
Model Systems to Study the Mechanism of Vascular Aging
 , ,
, ,
Abstract
:1. Introduction
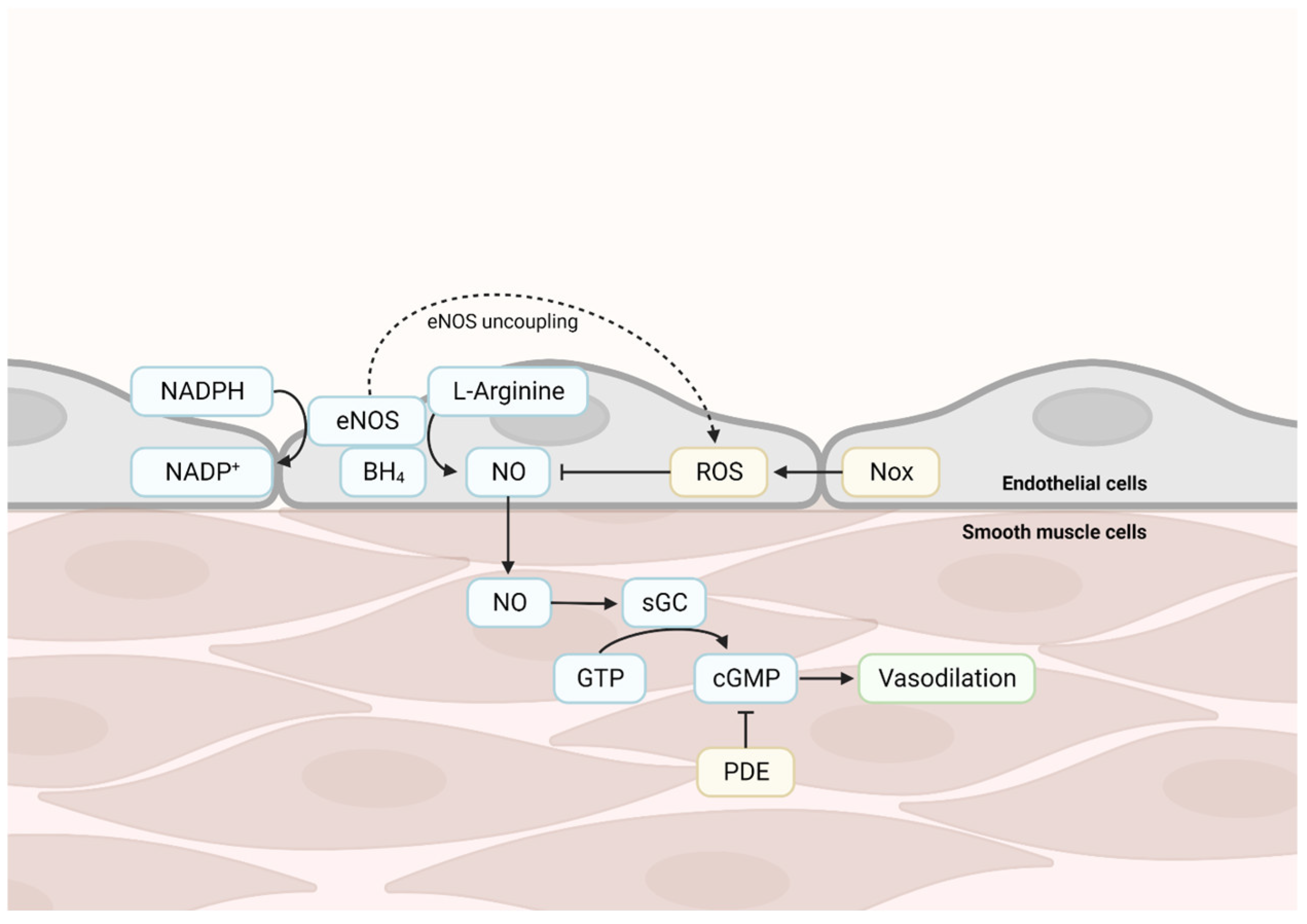
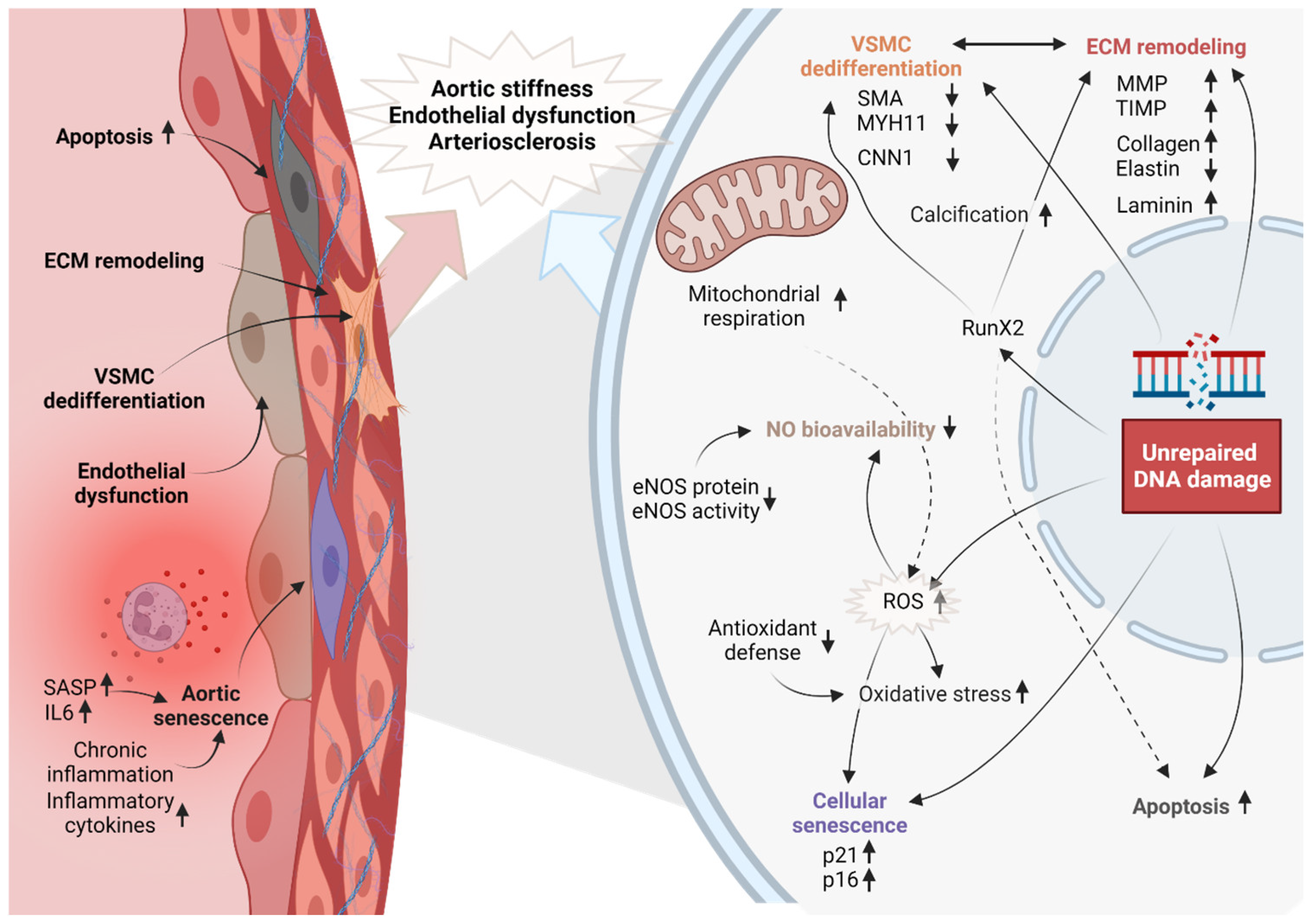
2. Structural, Functional, and Pathological Changes in the Aged Vascular System
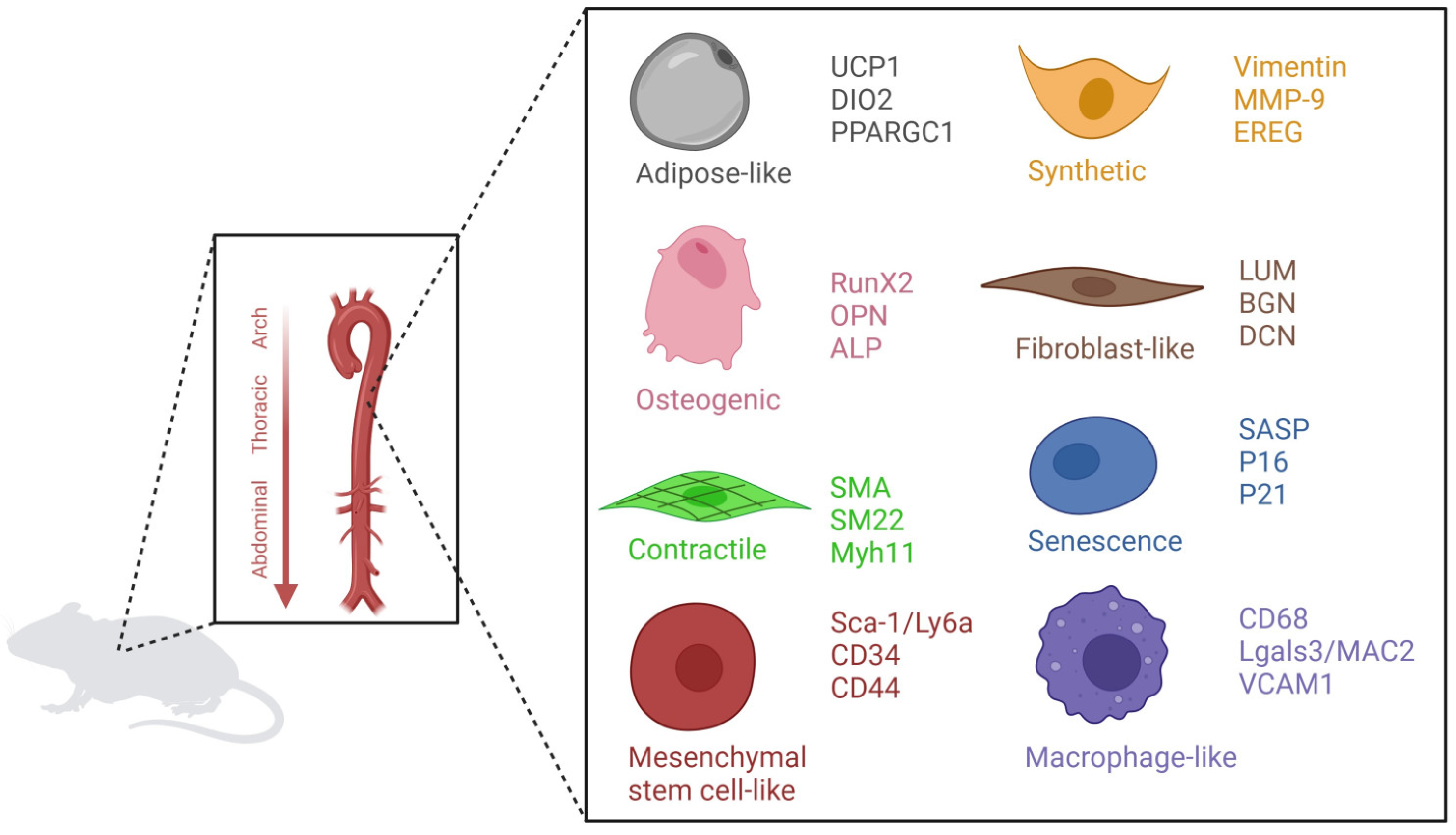
Cellular Changes in the Aged Vascular System
3. Vascular Aging in Human Progeria Syndromes
4. Vascular Aging Mouse Models
4.1. Nucleotide Excision Repair (NER) Deficient Mouse Models
4.2. LMNA
4.3. Bub1b
4.4. Telomere Attrition
4.5. Epigenetic Alterations
4.6. Limitations of Mouse Models for Vascular Aging

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Model | Gene Targeted | Cell Type | Vascular Phenotypes | Reference |
|---|---|---|---|---|
| ERCC1d/− | ERCC1 | Full body | Increased aortic stiffness, blood pressure, aortic senescence, endothelial dysfunction, and decreased reactive hyperemia | [39,40,41] |
| ERCC1-Tie2Cre | ERCC1 | EC | Decreased endothelium-derived NO, altered vasoconstriction, arterial remodeling, and arterial stiffness | [43] |
| ERCC1-SM22 | ERCC1 | SMCs | Diminished NO-mediated vasodilation, decreased reactive hyperemia, and arterial stiffness | [44] |
| LmnaG609G/G609G | LMNA | Full body | Vascular stiffening, ECM remodeling | [48,50] |
| Ldlr−/−LmnaG609G/G609G | LMNA | Full body | VSMC depletion, adventitial thickening, and changes to the elastin structure, atherosclerosis | [51] |
