2012, 31, 5038–5044. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Luo, Z.; Gong, Y.; Fu, Y.; Luo, Y. NAD+ Supplementation Limits Triple-Negative Breast Cancer Metastasis via SIRT1-P66Shc Signaling. Oncogene 2023, 42, 808–824. [Google Scholar] [CrossRef] [PubMed]
- Bhat, H.F.; Baba, R.A.; Adams, M.E.; Khanday, F.A. Role of SNTA1 in Rac1 Activation, Modulation of ROS Generation, and Migratory Potential of Human Breast Cancer Cells. Br. J. Cancer 2014, 110, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhu, S.; Shi, X.; Sha, W. The Silence of P66(Shc) in HCT8 Cells Inhibits the Viability Via PI3K/AKT/Mdm-2/p53 Signaling Pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 9097–9104. [Google Scholar]
- Galimov, E.R.; Sidorenko, A.S.; Tereshkova, A.V.; Pletiushkina, O.I.; Cherniak, B.V.; Chumakov, P.M. P66shc Action on Resistance of Colon Carcinoma RKO Cells to Oxidative Stress. Mol. Biol. 2012, 46, 139–146. [Google Scholar] [CrossRef]
- Yan, H.; Jihong, Y.; Feng, Z.; Xiaomei, X.; Xiaohan, Z.; Guangzhi, W.; Zhenhai, M.; Dongyan, G.; Xiaochi, M.; Qing, F.; et al. Sirtuin 1-Mediated Inhibition of P66shc Expression Alleviates Liver Ischemia/Reperfusion Injury. Crit. Care Med. 2014, 42, 246. [Google Scholar] [CrossRef]
- Huang, P.; Feng, X.; Zhao, Z.; Yang, B.; Fang, T.; Guo, M.; Xia, J. P66Shc Promotes HCC Progression in the Tumor Microenvironment via STAT3 Signaling. Exp. Cell Res. 2019, 383, 111550. [Google Scholar] [CrossRef]
- Fasolato, S.; Ruvoletto, M.; Nardo, G.; Rasola, A.; Sciacovelli, M.; Zanus, G.; Turato, C.; Quarta, S.; Terrin, L.; Fadini, G.P.; et al. Low P66shc with High SerpinB3 Levels Favors Necroptosis and Better Survival in Hepatocellular Carcinoma. Biology 2021, 10, 363. [Google Scholar] [CrossRef]
- Ortiz, G.G.; Huerta, M.; González-Usigli, H.A.; Torres-Sánchez, E.D.; Delgado-Lara, D.L.; Pacheco-Moisés, F.P.; Mireles-Ramírez, M.A.; Torres-Mendoza, B.M.; Moreno-Cih, R.I.; Velázquez-Brizuela, I.E. Cognitive Disorder and Dementia in Type 2 Diabetes Mellitus. World J. Diabetes 2022, 13, 319–337. [Google Scholar] [CrossRef] [PubMed]
- Marrano, N.; Biondi, G.; Borrelli, A.; Rella, M.; Zambetta, T.; Di Gioia, L.; Caporusso, M.; Logroscino, G.; Perrini, S.; Giorgino, F.; et al. Type 2 Diabetes and Alzheimer’s Disease: The Emerging Role of Cellular Lipotoxicity. Biomolecules 2023, 13, 183. [Google Scholar] [CrossRef] [PubMed]
- Muriach, M.; Flores-Bellver, M.; Romero, F.J.; Barcia, J.M. Diabetes and the Brain: Oxidative Stress, Inflammation, and Autophagy. Oxid. Med. Cell Longev. 2014, 2014, 102158. [Google Scholar] [CrossRef] [PubMed]
- Minami, Y.; Sonoda, N.; Hayashida, E.; Makimura, H.; Ide, M.; Ikeda, N.; Ohgidani, M.; Kato, T.A.; Seki, Y.; Maeda, Y.; et al. P66Shc Signaling Mediates Diabetes-Related Cognitive Decline. Sci. Rep. 2018, 8, 3213. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.S.; Ning, J.Q.; Chen, Z.Y.; Li, W.J.; Zhou, R.L.; Yan, R.Y.; Chen, M.J.; Ding, L.L. The Role of Mitochondrial Quality Control in Cognitive Dysfunction in Diabetes. Neurochem. Res. 2022, 47, 2158–2172. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef]
- Smith, W.W.; Norton, D.D.; Gorospe, M.; Jiang, H.; Nemoto, S.; Holbrook, N.J.; Finkel, T.; Kusiak, J.W. Phosphorylation of P66Shc and Forkhead Proteins Mediates Abeta Toxicity. J. Cell Biol. 2005, 169, 331–339. [Google Scholar] [CrossRef]
