
Inflammatory and Metabolic Signaling Interfaces of the Hypertrophic and Senescent Chondrocyte Phenotypes Associated with Osteoarthritis
 , and
, and
Abstract
:1. Introduction
2. Evidence for the Presence of Low-Grade Inflammation in Osteoarthritis
3. Sox-9, NF-kB and Proinflammatory Cytokines Mediate Hypertrophy and Aberrant Signal Transduction Leading to Cartilage Deterioration
4. Hypertrophic Differentiation and Chondrocyte Phenotype
5. Altered Carbohydrate Metabolism Is Linked to Catabolic Reprogramming with Common Features of Pro-Inflammatory Phenotype in Osteoarthritic Chondrocytes
6. Central Metabolic Pathways’ Reprogramming and Mitochondrial Dysfunction Are Hallmarks of Hypertrophy and Senescence
7. Imbalance of Hypoxia-Inducible Factors Regulate Inflammatory Signals and Cartilage Destruction
8. Senescent Chondrocytes Possess Remarkable Pro-Inflammatory and Catabolic Signatures
9. Mechanosensing, the YAP/TAZ Signaling, Oxidative Stress and Chondrocyte Senescence
10. The Energy Sensor mTOR Protein Complex Regulates Chondrocyte Senescence
11. Regulation of Senescence at the Post-Transcriptional Level with miR and Other Small Non-Coding RNAs Modulates the Inflammatory Signature
12. Senolysis Has Beneficial Effects in Experimental Conditions
13. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACAN | aggrecan |
| ACLT | Anterior Cruciate Ligament Transection |
| ADAMTS 4,5 | A Disintegrin and Metalloproteinase with Thrombospondin motifs |
| Akt | Protein kinase B |
| AMPKα | AMP-activated protein kinase alpha |
| BMP | Bone morphogenetic protein |
| CCL-27 | C-C motif chemokine ligand 27 |
| CD | Cluster of differentiation |
| cGAS | Cyclic GMP-AMP |
| COL10A1 | Collagen type X alpha chain 1 |
| COL2A1 | Collagen type II alpha chain 1 |
| CXCL8 | CXC motif chemokine ligand 8 |
| DMEM | Dulbecco’s Modified Eagle Medium |
| FADH2 | Flavin-adenine dinucleotide, reduced |
| FRZB | Frizzled-related protein |
| G-6-P DH | Glucose 6-phosphate dehydrogenase |
| GADD45 | Growth arrest and DNA damage inducible 45 |
| GLUT-1 | Glucose transporter 1 |
| GROα | Chemokine C-X-C motif ligand 1 |
| GSK3 | Glycogen synthase kinase 3 |
| HDAC | Histone deacetylase |
| HIF1α, 2α | Hypoxia inducible factor 1α, 2α |
| HK | Hexokinase |
| HMGB2 | High-mobility group protein 2 |
| IGFBP7 | Insulin-like growth factor (IGF)-binding protein |
| IHH | Indian hedgehog |
| IKK | Inhibitory-kB kinase |
| IL | Interleukin |
| JNK | Jun N-terminal kinase |
| LATS 1 | Large tumor suppressor kinase 1 |
| Lcn2 | Lipocalin 2 |
| LDH | Lactate dehydrogenase |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen activated protein kinase |
| MCP-1 | monocyte chemoattractant protein-1 |
| miR | micro RNA |
| MMP | Matrix metalloproteinase |
| MOB1A/B | Mps one binder kinase activator 1 A/B |
| mPGES-1 | Microsomal prostaglandin E synthase 1 |
| MST1/2 | Mammalian sterile 20-like kinase 1/2 |
| mTORc | Mammalian target of rapamycin complex 1 |
| NADH+ | Nicotinamide-adenine dinucleotide, reduced |
| NADPH+ | Nicotinamide-adenine dinucleotide phosphate, reduced |
| NDRG2 | NMYC downstream-regulated gene 2 |
| NFkB | Nuclear factor kappa B |
| NLRP3 | NLR family pyrin domain containing 3 |
| NO | Nitrogen monoxide |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| OARSI | Osteoarthritis Research Society International |
| OXPHOS | Oxidative phosphorylation |
| PAI-1 | Plasminogen activator inhibitor-1 |
| PGC1α | Peroxisome proliferator-activated receptor-gamma coactivator |
| PI3K | Phospho-inositole 3-kinase |
| PKM2 | Piruvate kinase M2 |
| PPAR | Peroxisome Proliferator-Activated Recpetor |
| PTHrP | Parathormone-related peptide |
| RAGE | Receptor of end-glycation products |
| RANKL | Receptor activator for nuclear factor kappa B ligand |
| ROS | Reactive oxygen species |
| RunX2 | Runt-related transcription factor 2 |
| SASP | Senescence-associated secretory pattern |
| SAV | Salvador |
| SIRT 1,6 | Sirtuin 1,6 |
| SMAD | Suppressor of mothers against Decapentaplegic |
| Sox-9 | Sex-determining region Y-type (SRY) high mobility group (HMG) box family of DNA binding protein 9 |
| STING | Stimulator of interferon genes |
| TAZ | Transcriptional coactivator with PDZ-binding motif |
| TCA | Tricarboxylic acid cycle |
| TGF-β | Transforming growth factor beta |
| TIMP-1 | Tissue inhibitor of metalloproteinases 1 |
| TLR-4 | Toll-like Receptor 4 |
| TNF-α | Tumor necrosis factor alpha |
| TRPV4 | Transient Receptor Potential Vanilloid 4 |
| TSPYL2 | Testis-specific Y-encoded-like protein 2 |
| VEGF | Vascular endothelial growth factor |
| YAP | Yes-associated protein |
References
- Chen, D.; Shen, J.; Zhao, W.W.; Wang, T.Y.; Han, L.; Hamilton, J.L.; Im, H.J. Osteoarthritis: Toward a comprehensive understanding of pathological mechanism. Bone Res. 2017, 5, 16044. [Google Scholar] [CrossRef]
- Jiang, Y.Z.; Tuan, R.S. Origin and function of cartilage stem/progenitor cells in osteoarthritis. Nat. Rev. Rheumatol. 2015, 11, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Aigner, T.; Soeder, S.; Gebhard, P.M.; McAlinden, A.; Haag, J. Mechanisms of Disease: Role of chondrocytes in the pathogenesis of osteoarthritis—Structure, chaos and senescence. Nat. Clin. Pract. Rheumatol. 2007, 3, 391–399. [Google Scholar] [CrossRef]
- Yoshioka, N.K.; Young, G.M.; Khajuria, D.K.; Karuppagounder, V.; Pinamont, W.J.; Fanburg-Smith, J.C.; Abraham, T.; Elbarbary, R.A.; Kamal, F. Structural changes in the collagen network of joint tissues in late stages of murine OA. Sci. Rep. 2022, 12, 9159. [Google Scholar] [CrossRef] [PubMed]
- Rim, Y.A.; Nam, Y.; Ju, J.H. The Role of Chondrocyte Hypertrophy and Senescence in Osteoarthritis Initiation and Progression. Int. J. Mol. Sci. 2020, 21, 2358. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, K.; Litherland, G.J.; Rai, T.S. Cellular senescence in osteoarthritis pathology. Aging Cell 2017, 16, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W.H.; Lepus, C.M.; Wang, Q.; Raghu, H.; Mao, R.; Lindstrom, T.M.; Sokolove, J. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 580–592. [Google Scholar] [CrossRef]