| LmnaLCS/LCSSM22αCre+/tg | LMNA | SMCs | Vascular stiffening and ECM remodeling | [50] |
| LmnaLCS/LCSTie2Cre+/tg | LMNA | EC | Unkown | [50] |
| Apoe−/−LmnaG609G/G609G | LMNA | Full body | VSMC depletion, adventitial thickening, and changes to the elastin structure | [55] |
| Apoe−/−LmnaLCS/LCSSM22αCre | LMNA | SMCs | VSMC depletion, adventitial thickening, and changes to the elastin structure | [55] |
| Apoe−/−LmnaLCS/LCSLysMCre | LMNA | Macrophage | Unkown | [55] |
| Bub1b | Bub1b | Full body | Endothelial dysfunction, decreased levels of elastin, fibrosis, thinning of both the arterial wall and the inner diameter, VSMC loss, and impaired angiogenesis | [56,58] |
| Terc | Terc | Full body | Endothelial dysfunction, senescence, and hypertension | [60,61,62] |
| Sirt6 | Sirt6 | Full body | Endothelial dysfunction and vascular calcification | [65,66,67] |
| SIRT6+/−;ApoE−/− | Sirt6 | Full body | Atherosclerosis and vascular inflammation | [68] |
5. The Role of Genomic Instability in Molecular and Cellular Mechanisms Underlying Vascular Aging
6. Models to Study Vascular Aging In Vitro
7. Cell Culture Models to Study Vascular Aging
Limitations Cell Culture Models to Study Vascular Aging
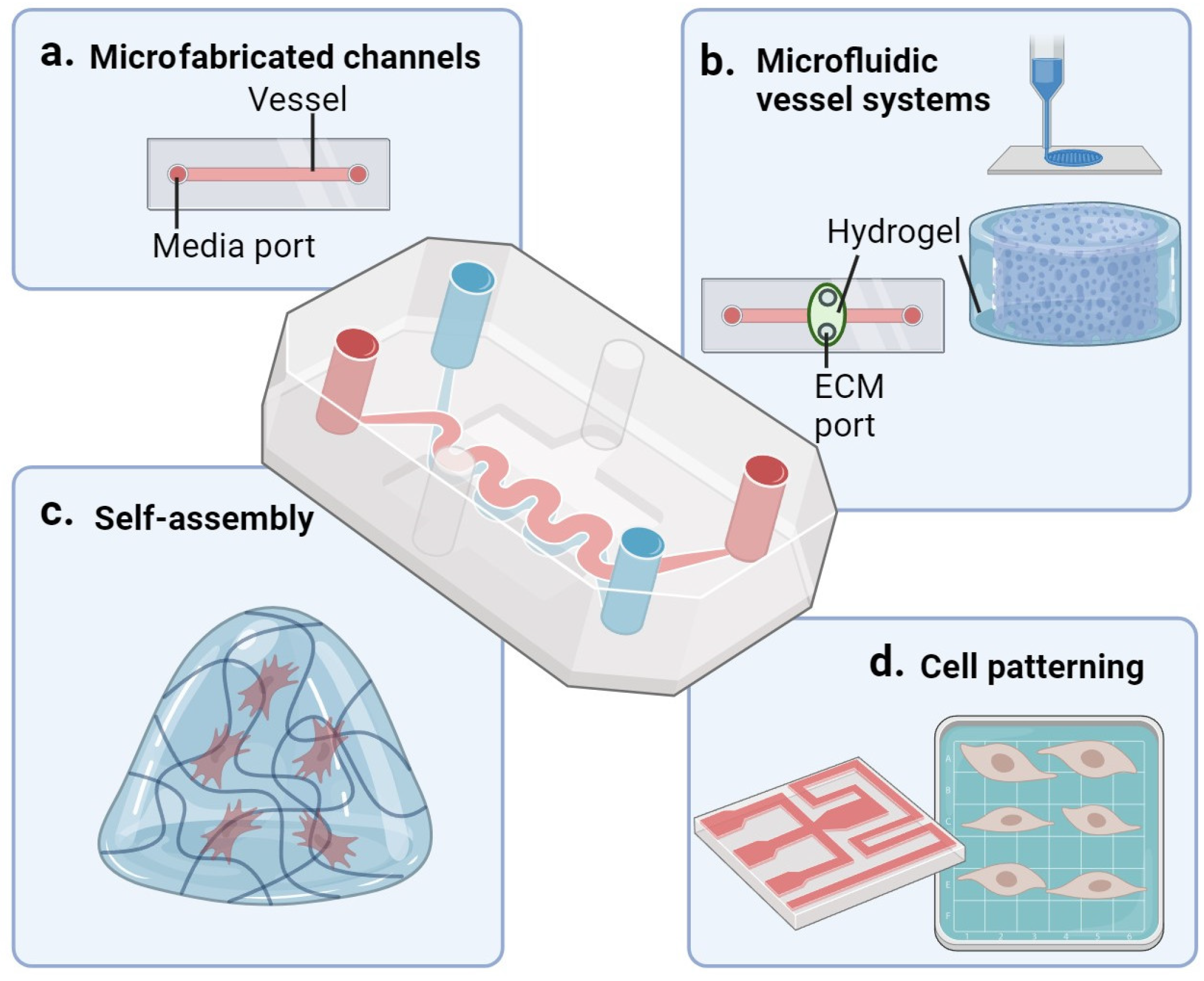
8. Microfluidic Cell Culture Systems and Vascular Aging
8.1. Microfabricated Channels
8.2. Microfluidic Vessel Models
8.3. Microfluidics Combined with Self-Assembly
8.4. Cell Patterning
8.5. Scientific Outcomes Microfluidic Systems
