
Changes in Lipoprotein(a) Levels in People after ST Elevation Myocardial Infarction—The STEMI-Lipids Study
 , and
, and
Abstract
:1. Introduction
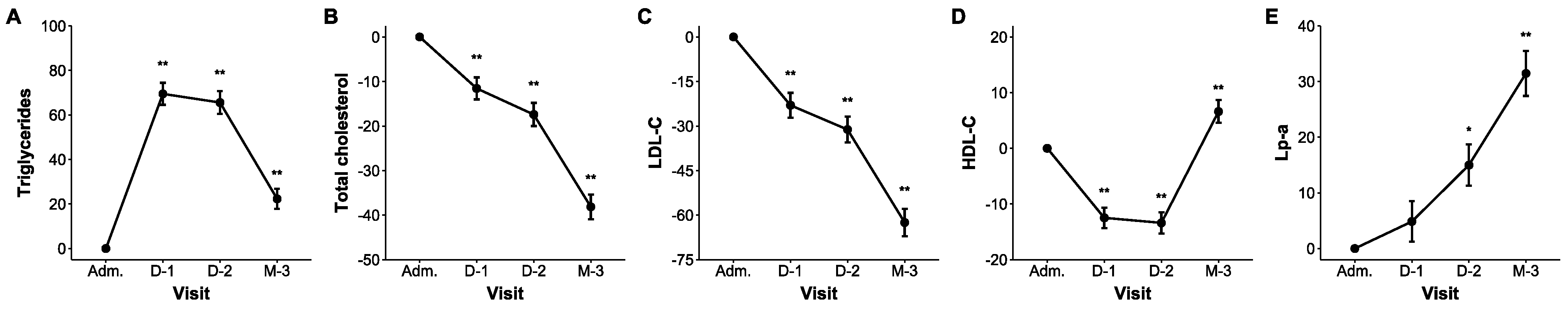
2. Results
3. Discussion
4. Materials and Methods
5. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bhatt, D.L.; Lopes, R.D.; Harrington, R.A. Diagnosis and Treatment of Acute Coronary Syndromes: A Review. JAMA 2022, 327, 662–675. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Eagle, K.A.; Ohman, E.M.; Hirsch, A.T.; Goto, S.; Mahoney, E.M.; Wilson, P.W.F.; Alberts, M.J.; D’Agostino, R.; Liau, C.-S.; et al. Comparative Determinants of 4-Year Cardiovascular Event Rates in Stable Outpatients at Risk of or With Atherothrombosis. JAMA 2010, 304, 1350–1357. [Google Scholar] [CrossRef]
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.-C.; Baum, S.J.; Steinhagen-Thiessen, E.; Shapiro, M.D.; Stroes, E.S.; Moriarty, P.M.; Nordestgaard, B.G.; et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N. Engl. J. Med. 2020, 382, 244–255. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Rosenson, R.S.; Gencer, B.; López, J.A.G.; Lepor, N.E.; Baum, S.J.; Stout, E.; Gaudet, D.; Knusel, B.; Kuder, J.F.; et al. Small Interfering RNA to Reduce Lipoprotein(a) in Cardiovascular Disease. N. Engl. J. Med. 2022, 387, 1855–1864. [Google Scholar] [CrossRef] [PubMed]
- Duarte Lau, F.; Giugliano, R.P. Lipoprotein(a) and its Significance in Cardiovascular Disease: A Review. JAMA Cardiol. 2022, 7, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S. A Test in Context: Lipoprotein(a): Diagnosis, Prognosis, Controversies, and Emerging Therapies. J. Am. Coll. Cardiol. 2017, 69, 692–711. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Pearson, G.J.; Thanassoulis, G.; Anderson, T.J.; Barry, A.R.; Couture, P.; Dayan, N.; Francis, G.A.; Genest, J.; Grégoire, J.; Grover, S.A.; et al. 2021 Canadian Cardiovascular Society Guidelines for the Management of Dyslipidemia for the Prevention of Cardiovascular Disease in Adults. Can. J. Cardiol. 2021, 37, 1129–1150. [Google Scholar] [CrossRef]
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.-M.; Capodanno, D.; et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice: Developed by the Task Force for cardiovascular disease prevention in clinical practice with representatives of the European Society of Cardiology and 12 medical societies With the special contribution of the European Association of Preventive Cardiology (EAPC). Eur. Heart J. 2021, 42, 3227–3337. [Google Scholar] [CrossRef]
- Fusco, S.A.D.; Arca, M.; Scicchitano, P.; Alonzo, A.; Perone, F.; Gulizia, M.M.; Gabrielli, D.; Oliva, F.; Imperoli, G.; Colivicchi, F. Lipoprotein(a): A risk factor for atherosclerosis and an emerging therapeutic target. Heart 2023, 109, 18–25. [Google Scholar] [CrossRef]
- Kronenberg, F.; Mora, S.; Stroes, E.S.G.; Ference, B.A.; Arsenault, B.J.; Berglund, L.; Dweck, M.R.; Koschinsky, M.; Lambert, G.; Mach, F.; et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: A European Atherosclerosis Society consensus statement. Eur. Heart J. 2022, 43, 3925–3946. [Google Scholar] [CrossRef] [PubMed]