- Sohn, D.H.; Sokolove, J.; Sharpe, O.; Erhart, J.C.; Chandra, P.E.; Lahey, L.J.; Lindstrom, T.M.; Hwang, I.; Boyer, K.A.; Andriacchi, T.P.; et al. Plasma proteins present in osteoarthritic synovial fluid can stimulate cytokine production via Toll-like receptor 4. Arthritis Res. Ther. 2012, 14, R7. [Google Scholar] [CrossRef]
- Terkawi, M.A.; Ebata, T.; Yokota, S.; Takahashi, D.; Endo, T.; Matsumae, G.; Shimizu, T.; Kadoya, K.; Iwasaki, N. Low-Grade Inflammation in the Pathogenesis of Osteoarthritis: Cellular and Molecular Mechanisms and Strategies for Future Therapeutic Intervention. Biomedicines 2022, 10, 1109. [Google Scholar] [CrossRef]
- van den Bosch, M.H.J. Inflammation in osteoarthritis: Is it time to dampen the alarm(in) in this debilitating disease? Clin. Exp. Immunol. 2019, 195, 153–166. [Google Scholar] [CrossRef]
- Gobezie, R.; Kho, A.; Krastins, B.; Sarracino, D.A.; Thornhill, T.S.; Chase, M.; Millett, P.J.; Lee, D.M. High abundance synovial fluid proteome: Distinct profiles in health and osteoarthritis. Arthritis Res. Ther. 2007, 9, R36. [Google Scholar] [CrossRef] [PubMed]
- Krenytska, D.; Strubchevska, K.; Kozyk, M.; Vovk, T.; Halenova, T.; Kot, L.; Raksha, N.; Savchuk, O.; Falalyeyeva, T.; Tsyryuk, O.; et al. Circulating levels of inflammatory cytokines and angiogenesis-related growth factors in patients with osteoarthritis after COVID-19. Front. Med. 2023, 10, 1168487. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.M.; Chen, P.C.; Lin, C.M.; Fang, M.L.; Chi, M.C.; Liu, J.F. CXCL1 contributes to IL-6 expression in osteoarthritis and rheumatoid arthritis synovial fibroblasts by CXCR2, c-Raf, MAPK, and AP-1 pathway. Arthritis Res. Ther. 2020, 22, 251. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.W.; Chen, L.; Hao, X.R.; Qu, Z.A.; Huang, S.B.; Ma, X.J.; Wang, J.C.; Wang, W.M. Elevated levels of interleukin-1 beta, interleukin-6, tumor necrosis factor-alpha and vascular endothelial growth factor in patients with knee articular cartilage injury. World J. Clin. Cases 2019, 7, 1262–1269. [Google Scholar] [CrossRef]
- Kuyinu, E.L.; Narayanan, G.; Nair, L.S.; Laurencin, C.T. Animal models of osteoarthritis: Classification, update, and measurement of outcomes. J. Orthop. Surg. Res. 2016, 11, 19. [Google Scholar] [CrossRef]
- Arra, M.; Swarnkar, G.; Alippe, Y.; Mbalaviele, G.; Abu-Amer, Y. I kappa B-zeta signaling promotes chondrocyte inflammatory phenotype, senescence, and erosive joint pathology. Bone Res. 2022, 10, 12. [Google Scholar] [CrossRef]
- Van Dalen, S.C.M.; Blom, A.B.; Sloetjes, A.W.; Helsen, M.M.A.; Roth, J.; Vogl, T.; de Loo, F.A.J.; Koenders, M.I.; Van der Kraan, P.M.; Van den Berg, W.B.; et al. Interleukin-1 is not involved in synovial inflammation and cartilage destruction in collagenase-induced osteoarthritis. Osteoarthr. Cartil. 2017, 25, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Nasi, S.; Ea, H.K.; So, A.; Busso, N. Revisiting the Role of Interleukin-1 Pathway in Osteoarthritis: Interleukin-1 alpha and-1 beta, and NLRP3 Inflammasome Are Not Involved in the Pathological Features of the Murine Menisectomy Model of osteoarthritis. Front. Pharmacol. 2017, 8, 282. [Google Scholar] [CrossRef]
- Stannus, O.; Jones, G.; Cicuttini, F.M.; Parameswaran, V.; Quinn, S.; Burgess, J.; Ding, C. Circulating levels of IL-6 and TNF-alpha are associated with knee radiographic osteoarthritis and knee cartilage loss in older adults. Osteoarthr. Cartil. 2010, 18, 1441–1447. [Google Scholar] [CrossRef]
- Barker, T.; Rogers, V.E.; Henriksen, V.T.; Trawick, R.H.; Momberger, N.G.; Lynn Rasmussen, G. Circulating IL-10 is compromised in patients predisposed to developing and in patients with severe knee osteoarthritis. Sci. Rep. 2021, 11, 1812. [Google Scholar] [CrossRef]
- Panina, S.B.; Krolevets, I.V.; Milyutina, N.P.; Sagakyants, A.B.; Kornienko, I.V.; Ananyan, A.A.; Zabrodin, M.A.; Plotnikov, A.A.; Vnukov, V.V. Circulating levels of proinflammatory mediators as potential biomarkers of post-traumatic knee osteoarthritis development. J. Orthop. Traumatol. 2017, 18, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Waszczykowski, M.; Fabis-Strobin, A.; Bednarski, I.; Narbutt, J.; Fabis, J. Serum and synovial fluid concentrations of interleukin-18 and interleukin-20 in patients with osteoarthritis of the knee and their correlation with other markers of inflammation and turnover of joint cartilage. Arch. Med. Sci. 2022, 18, 448–458. [Google Scholar] [CrossRef]
- Na, H.S.; Park, J.S.; Cho, K.H.; Kwon, J.Y.; Choi, J.; Jhun, J.; Kim, S.J.; Park, S.H.; Cho, M.L. Interleukin-1-Interleukin-17 Signaling Axis Induces Cartilage Destruction and Promotes Experimental Osteoarthritis. Front. Immunol. 2020, 11, 730. [Google Scholar] [CrossRef] [PubMed]
- Nasi, S.; So, A.; Combes, C.; Daudon, M.; Busso, N. Interleukin-6 and chondrocyte mineralisation act in tandem to promote experimental osteoarthritis. Ann. Rheum. Dis. 2016, 75, 1372–1379. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.Q.; Wang, W.J.; Tang, S.S.; Liu, Y.; Wang, J.L. Correlation between interleukin-6 expression in articular cartilage bone and osteoarthritis. Genet. Mol. Res. 2015, 14, 14189–14195. [Google Scholar] [CrossRef] [PubMed]
- Jhang, J.J.; Yen, G.C. The role of Nrf2 in NLRP3 inflammasome activation. Cell Mol. Immunol. 2017, 14, 1011–1012. [Google Scholar] [CrossRef]
- Warner, S.C.; Nair, A.; Marpadga, R.; Chubinskaya, S.; Doherty, M.; Valdes, A.M.; Scanzello, C.R. IL-15 and IL15RA in Osteoarthritis: Association With Symptoms and Protease Production, but Not Structural Severity. Front. Immunol. 2020, 11, 1385. [Google Scholar] [CrossRef]
- Scanzello, C.R.; Umoh, E.; Pessler, F.; Diaz-Torne, C.; Miles, T.; DiCarlo, E.; Potter, H.G.; Mand, L.; Marx, R.; Rodeo, S.; et al. Local cytokine profiles in knee osteoarthritis: Elevated synovial fluid interleukin-15 differentiates early from end-stage disease. Osteoarthr. Cartil. 2009, 17, 1040–1048. [Google Scholar] [CrossRef]