8.6. Comparison Microfluidic Systems
9. Personalized Medicine
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vos, T.; Lim, S.S.; Abbafati, C.; Abbas, K.M.; Abbasi, M.; Abbasifard, M.; Abbasi-Kangevari, M.; Abbastabar, H.; Abd-Allah, F.; Abdelalim, A.; et al. Global Burden of 369 Diseases and Injuries in 204 Countries and Territories, 1990–2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990-2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Reference Values for Arterial Stiffness’ Collaboration. Determinants of Pulse Wave Velocity in Healthy People and in the Presence of Cardiovascular Risk Factors: ‘Establishing Normal and Reference Values’. Eur. Heart. J. 2010, 31, 2338–2350. [Google Scholar] [CrossRef] [PubMed]
- Vlachopoulos, C.; Aznaouridis, K.; Stefanadis, C. Prediction of Cardiovascular Events and All-Cause Mortality with Arterial Stiffness: A Systematic Review and Meta-Analysis. J. Am. Coll. Cardiol. 2010, 55, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, F.D.; Whelton, P.K. High Blood Pressure and Cardiovascular Disease. Hypertension 2020, 75, 285–292. [Google Scholar] [CrossRef]
- Lakatta, E.G. Arterial and Cardiac Aging: Major Shareholders in Cardiovascular Disease Enterprises: Part III: Cellular and Molecular Clues to Heart and Arterial Aging. Circulation 2003, 107, 490–497. [Google Scholar] [CrossRef]