- Lone, A.; Harris, R.A.; Singh, O.; Betts, D.H.; Cumming, R.C. P66Shc Activation Promotes Increased Oxidative Phosphorylation and Renders CNS Cells More Vulnerable to Amyloid Beta Toxicity. Sci. Rep. 2018, 8, 17081. [Google Scholar] [CrossRef]
- Derungs, R.; Camici, G.G.; Spescha, R.D.; Welt, T.; Tackenberg, C.; Späni, C.; Wirth, F.; Grimm, A.; Eckert, A.; Nitsch, R.M.; et al. Genetic Ablation of the P66Shc Adaptor Protein Reverses Cognitive Deficits and Improves Mitochondrial Function in an APP Transgenic Mouse Model of Alzheimer’s Disease. Mol. Psychiatry 2017, 22, 605–614. [Google Scholar] [CrossRef]
- Cheng, H.; Gang, X.; Liu, Y.; Wang, G.; Zhao, X.; Wang, G. Mitochondrial Dysfunction Plays a Key Role in the Development of Neurodegenerative Diseases in Diabetes. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E750–E764. [Google Scholar] [CrossRef]
- Maj, M.C.; Tkachyova, I.; Patel, P.; Addis, J.B.; Mackay, N.; Levandovskiy, V.; Lee, J.; Lang, A.E.; Cameron, J.M.; Robinson, B.H. Oxidative Stress Alters the Regulatory Control of P66Shc and Akt in PINK1 Deficient Cells. Biochem. Biophys. Res. Commun. 2010, 399, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.H.; Li, C.Y.; Chang, H.H.; Sun, Y.; Tsai, C.C. A Population-Based Cohort Study Suggests an Increased Risk of Multiple Sclerosis Incidence in Patients with Type 2 Diabetes Mellitus. J. Epidemiol. 2017, 27, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Savino, C.; Pelicci, P.; Giorgio, M. The P66Shc/Mitochondrial Permeability Transition Pore Pathway Determines Neurodegeneration. Oxid. Med. Cell Longev. 2013, 2013, 719407. [Google Scholar] [CrossRef] [PubMed]
- Wium-Andersen, I.K.; Jørgensen, T.S.H.; Jørgensen, M.B.; Osler, M.; Wium-Andersen, M.K. Diabetes, Sleep Disorders and Risk of Depression—A Danish Register-Based Cohort Study. J. Diabetes Complicat. 2022, 36, 108266. [Google Scholar] [CrossRef]
- Schipper, S.B.J.; Van Veen, M.M.; Elders, P.J.M.; van Straten, A.; Van Der Werf, Y.D.; Knutson, K.L.; Rutters, F. Sleep Disorders in People with Type 2 Diabetes and Associated Health Outcomes: A Review of the Literature. Diabetologia 2021, 64, 2367–2377. [Google Scholar] [CrossRef]
- Lyu, X.; Cai, J.; Yan, R.; Huang, P.; Gong, H.; Peng, J.; Liu, Y.; Li, S.; Tan, S.; Hu, M.; et al. P66Shc Is Increased in Peripheral Blood Mononuclear Cells of the Patients with Obstructive Sleep Apnea. Int. J. Med. Sci. 2023, 20, 455–462. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biondi, G.; Marrano, N.; Borrelli, A.; Rella, M.; D’Oria, R.; Genchi, V.A.; Caccioppoli, C.; Cignarelli, A.; Perrini, S.; Laviola, L.; et al. The p66Shc Redox Protein and the Emerging Complications of Diabetes. Int. J. Mol. Sci. 2024, 25, 108. https://doi.org/10.3390/ijms25010108
Biondi G, Marrano N, Borrelli A, Rella M, D’Oria R, Genchi VA, Caccioppoli C, Cignarelli A, Perrini S, Laviola L, et al. The p66Shc Redox Protein and the Emerging Complications of Diabetes. International Journal of Molecular Sciences. 2024; 25(1):108. https://doi.org/10.3390/ijms25010108
Chicago/Turabian StyleBiondi, Giuseppina, Nicola Marrano, Anna Borrelli, Martina Rella, Rossella D’Oria, Valentina Annamaria Genchi, Cristina Caccioppoli, Angelo Cignarelli, Sebastio Perrini, Luigi Laviola, and et al. 2024. "The p66Shc Redox Protein and the Emerging Complications of Diabetes" International Journal of Molecular Sciences 25, no. 1: 108. https://doi.org/10.3390/ijms25010108