- Iannone, F.; De Bari, C.; Dell’Accio, F.; Covelli, M.; Cantatore, F.P.; Patella, V.; Lo Bianco, G.; Lapadula, G. Interleukin-10 and interleukin-10 receptor in human osteoarthritic and healthy chondrocytes. Clin. Exp. Rheumatol. 2001, 19, 139–146. [Google Scholar]
- Nagy, E.; Vajda, E.; Vari, C.; Sipka, S.; Farr, A.M.; Horvath, E. Meloxicam ameliorates the cartilage and subchondral bone deterioration in monoiodoacetate-induced rat osteoarthritis. PeerJ 2017, 5, e3185. [Google Scholar] [CrossRef]
- Csifo, E.; Nagy, E.E.; Horvath, E.; Farr, A.; Muntean, D.L. Mid-term effects of Meloxicam on collagen type II degradation in a rat osteoarthritis model induced by iodoacetate. Farmacia 2015, 63, 556–560. [Google Scholar]
- Koskinen-Kolasa, A.; Vuolteenaho, K.; Korhonen, R.; Moilanen, T.; Moilanen, E. Catabolic and proinflammatory effects of leptin in chondrocytes are regulated by suppressor of cytokine signaling-3. Arthritis Res. Ther. 2016, 18, 215. [Google Scholar] [CrossRef] [PubMed]
- Lambova, S.N.; Batsalova, T.; Moten, D.; Stoyanova, S.; Georgieva, E.; Belenska-Todorova, L.; Kolchakova, D.; Dzhambazov, B. Serum Leptin and Resistin Levels in Knee Osteoarthritis-Clinical and Radiologic Links: Towards Precise Definition of Metabolic Type Knee Osteoarthritis. Biomedicines 2021, 9, 1019. [Google Scholar] [CrossRef]
- Vuolteenaho, K.; Koskinen, A.; Kukkonen, M.; Nieminen, R.; Päivärinta, U.; Moilanen, T.; Moilanen, E. Leptin Enhances Synthesis of Proinflammatory Mediators in Human Osteoarthritic Cartilage-Mediator Role of NO in Leptin-Induced PGE2, IL-6, and IL-8 Production. Mediat. Inflamm. 2009, 2009, 345838. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Marcu, K.B.; Goldring, M.B.; Otero, M. Phenotypic instability of chondrocytes in osteoarthritis: On a path to hypertrophy. Ann. N. Y. Acad. Sci. 2019, 1442, 17–34. [Google Scholar] [CrossRef]
- Wang, X.; Li, F.F.; Fan, C.Y.; Wang, C.Y.; Ruan, H.J. Effects and relationship of ERK1 and ERK2 in interleukin-1 beta-induced alterations in MMP3, MMP13, type II collagen and aggrecan expression in human chondrocytes. Int. J. Mol. Med. 2011, 27, 583–589. [Google Scholar] [CrossRef]
- Molnar, V.; Matisic, V.; Kodvanj, I.; Bjelica, R.; Jelec, Z.; Hudetz, D.; Rod, E.; Cukelj, F.; Vrdoljak, T.; Vidovic, D.; et al. Cytokines and Chemokines Involved in Osteoarthritis Pathogenesis. Int. J. Mol. Sci. 2021, 22, 9208. [Google Scholar] [CrossRef]
- Tsuda, M.; Takahashi, S.; Takahashi, Y.; Asahara, H. Transcriptional co-activators CREB-binding protein and p300 regulate chondrocyte-specific gene expression via association with Sox9. J. Biol. Chem. 2003, 278, 27224–27229. [Google Scholar] [CrossRef]
- Muramatsu, S.; Wakabayashi, M.; Ohno, T.; Amano, K.; Ooishi, R.; Sugahara, T.; Shiojiri, S.; Tashiro, K.; Suzuki, Y.; Nishimura, R.; et al. Functional gene screening system identified TRPV4 as a regulator of chondrogenic differentiation. J. Biol. Chem. 2007, 282, 32158–32167. [Google Scholar] [CrossRef] [PubMed]
- Trompeter, N.; Gardinier, J.D.; DeBarros, V.; Boggs, M.; Gangadharan, V.; Cain, W.J.; Hurd, L.; Duncan, R.L. Insulin-like growth factor-1 regulates the mechanosensitivity of chondrocytes by modulating TRPV4. Cell Calcium 2021, 99, 102467. [Google Scholar] [CrossRef]
- Takeda, Y.; Niki, Y.; Fukuhara, Y.; Fukuda, Y.; Udagawa, K.; Shimoda, M.; Kikuchi, T.; Kobayashi, S.; Harato, K.; Miyamoto, T.; et al. Compressive mechanical stress enhances susceptibility to interleukin-1 by increasing interleukin-1 receptor expression in 3D-cultured ATDC5 cells. BMC Musculoskelet. Disord. 2021, 22, 238. [Google Scholar] [CrossRef] [PubMed]
- Dudakovic, A.; Camilleri, E.T.; Xu, F.H.; Riester, S.M.; McGee-Lawrence, M.E.; Bradley, E.W.; Paradise, C.R.; Lewallen, E.A.; Thaler, R.; Deyle, D.R.; et al. Epigenetic Control of Skeletal Development by the Histone Methyltransferase Ezh2. J. Biol. Chem. 2015, 290, 27604–27617. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Gee, A.O.; Zielinska-Kwiatkowska, A.; Chansky, H.A. Generation and characterization of mice with mesenchyme-specific deletion of the entire ESET histone methyltransferase protein. Genesis 2018, 56, e23088. [Google Scholar] [CrossRef] [PubMed]
- Furumatsu, T.; Asahara, H. Histone Acetylation Influences the Activity of Sox9-related Transcriptional Complex. Acta Medica Okayama 2010, 64, 351–357. [Google Scholar] [PubMed]
- Dong, Z.Q.; Ma, Z.; Yang, M.J.; Cong, L.L.; Zhao, R.P.; Cheng, L.Y.; Sun, J.; Wang, Y.F.; Yang, R.J.; Wei, X.C.; et al. The Level of Histone Deacetylase 4 is Associated with Aging Cartilage Degeneration and Chondrocyte Hypertrophy. J. Inflamm. Res. 2022, 15, 3547–3560. [Google Scholar] [CrossRef]
- Lafont, J.E.; Moustaghfir, S.; Durand, A.L.; Mallein-Gerin, F. The epigenetic players and the chromatin marks involved in the articular cartilage during osteoarthritis. Front. Physiol. 2023, 14, 1070241. [Google Scholar] [CrossRef]
- Zhong, L.L.; Huang, X.B.; Karperien, M.; Post, J.N. Correlation between Gene Expression and Osteoarthritis Progression in Human. Int. J. Mol. Sci. 2016, 17, 1126. [Google Scholar] [CrossRef] [PubMed]
- Mattiuzzo, E.; Faggian, A.; Venerando, R.; Benetti, A.; Belluzzi, E.; Abatangelo, G.; Ruggieri, P.; Brun, P. In Vitro Effects of Low Doses of β-Caryophyllene, Ascorbic Acid and d-Glucosamine on Human Chondrocyte Viability and Inflammation. Pharmaceuticals 2021, 14, 286. [Google Scholar] [CrossRef]
- Picciolo, G.; Pallio, G.; Altavilla, D.; Vaccaro, M.; Oteri, G.; Irrera, N.; Squadrito, F. β-Caryophyllene Reduces the Inflammatory Phenotype of Periodontal Cells by Targeting CB2 Receptors. Biomedicines 2020, 8, 164. [Google Scholar] [CrossRef]
- Chawla, S.; Mainardi, A.; Majumder, N.; Donges, L.; Kumar, B.; Occhetta, P.; Martin, I.; Egloff, C.; Ghosh, S.; Bandyopadhyay, A.; et al. Chondrocyte Hypertrophy in Osteoarthritis: Mechanistic Studies and Models for the Identification of New Therapeutic Strategies. Cells 2022, 11, 4034. [Google Scholar] [CrossRef]