- AlGhatrif, M.; Lakatta, E.G. The Conundrum of Arterial Stiffness, Elevated Blood Pressure, and Aging. Curr. Hypertens. Rep. 2015, 17, 12. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Tarantini, S.; Donato, A.J.; Galvan, V.; Csiszar, A. Mechanisms of Vascular Aging. Circ. Res. 2018, 123, 849–867. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of Aging: An Expanding Universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Thijssen, D.H.J.; Carter, S.E.; Green, D.J. Arterial Structure and Function in Vascular Ageing: Are You as Old as Your Arteries? J. Physiol. 2016, 594, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Hummel, S.L.; Salazar, J.B.; Taffet, G.E.; Zieman, S.; Schwartz, J.B. Cardiovascular Physiology in the Older Adults. J. Geriatr. Cardiol. 2015, 12, 196–201. [Google Scholar] [CrossRef]
- Lüscher, T.F.; Boulanger, C.M.; Dohi, Y.; Yang, Z.H. Endothelium-Derived Contracting Factors. Hypertension 1992, 19, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, V.; Vickneson, K.; Kofidis, T.; Woo, C.C.; Lin, X.Y.; Foo, R.; Shanahan, C.M. Role of Vascular Smooth Muscle Cell Plasticity and Interactions in Vessel Wall Inflammation. Front. Immunol. 2020, 11, 599415. [Google Scholar] [CrossRef]
- Cao, G.; Xuan, X.; Hu, J.; Zhang, R.; Jin, H.; Dong, H. How Vascular Smooth Muscle Cell Phenotype Switching Contributes to Vascular Disease. Cell. Commun. And. Signal. 2022, 20, 180. [Google Scholar] [CrossRef]