- Forbang, N.I.; Criqui, M.H.; Allison, M.A.; Ix, J.H.; Steffen, B.T.; Cushman, M.; Tsai, M.Y. Sex and ethnic differences in the associations between lipoprotein(a) and peripheral arterial disease in the Multi-Ethnic Study of Atherosclerosis. J. Vasc. Surg. 2016, 63, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Varvel, S.; McConnell, J.P.; Tsimikas, S. Prevalence of Elevated Lp(a) Mass Levels and Patient Thresholds in 532 359 Patients in the United States. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2239–2245. [Google Scholar] [CrossRef] [PubMed]
- Ziogos, E.; Vavuranakis, M.A.; Harb, T.; Foran, P.L.; Blaha, M.J.; Jones, S.R.; Lai, S.; Gerstenblith, G.; Leucker, T.M. Lipoprotein(a) concentrations in acute myocardial infarction patients are not indicative of levels at six month follow-up. Eur. Heart J. Open 2023, 3, oead035. [Google Scholar] [CrossRef]
- Mooser, V.; Berger, M.M.; Tappy, L.; Cayeux, C.; Marcovina, S.M.; Darioli, R.; Nicod, P.; Chioléro, R. Major Reduction in Plasma Lp(a) Levels During Sepsis and Burns. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1137–1142. [Google Scholar] [CrossRef]
- Tsimikas, S.; Gordts, P.L.S.M.; Nora, C.; Yeang, C.; Witztum, J.L. Statin therapy increases lipoprotein(a) levels. Eur. Heart J. 2020, 41, 2275–2284. [Google Scholar] [CrossRef]
- Hermel, M.; Lieberman, M.; Slipczuk, L.; Rana, J.S.; Virani, S.S. Monoclonal Antibodies, Gene Silencing and Gene Editing (CRISPR) Therapies for the Treatment of Hyperlipidemia—The Future Is Here. Pharmaceutics 2023, 15, 459. [Google Scholar] [CrossRef]
- Yeang, C.; Karwatowska-Prokopczuk, E.; Su, F.; Dinh, B.; Xia, S.; Witztum, J.L.; Tsimikas, S. Effect of Pelacarsen on Lipoprotein(a) Cholesterol and Corrected Low-Density Lipoprotein Cholesterol. J. Am. Coll. Cardiol. 2022, 79, 1035–1046. [Google Scholar] [CrossRef]
- Fusco, S.A.D.; Maggioni, A.P.; Bernelli, C.; Perone, F.; Marzo, V.D.; Conte, E.; Musella, F.; Uccello, G.; Luca, L.D.; Gabrielli, D.; et al. Inclisiran: A New Pharmacological Approach for Hypercholesterolemia. Rev. Cardiovasc. Med. 2022, 23, 375. [Google Scholar] [CrossRef]
- Yu, Z.; Hu, L.; Sun, C.; Wang, Z.; Zhang, X.; Wu, M.; Liu, L. Effect of Different Types and Dosages of Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitors on Lipoprotein(a) Levels: A Network Meta-analysis. J. Cardiovasc. Pharmacol. 2023, 81, 445–453. [Google Scholar] [CrossRef]
- Kim, K.A.; Park, H.-J. New Therapeutic Approaches to the Treatment of Dyslipidemia 2: LDL-C and Lp(a). J. Lipid Atheroscler. 2023, 12, 37. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Characteristic | |
|---|---|
| Age at admission (years), median (IQR) | 61 (55–70) |
| BMI (kg/m2), median (IQR) | 27.2 (25.0–30.7) |
| Triglycerides (mg/dL), median (IQR) | 74 (53–101) |
| Total cholesterol (mg/dL), median (IQR) | 195 (169–228) |
| LDL-C (mg/dL), median (IQR) | 130 (102–154) |
| HDL-C (mg/dL), median (IQR) | 47 (41–57) |
| Lp(a) (mg/dL), median (IQR) | 7.85 (3.70–37.10) |
| Maximum CK (U/L), median (IQR) | 1149 (575–2107) |
| Maximum Troponin T (pg/mL), median (IQR) | 3773 (1259–6216) |
| Systolic blood pressure (mmHg), median (IQR) | 126 (108–140) |