- Fukui, N.; Ikeda, Y.; Ohnuki, T.; Hikita, A.; Tanaka, S.; Yamane, S.; Suzuki, R.; Sandell, L.J.; Ochi, T. Pro-inflammatory cytokine tumor necrosis factor-alpha induces bone morphogenetic protein-2 in chondrocytes via mRNA stabilization and transcriptional up-regulation. J. Biol. Chem. 2006, 281, 27229–27241. [Google Scholar] [CrossRef] [PubMed]
- Ijiri, K.; Zerbini, L.F.; Peng, H.B.; Correa, R.G.; Lu, B.F.; Walsh, N.; Zhao, Y.N.; Taniguchi, N.; Huang, X.L.; Otu, H.; et al. A novel role for GADD45 beta as a mediator of MMP-13 gene expression during chondrocyte terminal differentiation. J. Biol. Chem. 2005, 280, 38544–38555. [Google Scholar] [CrossRef] [PubMed]
- Marcu, K.B.; Otero, M.; Olivotto, E.; Borzi, R.M.; Goldring, M.B. NF-kappa B Signaling: Multiple Angles to Target OA. Curr. Drug Targets 2010, 11, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Arra, M.; Swarnkar, G.; Ke, K.; Otero, J.E.; Ying, J.; Duan, X.; Maruyama, T.; Rai, M.F.; O’Keefe, R.J.; Mbalaviele, G.; et al. LDHA-mediated ROS generation in chondrocytes is a potential therapeutic target for osteoarthritis. Nat. Commun. 2020, 11, 3427. [Google Scholar] [CrossRef]
- Olivotto, E.; Otero, M.; Astolfi, A.; Platano, D.; Facchini, A.; Pagani, S.; Flamigni, F.; Goldring, M.B.; Borzi, R.M.; Marcu, K.B. IKK alpha/CHUK Regulates Extracellular Matrix Remodeling Independent of Its Kinase Activity to Facilitate Articular Chondrocyte Differentiation. PLoS ONE 2013, 8, e73024. [Google Scholar] [CrossRef] [PubMed]
- Chawla, S.; Berkelaar, M.H.M.; Dasen, B.; Halleux, C.; Guth-Gundel, S.; Kramer, I.; Ghosh, S.; Martin, I.; Barbero, A.; Occhetta, P. Blockage of bone morphogenetic protein signalling counteracts hypertrophy in a human osteoarthritic micro-cartilage model. J. Cell Sci. 2020, 133, jcs249094. [Google Scholar] [CrossRef]
- Hallett, S.A.; Ono, W.; Ono, N. The hypertrophic chondrocyte: To be or not to be. Histol. Histopathol. 2021, 36, 1021–1036. [Google Scholar] [CrossRef]
- van der Kraan, P.M.; van den Berg, W.B. Chondrocyte hypertrophy and osteoarthritis: Role in initiation and progression of cartilage degeneration? Osteoarthr. Cartil. 2012, 20, 223–232. [Google Scholar] [CrossRef]
- Majumder, N.; Ghosh, S. Unfolding the Mystery Behind the Onset of Chondrocyte Hypertrophy during Chondrogenesis: Toward Designing Advanced Permanent Cartilage-mimetic Biomaterials. Adv. Funct. Mater. 2023, 33, 2300651. [Google Scholar] [CrossRef]
- Parreno, J.; Raju, S.; Niaki, M.N.; Andrejevic, K.; Jiang, A.; Delve, E.; Kandel, R. Expression of type I collagen and tenascin C is regulated by actin polymerization through MRTF in dedifferentiated chondrocytes. Febs Lett. 2014, 588, 3677–3684. [Google Scholar] [CrossRef]
- Ortega, N.; Behonick, D.J.; Werb, Z. Matrix remodeling during endochondral ossification. Trends Cell Biol. 2004, 14, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.M.; Clifton, K.B.; Lorenzo, J.; Hansen, M.F.; Drissi, H. Comparative transcriptomic analysis identifies distinct molecular signatures and regulatory networks of chondroclasts and osteoclasts. Arthritis Res. Ther. 2020, 22, 168. [Google Scholar] [CrossRef] [PubMed]
- Biswas, T.; Jaswal, A.P.; Yadav, U.S.; Bandyopadhyay, A. Simultaneous differentiation of articular and transient cartilage: WNT-BMP interplay and its therapeutic implication. Int. J. Dev. Biol. 2020, 64, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Catheline, S.E.; Hoak, D.; Chang, M.; Ketz, J.P.; Hilton, M.J.; Zuscik, M.J.; Jonason, J.H. Chondrocyte-Specific RUNX2 Overexpression Accelerates Post-traumatic Osteoarthritis Progression in Adult Mice. J. Bone Miner. Res. 2019, 34, 1676–1689. [Google Scholar] [CrossRef] [PubMed]
- Ripmeester, E.G.J.; Timur, U.T.; Caron, M.M.J.; Welting, T.J.M. Recent Insights into the Contribution of the Changing Hypertrophic Chondrocyte Phenotype in the Development and Progression of Osteoarthritis. Front. Bioeng. Biotechnol. 2018, 6, 18. [Google Scholar] [CrossRef]
- Bradley, E.W.; Drissi, M.H. WNT5A Regulates Chondrocyte Differentiation through Differential Use of the CaN/NFAT and IKK/NF-kappa B Pathways. Mol. Endocrinol. 2010, 24, 1581–1593. [Google Scholar] [CrossRef]
- Ferrao Blanco, M.N.; Bastiaansen-Jenniskens, Y.M.; Chambers, M.G.; Pitsillides, A.A.; Narcisi, R.; van Osch, G.J.V.M. Effect of Inflammatory Signaling on Human Articular Chondrocyte Hypertrophy: Potential Involvement of Tissue Repair Macrophages. Cartilage 2021, 13 (Suppl. S2), 168S–174S. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, J.; Blom, A.B.; Wainwright, S.; Hughes, C.; Caterson, B.; van den Berg, W.B. The Role of Synovial Macrophages and Macrophage-Produced Mediators in Driving Inflammatory and Destructive Responses in Osteoarthritis. Arthritis Rheum. 2010, 62, 647–657. [Google Scholar] [CrossRef]
- Hollander, J.M.; Zeng, L. The Emerging Role of Glucose Metabolism in Cartilage Development. Curr. Osteoporos. Rep. 2019, 17, 59–69. [Google Scholar] [CrossRef]
- Mobasheri, A.; Bondy, C.A.; Moley, K.; Mendes, A.F.; Rosa, S.C.; Richardson, S.M.; Hoyland, J.A.; Barrett-Jolley, R.; Shakibaei, M. Facilitative Glucose Transporters in Articular Chondrocytes Expression, Distribution and Functional Regulation of GLUT Isoforms by Hypoxia, Hypoxia Mimetics, Growth Factors and Pro-Inflammatory Cytokines. Adv. Anat. Embryol. Cell Biol. 2008, 200, 1–84. [Google Scholar]
- Li, Q.; Wang, L.; Wang, Y.; Liu, L.; Han, H.; Chen, L.; Wang, H. Programming changes in GLUT1 mediated the accumulation of AGEs and matrix degradation in the articular cartilage of female adult rats after prenatal caffeine exposure. Pharmacol. Res. 2020, 151, 104555. [Google Scholar] [CrossRef]
- Cecil, D.L.; Johnson, K.; Rediske, J.; Lotz, M.; Schmidt, A.M.; Terkeltaub, R. Inflammation-induced chondrocyte hypertrophy is driven by receptor for advanced glycation end products. J. Immunol. 2005, 175, 8296–8302. [Google Scholar] [CrossRef]