- Yap, C.; Mieremet, A.; de Vries, C.J.M.; Micha, D.; de Waard, V. Six Shades of Vascular Smooth Muscle Cells Illuminated by KLF4 (Krüppel-Like Factor 4). Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2693–2707. [Google Scholar] [CrossRef]
- Tang, H.-Y.; Chen, A.-Q.; Zhang, H.; Gao, X.-F.; Kong, X.-Q.; Zhang, J.-J. Vascular Smooth Muscle Cells Phenotypic Switching in Cardiovascular Diseases. Cells 2022, 11, 4060. [Google Scholar] [CrossRef] [PubMed]
- Rensen, S.S.M.; Doevendans, P.A.F.M.; van Eys, G.J.J.M. Regulation and Characteristics of Vascular Smooth Muscle Cell Phenotypic Diversity. Neth. Heart J. 2007, 15, 100–108. [Google Scholar] [CrossRef]
- Zhao, D.; Li, J.; Xue, C.; Feng, K.; Liu, L.; Zeng, P.; Wang, X.; Chen, Y.; Li, L.; Zhang, Z.; et al. TL1A Inhibits Atherosclerosis in ApoE-Deficient Mice by Regulating the Phenotype of Vascular Smooth Muscle Cells. J. Biol. Chem. 2020, 295, 16314–16327. [Google Scholar] [CrossRef]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of Smooth Muscle Cells in Vascular Calcification: Implications in Atherosclerosis and Arterial Stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef]
- Depuydt, M.A.C.; Prange, K.H.M.; Slenders, L.; Örd, T.; Elbersen, D.; Boltjes, A.; de Jager, S.C.A.; Asselbergs, F.W.; de Borst, G.J.; Aavik, E.; et al. Microanatomy of the Human Atherosclerotic Plaque by Single-Cell Transcriptomics. Circ. Res. 2020, 127, 1437–1455. [Google Scholar] [CrossRef] [PubMed]