| Diastolic blood pressure (mmHg), median (IQR) | 76 (66–83) |
| eGFR (ml/min/1.73 m2), median (IQR) | 83.93 (69.48–96.53) |
| Sex | |
| Men, n (%) | 61 (70.9) |
| Women, n (%) | 25 (29.1) |
| BMI | |
| Underweight (<18.5), (%) | 1.2 |
| Normal range (18.5–24.9), (%) | 23.3 |
| Overweight (25–29.9), (%) | 50 |
| Obesity class I (30–34.9), (%) | 17.4 |
| Obesity class II (35–39.9), (%) | 6.9 |
| Obesity class III (>40), (%) | 1.2 |
| Diabetes (%) | 18.8 |
| Arterial hypertension (%) | 75.3 |
| Current smoker (%) | 40.5 |
| Past smoker (%) | 21.4 |
| eGFR categories (%) | |
| >90 | 36.1 |
| <90 | 52.3 |
| ≤60 | 3.5 |
| ≤45 | 7.0 |
| ≤30 | 1.2 |
| Medication at hospital discharge | |
| Any statin (%) | 98.8 |
| Simvastatin (%) | 1.2 |
| Atorvastatin (%) | 96.4 |
| Rosuvastatin (%) | 2.4 |
| Ezetimibe 10 mg (%) | 8.5 |
| PCSK9 inhibitor (%) | 0.0 |
| ASA (%) | 96.4 |
| Ticagrelor (%) | 53.0 |
| Prasugrel (%) | 34.9 |
| Clopidogrel (%) | 10.8 |
| ACE inhibitors/ARB (%) | 89.2 |
| Betablocker (%) | 89.2 |
| MRA (%) | 29.0 |
| NOAC (%) | 7.2 |
| OAC (%) | 0.0 |
| Outcomes | Admission | Day 1 | Day 2 | Post-Discharge Follow-Up | |||
|---|---|---|---|---|---|---|---|
| Median (IQR) | Median (IQR) | ∆ (95% CI) | Median (IQR) | ∆ (95% CI) | Median (IQR) | ∆ (95% CI) | |
| Triglycerides (mg/dL) | 74 (53–101) | 134 (97–168) | 69.5 (54.2 to 86.3) ** | 126 (99–169) | 65.5 (50.1 to 82.6) ** | 97 (75–124) | 22.3 (10.2 to 35.8) ** |
| Total cholesterol (mg/dL) | 195 (169–228) | 172 (142–204) | −11.6 (−15.7 to −7.2) ** | 161 (134–194) | −17.4 (−21.5 to −13.2) ** | 118 (108–142) | −38.1 (−41.4 to −34.8) ** |
| LDL-C (mg/dL) | 130 (102–154) | 98 (75–125) | −23.0 (−29.0 to −16.5) ** | 88 (65–112) | −31.2 (−36.7 to −25.2) ** | 49 (40–61) | −62.5 (−65.7 to −59.1) ** |
| HDL-C (mg/dL) | 47 (41–57) | 40 (36–49) | −12.5 (−15.6 to −9.3) ** | 40 (35–48) | −13.4 (−16.6 to −10.1) ** | 50 (41–60) | 6.6 (2.5 to 11.0) * |
| Lp(a) (mg/dL) | 7.9 (3.8–37.1) | 8.4 (3.9–35.4) | 4.9 (−2.2 to −12.4) | 9.3 (3.7–39.1) | 15.0 (7.0 to 23.5) * | 11.2 (4.4–59.6) | 31.4 (21.6 to 42.0) ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sourij, C.; Aziz, F.; Krappinger, S.; Praschk, A.; Metzner, T.; Kojzar, H.; Zirlik, A.; Stojakovic, T.; Pätzold, D.; von Lewinski, D.; et al. Changes in Lipoprotein(a) Levels in People after ST Elevation Myocardial Infarction—The STEMI-Lipids Study. Int. J. Mol. Sci. 2023, 24, 15531. https://doi.org/10.3390/ijms242115531
Sourij C, Aziz F, Krappinger S, Praschk A, Metzner T, Kojzar H, Zirlik A, Stojakovic T, Pätzold D, von Lewinski D, et al. Changes in Lipoprotein(a) Levels in People after ST Elevation Myocardial Infarction—The STEMI-Lipids Study. International Journal of Molecular Sciences. 2023; 24(21):15531. https://doi.org/10.3390/ijms242115531
Chicago/Turabian StyleSourij, Caren, Faisal Aziz, Sarah Krappinger, Andreas Praschk, Thomas Metzner, Harald Kojzar, Andreas Zirlik, Tatjana Stojakovic, Dieter Pätzold, Dirk von Lewinski, and et al. 2023. "Changes in Lipoprotein(a) Levels in People after ST Elevation Myocardial Infarction—The STEMI-Lipids Study" International Journal of Molecular Sciences 24, no. 21: 15531. https://doi.org/10.3390/ijms242115531