- Zheng, L.L.; Zhang, Z.J.; Sheng, P.Y.; Mobasheri, A. The role of metabolism in chondrocyte dysfunction and the progression of osteoarthritis. Ageing Res. Rev. 2021, 66, 101249. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.W.; Huang, T.C.; Hu, Y.C.; Hsieh, B.S.; Chiu, P.R.; Cheng, H.L.; Chang, K.L. Zinc protects chondrocytes from monosodium iodoacetate-induced damage by enhancing ATP and mitophagy. Biochem. Biophys. Res. Commun. 2020, 521, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Tan, C.; Li, L.; Han, J.; Xu, K.; Liu, X. A new strategy for osteoarthritis therapy: Inhibition of glycolysis. Front. Pharmacol. 2022, 13, 1057229. [Google Scholar] [CrossRef]
- Qu, J.N.; Lu, D.G.; Guo, H.; Miao, W.S.; Wu, G.; Zhou, M.F. PFKFB3 modulates glycolytic metabolism and alleviates endoplasmic reticulum stress in human osteoarthritis cartilage. Clin. Exp. Pharmacol. Physiol. 2016, 43, 312–318. [Google Scholar] [CrossRef]
- Yang, X.B.; Chen, W.P.; Zhao, X.; Chen, L.W.; Li, W.L.; Ran, J.S.; Wu, L.D. Pyruvate Kinase M2 Modulates the Glycolysis of Chondrocyte and Extracellular Matrix in Osteoarthritis. DNA Cell Biol. 2018, 37, 271–277. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.Y.; Huang, H.; Zhou, G.L.; Cui, C.; Weng, Y.J.; Liu, W.C.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, Y.Q.; Ying, M.F.; Jin, C.M.; Li, J.T.; Hu, X. Lactate dehydrogenases amplify reactive oxygen species in cancer cells in response to oxidative stimuli. Signal Transduct. Target. Ther. 2021, 6, 242. [Google Scholar] [CrossRef]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Jagt, D.L.V.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Fan, X.; Crawford, R.; Xiao, Y.; Prasadam, I. The Metabolic Landscape in Osteoarthritis. Aging Dis. 2022, 13, 1166–1182. [Google Scholar] [CrossRef] [PubMed]
- Zainal, Z.; Longman, A.J.; Hurst, S.; Duggan, K.; Caterson, B.; Hughes, C.E.; Harwood, J.L. Relative efficacies of omega-3 polyunsaturated fatty acids in reducing expression of key proteins in a model system for studying osteoarthritis. Osteoarthr. Cartil. 2009, 17, 896–905. [Google Scholar] [CrossRef]
- Choi, W.S.; Lee, G.; Song, W.H.; Koh, J.T.; Yang, J.; Kwak, J.S.; Kim, H.E.; Kim, S.K.; Son, Y.O.; Nam, H.; et al. The CH25H-CYP7B1-RORα axis of cholesterol metabolism regulates osteoarthritis. Nature 2019, 566, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.S.; Yang, J.I.; Kim, W.; Kim, H.E.; Kim, S.K.; Won, Y.; Son, Y.O.; Chun, C.H.; Chun, J.S. Critical role for arginase II in osteoarthritis pathogenesis. Ann. Rheum. Dis. 2019, 78, 421–428. [Google Scholar] [CrossRef]
- Maneiro, E.; Martin, M.A.; de Andres, M.C.; Lopez-Armada, M.J.; Fernandez-Sueiro, J.L.; del Hoyo, P.; Galdo, F.; Arenas, J.; Blanco, F.J. Mitochondrial respiratory activity is altered in osteoarthritic human articular chondrocytes. Arthritis Rheum. 2003, 48, 700–708. [Google Scholar] [CrossRef]
- Haseeb, A.; Makki, M.S.; Haqqi, T.M. Modulation of Ten-Eleven Translocation 1 (TET1), Isocitrate Dehydrogenase (IDH) Expression, alpha-Ketoglutarate (alpha-KG), and DNA Hydroxymethylation Levels by Interleukin-1 beta in Primary Human Chondrocytes. J. Biol. Chem. 2014, 289, 6877–6885. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.T.; Guo, X.; Ma, W.J.; Zhang, Y.G.; Xu, P.; Yao, J.F.; Bai, Y.D. Mitochondrial function is altered in articular chondrocytes of an endemic osteoarthritis, Kashin-Beck disease. Osteoarthr. Cartil. 2010, 18, 1218–1226. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, H.; Jia, S.S.; Wang, Q.; Yao, W.Y.; Yang, Y.; Bai, L.H. Senomorphic agent pterostilbene ameliorates osteoarthritis through the PI3K/AKT/NF-?B axis: An in vitro and in vivo study. Am. J. Transl. Res. 2022, 14, 5243. [Google Scholar]
- Hollander, J.M.; Li, L.Y.; Rawal, M.; Wang, S.K.; Shu, Y.; Zhang, M.; Nielsen, H.C.; Rosen, C.J.; Zeng, L. A critical bioenergetic switch is regulated by IGF2 during murine cartilage development. Commun. Biol. 2022, 5, 1230. [Google Scholar] [CrossRef]
- Cooper, K.L.; Oh, S.; Sung, Y.; Dasari, R.R.; Kirschner, M.W.; Tabin, C.J. Multiple phases of chondrocyte enlargement underlie differences in skeletal proportions. Nature 2013, 495, 375–378. [Google Scholar] [CrossRef]
- Jeon, O.H.; David, N.; Campisi, J.; Elisseeff, J.H. Senescent cells and osteoarthritis: A painful connection. J. Clin. Investig. 2018, 128, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Tarricone, E.; Mattiuzzo, E.; Belluzzi, E.; Elia, R.; Benetti, A.; Venerando, R.; Vindigni, V.; Ruggieri, P.; Brun, P. Anti-Inflammatory Performance of Lactose-Modified Chitosan and Hyaluronic Acid Mixtures in an In Vitro Macrophage-Mediated Inflammation Osteoarthritis Model. Cells 2020, 9, 1328. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, S.; Minguzzi, M.; Platano, D.; Cattini, L.; Trisolino, G.; Mariani, E.; Borzi, R.M. Lithium Chloride Dependent Glycogen Synthase Kinase 3 Inactivation Links Oxidative DNA Damage, Hypertrophy and Senescence in Human Articular Chondrocytes and Reproduces Chondrocyte Phenotype of Obese Osteoarthritis Patients. PLoS ONE 2015, 10, e0143865. [Google Scholar] [CrossRef]
- Kovács, B.; Vajda, E.; Nagy, E.E. Regulatory Effects and Interactions of the Wnt and OPG-RANKL-RANK Signaling at the Bone-Cartilage Interface in Osteoarthritis. Int. J. Mol. Sci. 2019, 20, 4653. [Google Scholar] [CrossRef]
- Mennan, C.; Garcia, J.; McCarthy, H.; Owen, S.; Perry, J.; Wright, K.; Banerjee, R.; Richardson, J.B.; Roberts, S. Human Articular Chondrocytes Retain Their Phenotype in Sustained Hypoxia While Normoxia Promotes Their Immunomodulatory Potential. Cartilage 2019, 10, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.W.; Zhou, M.; Wang, Q.; Zhu, M.J.; Chen, S.; Li, H. Mechanical and hypoxia stress can cause chondrocytes apoptosis through over-activation of endoplasmic reticulum stress. Arch. Oral Biol. 2017, 84, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Bouaziz, W.; Sigaux, J.; Modrowski, D.; Devignes, C.S.; Funck-Brentano, T.; Richette, P.; Ea, H.K.; Provot, S.; Cohen-Solal, M.; Hay, E. Interaction of HIF1 alpha and beta-catenin inhibits matrix metalloproteinase 13 expression and prevents cartilage damage in mice. Proc. Natl. Acad. Sci. USA 2016, 113, 5453–5458. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Mori, D.; Makii, Y.; Nakamoto, H.; Murahashi, Y.; Yano, F.; Chang, S.H.; Taniguchi, Y.; Kobayashi, H.; Semba, H.; et al. Hypoxia-inducible factor-1 alpha maintains mouse articular cartilage through suppression of NF-kappa B signaling. Sci. Rep. 2020, 10, 5425. [Google Scholar] [CrossRef] [PubMed]