- Rzucidlo, E.M.; Martin, K.A.; Powell, R.J. Regulation of Vascular Smooth Muscle Cell Differentiation. J. Vasc. Surg. 2007, 45, A25–A32. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Vascular Smooth Muscle Cell in Atherosclerosis. Acta Physiol. 2015, 214, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Gorog, P.; Kovacs, I.B. Inhibition of Vascular Smooth Muscle Cell Migration by Intact Endothelium Is Nitric Oxide-Mediated: Interference by Oxidised Low Density Lipoproteins. J. Vasc. Res. 1998, 35, 165–169. [Google Scholar] [CrossRef]
- Sarkar, R.; Meinberg, E.G.; Stanley, J.C.; Gordon, D.; Clinton Webb, R. Nitric Oxide Reversibly Inhibits the Migration of Cultured Vascular Smooth Muscle Cells. Circ. Res. 1996, 78, 225–230. [Google Scholar] [CrossRef] [PubMed]
- van den Oever, I.A.M.; Raterman, H.G.; Nurmohamed, M.T.; Simsek, S. Endothelial Dysfunction, Inflammation, and Apoptosis in Diabetes Mellitus. Mediators. Inflamm. 2010, 2010, 792393. [Google Scholar] [CrossRef]
- Ataei Ataabadi, E.; Golshiri, K.; Jüttner, A.; Krenning, G.; Danser, A.H.J.; Roks, A.J.M. Nitric Oxide-CGMP Signaling in Hypertension. Hypertension 2020, 76, 1055–1068. [Google Scholar] [CrossRef]
- Graziano, S.; Kreienkamp, R.; Coll-Bonfill, N.; Gonzalo, S. Causes and Consequences of Genomic Instability in Laminopathies: Replication Stress and Interferon Response. Nucleus 2018, 9, 258–275. [Google Scholar] [CrossRef]
- Gonzalo, S. DNA Damage and Lamins. In Cancer Biology and the Nuclear Envelope; Springer: Berlin/Heidelberg, Germany, 2014; pp. 377–399. [Google Scholar] [CrossRef]