- Grimmer, C.; Pfander, D.; Swoboda, B.; Aigner, T.; Mueller, L.; Hennig, F.F.; Gelse, K. Hypoxia-inducible factor 1a is involved in the prostaglandin metabolism of osteoarthritic cartilage through up-regulation of microsomal prostaglandin E synthase 1 in articular chondrocytes. Arthritis Rheum. 2007, 56, 4084–4094. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Moon, Y.; Shin, J.S.; Lee, J.H.; Kim, T.W.; Jang, C.; Park, C.; Lee, J.; Kim, Y.; Werz, O.; et al. A novel mPGES-1 inhibitor alleviates inflammatory responses by downregulating PGE(2) in experimental models. Prostaglandins Other Lipid Mediat. 2019, 144, 106347. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.; Kang, L.J.; Jang, D.; Jeon, J.; Lee, H.; Choi, S.; Han, S.J.; Oh, E.; Nam, J.; Kim, C.S.; et al. Cirsium japonicum var. maackii and apigenin block Hif-2 alpha-induced osteoarthritic cartilage destruction. J. Cell. Mol. Med. 2019, 23, 5369–5379. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Kim, J.; Ryu, J.H.; Oh, H.; Chun, C.H.; Kim, B.J.; Min, B.H.; Chun, J.S. Hypoxia-inducible factor-2 alpha is a catabolic regulator of osteoarthritic cartilage destruction. Nat. Med. 2010, 16, 687–691. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F. Aging and osteoarthritis: The role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthr. Cartil. 2009, 17, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Jallali, N.; Ridha, H.; Thrasivoulou, C.; Underwood, C.; Butler, P.E.M.; Cowen, T. Vulnerability to ROS-induced cell death in ageing articular cartilage: The role of antioxidant enzyme activity. Osteoarthr. Cartil. 2005, 13, 614–622. [Google Scholar] [CrossRef]
- Dai, S.M.; Shan, Z.Z.; Nakamura, H.; Masuko-Hongo, K.; Kato, T.; Nishioka, K.; Yudoh, K. Catabolic stress induces features of chondrocyte senescence through overexpression of caveolin 1—Possible involvement of caveolin 1-induced down-regulation of articular chondrocytes in the pathogenesis of osteoarthritis. Arthritis Rheum. 2006, 54, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Vinatier, C.; Dominguez, E.; Guicheux, J.; Carames, B. Role of the Inflammation-Autophagy-Senescence Integrative Network in Osteoarthritis. Front. Physiol. 2018, 9, 706. [Google Scholar] [CrossRef]
- Guang, Z.; Min, Z.; Li, J.T.; Dou, T.X.; Xiang, G. Single-cell protein activity analysis reveals a novel subpopulation of chondrocytes and the corresponding key master regulator proteins associated with anti-senescence and OA progression. Front. Immunol. 2023, 14, 1077003. [Google Scholar] [CrossRef]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1416. [Google Scholar] [CrossRef]
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell surface-bound IL-1 alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. USA 2009, 106, 17031–17036. [Google Scholar] [CrossRef]
- Ji, M.L.; Jiang, H.; Li, Z.; Geng, R.; Hu, J.Z.; Lin, Y.C.; Lu, J. Sirt6 attenuates chondrocyte senescence and osteoarthritis progression. Nat. Commun. 2022, 13, 7658. [Google Scholar] [CrossRef]
- Cao, X.; Wang, W.; Zhao, B. YAP/TAZ links mechanosensing to aging. Life Med. 2023, 2, lnac039. [Google Scholar] [CrossRef]
- Goto, H.; Nishio, M.; To, Y.; Oishi, T.; Miyachi, Y.; Maehama, T.; Nishina, H.; Akiyama, H.; Mak, T.W.; Makii, Y.; et al. Loss of Mob1a/b in mice results in chondrodysplasia due to YAP1/TAZ-TEAD-dependent repression of SOX9. Development 2018, 145, dev159244. [Google Scholar] [CrossRef]
- Deng, Y.J.; Wu, A.L.; Li, P.S.; Li, G.; Qin, L.; Song, H.; Mak, K.K. Yap1 Regulates Multiple Steps of Chondrocyte Differentiation during Skeletal Development and Bone Repair. Cell Rep. 2016, 14, 2224–2237. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, E.; Despot-Slade, E.; Pichler, M.; Zenobi-Wong, M. RhoA activation and nuclearization marks loss of chondrocyte phenotype in crosstalk with Wnt pathway. Exp. Cell Res. 2017, 360, 113–124. [Google Scholar] [CrossRef]
- Yang, B.N.; Sun, H.L.; Song, F.F.; Yu, M.; Wu, Y.R.; Wang, J.W. YAP1 negatively regulates chondrocyte differentiation partly by activating the beta-catenin signaling pathway. Int. J. Biochem. Cell Biol. 2017, 87, 104–113. [Google Scholar] [CrossRef]
- Deng, Y.J.; Lu, J.Q.; Li, W.L.; Wu, A.L.; Zhang, X.; Tong, W.X.; Ho, K.K.; Qin, L.; Song, H.; Mak, K.K. Reciprocal inhibition of YAP/TAZ and NF-kappa B regulates osteoarthritic cartilage degradation. Nat. Commun. 2018, 9, 4564. [Google Scholar] [CrossRef] [PubMed]
- Zarka, M.; Hay, E.; Cohen-Solal, M. YAP/TAZ in Bone and Cartilage Biology. Front. Cell Dev. Biol. 2022, 9, 788773. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef]
- Jiang, L.J.; Lin, J.C.; Zhao, S.; Wu, J.Q.; Jin, Y.M.; Yu, L.; Wu, N.; Wu, Z.H.; Wang, Y.; Lin, M. ADAMTS5 in Osteoarthritis: Biological Functions, Regulatory Network, and Potential Targeting Therapies. Front. Mol. Biosci. 2021, 8, 703110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.T.; Gui, T.; Liu, R.X.; Tong, K.L.; Wu, C.J.; Li, Z.Y.; Huang, X.; Xu, Q.T.; Yang, J.; Tang, W.; et al. Sequential targeting of YAP1 and p21 enhances the elimination of senescent cells induced by the BET inhibitor JQ1. Cell Death Dis. 2021, 12, 121. [Google Scholar] [CrossRef]
- Zhang, W.; Hsu, P.C.; Zhong, B.; Guo, S.; Zhang, C.; Wang, Y.K.; Luo, C.F.; Zhan, Y.L.; Zhang, C.Q. MiR-34a Enhances Chondrocyte Apoptosis, Senescence and Facilitates Development of Osteoarthritis by Targeting DLL1 and Regulating PI3K/AKT Pathway. Cell. Physiol. Biochem. 2018, 48, 1304–1316. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.N.; Hu, Y.Q.; Song, M.S.; Liu, Z.P.; Zhang, W.Q.; Yu, F.X.; Wu, J.; Wang, S.; Belmonte, J.C.I.; Chan, P.; et al. Up-regulation of FOXD1 by YAP alleviates senescence and osteoarthritis. PLoS Biol. 2019, 17, e3000201. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Hu, Z.L.; Qi, Y.H.; Li, H.Y.; Chang, X.; Gao, X.X.; Liu, C.H.; Li, Y.Y.; Lou, J.H.; Zhai, Y.; et al. Pretreatment of nucleus pulposus mesenchymal stem cells with appropriate concentration of H2O2 enhances their ability to treat intervertebral disc degeneration. Stem Cell Res. Ther. 2022, 13, 340. [Google Scholar] [CrossRef]
- Jin, J.Q.; Zhang, L.; Li, X.Y.; Xu, W.Z.; Yang, S.Y.; Song, J.G.; Zhang, W.H.; Zhan, J.; Luo, J.Y.; Zhang, H.Q. Oxidative stress-CBP axis modulates MOB1 acetylation and activates the Hippo signaling pathway. Nucleic Acids Res. 2022, 50, 3817–3834. [Google Scholar] [CrossRef]
- Wang, K.C.; Yeh, Y.T.; Nguyen, P.; Limqueco, E.; Lopez, J.; Thorossian, S.; Guan, K.L.; Li, Y.S.J.; Chien, S. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 11525–11530. [Google Scholar] [CrossRef]
- Yu, F.F.; Zuo, J.; Fu, X.L.; Gao, M.H.; Sun, L.; Yu, S.Y.; Li, Z.Y.; Zhou, G.Y.; Ba, Y. Role of the hippo signaling pathway in the extracellular matrix degradation of chondrocytes induced by fluoride exposure. Ecotoxicol. Environ. Saf. 2021, 225, 112796. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Li, S.J.; Liu, R.; Zhan, J.F.; Tan, C.; Fang, Y.F.; Chen, Y.; Yu, B. Inhibition of YAP with siRNA prevents cartilage degradation and ameliorates osteoarthritis development. J. Mol. Med. 2019, 97, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.P.; Chen, Z.G.; Xu, L.H.; Wu, L.D.; Xiong, Y. Rapamycin protects chondrocytes against IL-18-induced apoptosis and ameliorates rat osteoarthritis. Aging 2020, 12, 5152–5167. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Jia, C.; Wu, D.; Jin, H.; Lin, Z.; Pan, J.; Li, X.; Wang, W. Fibroblast growth factor 21 (FGF21) alleviates senescence, apoptosis, and extracellular matrix degradation in osteoarthritis via the SIRT1-mTOR signaling pathway. Cell Death Dis. 2021, 12, 865. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.F.; Pan, J.Y.; Li, J.Y.; Zeng, C.; Qi, W.Z.; Shao, Y.; Liu, X.; Liu, L.L.; Xiao, G.Z.; Zhang, H.Y.; et al. Metformin attenuates cartilage degeneration in an experimental osteoarthritis model by regulating AMPK/mTOR. Aging 2020, 12, 1087–1103. [Google Scholar] [CrossRef]
- Zhou, X.D.; Li, J.; Zhou, Y.S.; Yang, Z.C.; Yang, H.Y.; Li, D.; Zhang, J.J.; Zhang, Y.; Xu, N.W.; Huang, Y.; et al. Down-regulated ciRS-7/up-regulated miR-7 axis aggravated Cartilage degradation and autophagy defection by PI3K/AKT/mTOR activation mediated by IL-17A in osteoarthritis. Aging 2020, 12, 20163–20183. [Google Scholar] [CrossRef]
- Swingler, T.E.; Niu, L.; Smith, P.; Paddy, P.; Le, L.; Barter, M.J.; Young, D.A.; Clark, I.M. The function of microRNAs in cartilage and osteoarthritis. Clin. Exp. Rheumatol. 2019, 37, 40–47. [Google Scholar] [PubMed]
- Liu, L.L.; Zhao, C.; Zhang, H.Y.; Lu, Y.H.; Luo, B.S.; Yao, Z.H.; Shao, Y.; Zeng, H.; Zeng, C.; Zhang, R.K.; et al. Asporin regulated by miR-26b-5p mediates chondrocyte senescence and exacerbates osteoarthritis progression via TGF-beta 1/Smad2 pathway. Rheumatology 2022, 61, 2631–2643. [Google Scholar] [CrossRef]
- Liu, Y.K.; Zhang, Z.; Lu, X.Z.; Liu, C.; Zhang, H.N. Senescence-responsive miR-33-5p promotes chondrocyte senescence and osteoarthritis progression by targeting SIRT6. Int. Immunopharmacol. 2023, 121, 110506. [Google Scholar] [CrossRef]
- Xiao, F.; Wang, C.L.; Peng, J.P.; Zhou, X.; Ma, D.; Wang, Y.; Li, Y.P.; Chen, X.D.; Wang, C.D. Changes in Small Noncoding RNA Expression during Chondrocyte Senescence. Cartilage 2022, 13, 19476035221118165. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.J.; Dong, W.J.; Li, Z.R.; Wei, J.Q.; Han, T.; Wang, J.L.; Lin, F. Metformin regulates chondrocyte senescence and proliferation through microRNA-34a/SIRT1 pathway in osteoarthritis. J. Orthop. Surg. Res. 2023, 18, 198. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.Y.; Li, C.J.; Qin, T.; Jin, Y.X.; He, R.D.; Sun, Y.; Liu, Z.D.; Wu, T.D.; Duan, C.Y.; Cao, Y.; et al. Mechanical overloading-induced miR-325-3p reduction promoted chondrocyte senescence and exacerbated facet joint degeneration. Arthritis Res. Ther. 2023, 25, 54. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.H.; Liu, L.L.; Pan, J.Y.; Luo, B.S.; Zeng, H.; Shao, Y.; Zhang, H.B.; Guan, H.; Guo, D.; Zeng, C.; et al. MFG-E8 regulated by miR-99b-5p protects against osteoarthritis by targeting chondrocyte senescence and macrophage reprogramming via the NF-kappa B pathway. Cell Death Dis. 2021, 12, 533. [Google Scholar] [CrossRef]
- Lu, G.H.; Li, L.; Wang, B.; Kuang, L. LINC00623/miR-101/HRAS axis modulates IL-1 beta-mediated ECM degradation, apoptosis and senescence of osteoarthritis chondrocytes. Aging 2020, 12, 3218–3237. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Wang, T.J.; Cai, B.; Wang, X.N.; Feng, W.; Han, Y.; Li, D.S.; Li, S.Q.; Liu, J.G. MicroRNA-495 enhances chondrocyte apoptosis, senescence and promotes the progression of osteoarthritis by targeting AKT1. Am. J. Transl. Res. 2019, 11, 2232–2244. [Google Scholar] [PubMed]
- Philipot, D.; Guerit, D.; Platano, D.; Chuchana, P.; Olivotto, E.; Espinoza, F.; Dorandeu, A.; Pers, Y.M.; Piette, J.; Borzi, R.M.; et al. p16(INK4a) and its regulator miR-24 link senescence and chondrocyte terminal differentiation-associated matrix remodeling in osteoarthritis. Arthritis Res. Ther. 2014, 16, R58. [Google Scholar] [CrossRef]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef]
- Chen, H.T.; Tu, M.; Liu, S.Y.; Wen, Y.X.; Chen, L.B. Dendrobine Alleviates Cellular Senescence and Osteoarthritis via the ROS/NF-kappa B Axis. Int. J. Mol. Sci. 2023, 24, 2365. [Google Scholar] [CrossRef]
- Nogueira-Recalde, U.; Lorenzo-Gomez, I.; Blanco, F.J.; Loza, M.I.; Grassi, D.; Shirinsky, V.; Shirinsky, I.; Lotz, M.; Robbins, P.D.; Dominguez, E.; et al. Fibrates as drugs with senolytic and autophagic activity for osteoarthritis therapy. Ebiomedicine 2019, 45, 588–605. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Jiang, S.L.; Lu, Y.; Yuan, F. Butorphanol tartrate mitigates cellular senescence against tumor necrosis factor -alpha (TNF-alpha) in human HC-A chondrocytes. Bioengineered 2022, 13, 5434–5442. [Google Scholar] [CrossRef]
- Zhu, J.J.; Yang, S.H.; Qi, Y.D.; Gong, Z.; Zhang, H.T.; Liang, K.Y.; Shen, P.Y.; Huang, Y.Y.; Zhang, Z.; Ye, W.L.; et al. Stem cell-homing hydrogel-based miR-29b-5p delivery promotes cartilage regeneration by suppressing senescence in an osteoarthritis rat model. Sci. Adv. 2022, 8, eabk0011. [Google Scholar] [CrossRef] [PubMed]
- Attwaters, M. Targeting senescence in OA. Nat. Rev. Rheumatol. 2022, 18, 305. [Google Scholar] [CrossRef] [PubMed]



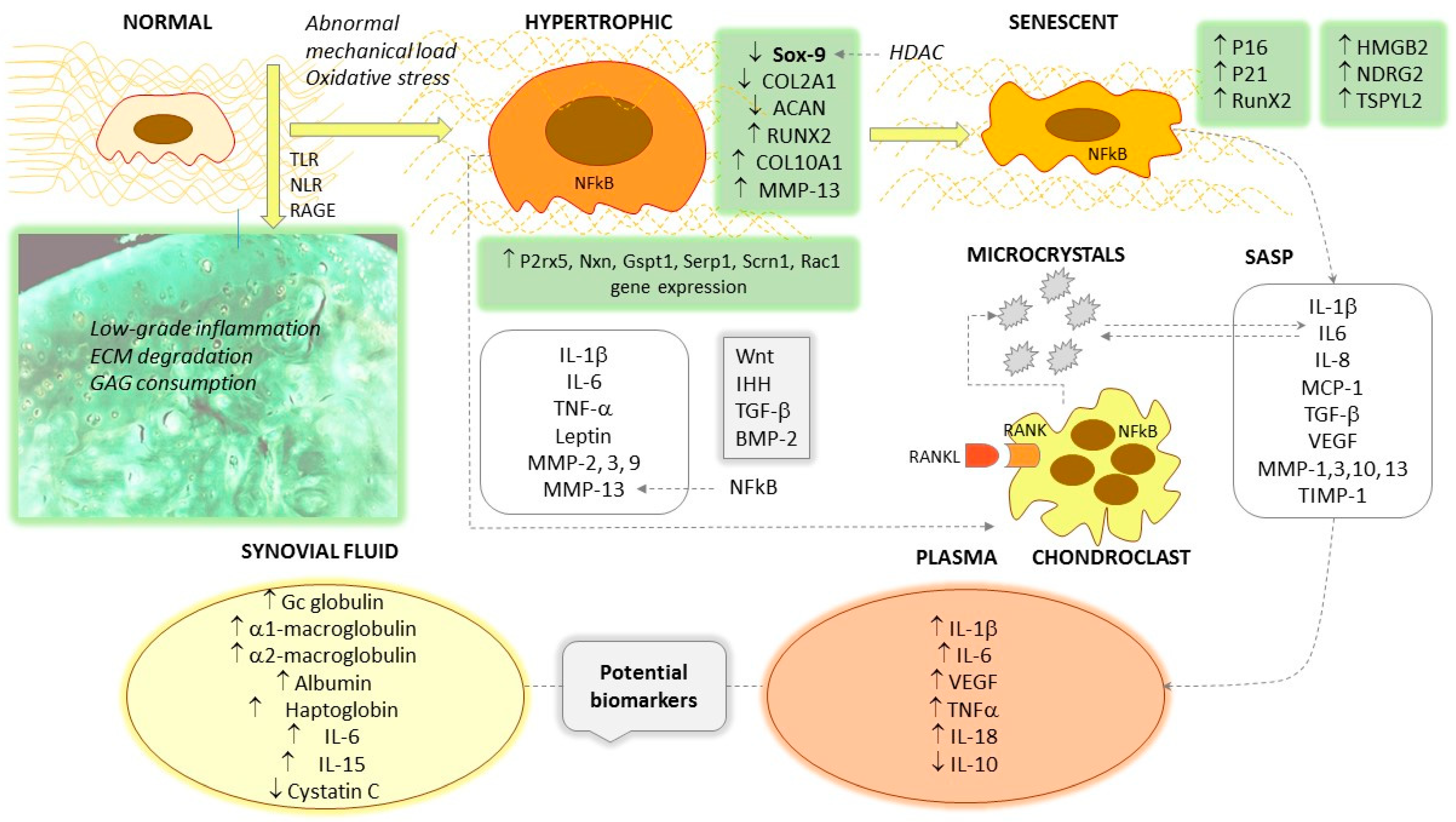{kind=link}
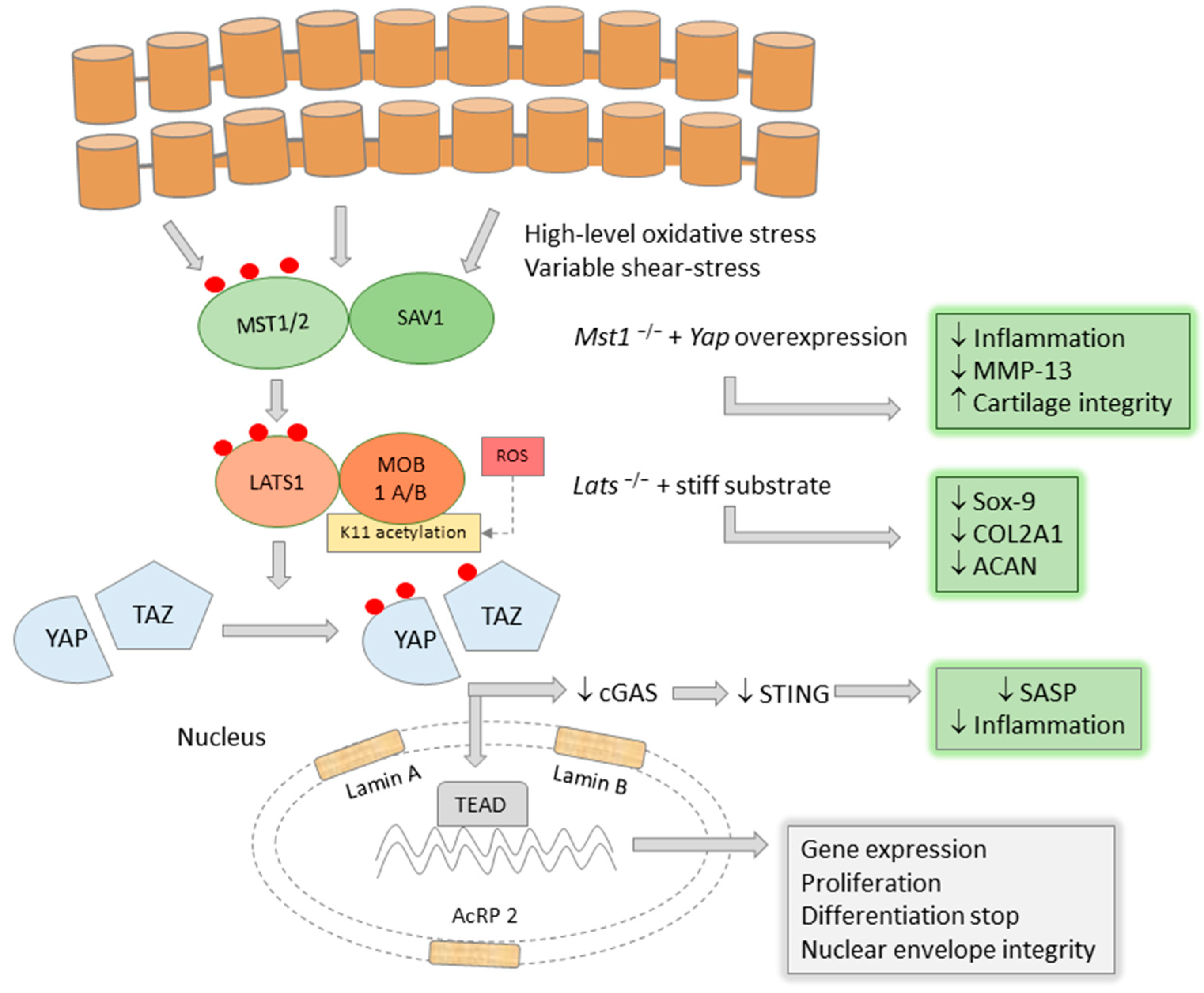{kind=link}
| Reference | Study Goals | Change of Mediators | Other Results |
|---|---|---|---|
| Circulating mediators of inflammation | |||
| Sohn D.H. et al. [8] | Proteomic analysis of serum/synovial fluid (SF) | Newly identified NF-kB-related proteins, cytokine receptors (IL-12R, IL-18R, IL-20R) and macrophage-derived inflammatory proteins in synovial fluid High levels of IL-6 in SF | Upregulation of histone deacetylase Upregulated plasma proteins, protease inhibitors, NF-kB subunits and regulators, cytokines (IL-6, MCP-1, VEGF), complement fragments in OA sera |
| Gobezie R. et al. [11] | Synovial protein analysis in OA patients and healthy | ↑ Albumin, fibrinogen, α1-microglobulin/bikunin precursor, α2-macroglobulin, haptoglobin, complement C3 ↓ Cystatin C, aggrecan | 18 Proteins differentially expressed between OA and healthy |
| Krenytska D. et al. [12] | Comparative analysis of plasma cytokines and growth factors in OA, OA + COVID-19 and controls | ↑ IL-1β in OA ↓ TNF-α, NF-kB in OA ↓ VEGF, PDGF, FGF2 in OA No significant changes in IL-6 and HIF-1α | Maximum values of IL-1β, less pronounced decrease in TNF-α and NF-kB |
| Wang Z.W. et al. [14] | Serum protein analysis in OA patients | ↑ IL-1β, IL-6, TNFα, VEGF in OA patients vs. controls | Increased expression of IL-1β, IL-6, TNFα, VEGF in synoviocytes |
| Stannus O. et al. [19] | Follow-up study of serum cytokines in an elderly cohort | Quartiles of IL-6 and TNF-α are associated with joint space narrowing | Baseline and changes of IL-6 predicts medial and lateral cartilage volume loss |
| Barker T. et al. [20] | Comparative study of serum IL-10 and TNF-α in different OA stages and controls | ↓ IL-10 and IL-10/TNFα in subjects who underwent ACL or TJR surgery No significant changes in TNFα | IL-10 and IL-10/TNFα significantly differ between Kellgren–Lawrence scores 3 vs. 4 Low IL-10 suggests predisposition for development of severe knee OA |
| Panina S.B. et al. [21] | Comparative study of plasma and SF mediators in post-traumatic OA patients and controls | ↑ plasma leptin, IL-1β and IL-6 | Plasma leptin and SF IL-18 correlate with the Kellgren–Lawrence score in PTOA Significant correlation between plasma and SF leptin, IL-6 and IL-18 |
| Waszczykowski M. et al. [22] | Comparative study of serum and SF cytokines in OA vs. healthy controls | ↑ serum IL-6, IL-18 and IL-20 in OA vs. control group | IL-18 correlates with MMP-3 in OA patients ROC curve of IL-20 suggests diagnostic potential |
| Histological markers of inflammation | |||
| Wang Z.W. et al. [14] | Synovial membrane immunohistochemistry analysis | ↑ IL-1β, IL-6, TNF-α and VEGF synovial membrane expression in moderate/advanced OA compared to mild OA | ↑ serum IL-1β, IL-6, TNFα, VEGF in OA patients vs. controls |
| Qu X.Q. et al. [25] | Comparative genetic and immunohistochemistry study of OA vs. normal cartilage specimens (knee arthroplasty and meniscus surgery) | ↑ IL-6 and MMP-9 expression in OA specimens | ↑ IL-6 and MMP-9 gene expression in OA cartilage |
| Warner S.C. et al. [27] | Immunohistochemistry and explant culture studies of OA patients | ↑ IL-15Rα in OA samples | IL-15 treatment induction of MMP-1 and MMP-3 |
| Scanzello C.R. et al. [28] | Synovial membrane immunohistochemistry in early vs. advanced OA patient cohort | ↑ IL-15Rα in synovial membrane cells from patients with advanced OA | Increased IL-15 in SF, correlating with IL-6 |
| Iannone F. et al. [29] | Comparative immunohistochemistry study of OA vs. healthy subjects | ↑ IL-10 protein and mRNA expression in high-intensity cartilage lesions | No relationship between IL-10R expression and cartilage lesion degree |
| Vuolteenaho et al. [34] | OA patients cohort study of phenotype-explanted cartilage | ↑ IL-6, IL-8, PGE2, Cox-2 expression in cartilage cultures obtained by joint replacement, induced by leptin/IL-1β | Selective inhibition of iNOS suppressed IL-6, IL-8, PGE2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horváth, E.; Sólyom, Á.; Székely, J.; Nagy, E.E.; Popoviciu, H. Inflammatory and Metabolic Signaling Interfaces of the Hypertrophic and Senescent Chondrocyte Phenotypes Associated with Osteoarthritis. Int. J. Mol. Sci. 2023, 24, 16468. https://doi.org/10.3390/ijms242216468
Horváth E, Sólyom Á, Székely J, Nagy EE, Popoviciu H. Inflammatory and Metabolic Signaling Interfaces of the Hypertrophic and Senescent Chondrocyte Phenotypes Associated with Osteoarthritis. International Journal of Molecular Sciences. 2023; 24(22):16468. https://doi.org/10.3390/ijms242216468
Chicago/Turabian StyleHorváth, Emőke, Árpád Sólyom, János Székely, Előd Ernő Nagy, and Horațiu Popoviciu. 2023. "Inflammatory and Metabolic Signaling Interfaces of the Hypertrophic and Senescent Chondrocyte Phenotypes Associated with Osteoarthritis" International Journal of Molecular Sciences 24, no. 22: 16468. https://doi.org/10.3390/ijms242216468