- Olive, M.; Harten, I.; Mitchell, R.; Beers, J.K.; Djabali, K.; Cao, K.; Erdos, M.R.; Blair, C.; Funke, B.; Smoot, L.; et al. Cardiovascular Pathology in Hutchinson-Gilford Progeria: Correlation with the Vascular Pathology of Aging. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Ragnauth, C.D.; Warren, D.T.; Liu, Y.; McNair, R.; Tajsic, T.; Figg, N.; Shroff, R.; Skepper, J.; Shanahan, C.M. Prelamin A Acts to Accelerate Smooth Muscle Cell Senescence and Is a Novel Biomarker of Human Vascular Aging. Circulation 2010, 121, 2200–2210. [Google Scholar] [CrossRef]
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.M.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and Course of Hutchinson–Gilford Progeria Syndrome. N. Engl. J. Med. 2008, 358, 592–604. [Google Scholar] [CrossRef]
- Brosh, R.M. DNA Helicases Involved in DNA Repair and Their Roles in Cancer. Nat. Rev. Cancer 2013, 13, 542–558. [Google Scholar] [CrossRef]
- Valero, A.; Gellei, B. Retinitis Pigmentosa, Hypertension, and Uraemia in Werner’s Syndrome. BMJ 1960, 2, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Folgueras, A.R.; Freitas-Rodríguez, S.; Velasco, G.; López-Otín, C. Mouse Models to Disentangle the Hallmarks of Human Aging. Circ. Res. 2018, 123, 905–924. [Google Scholar] [CrossRef] [PubMed]
- Vanhooren, V.; Libert, C. The Mouse as a Model Organism in Aging Research: Usefulness, Pitfalls and Possibilities. Ageing Res. Rev 2013, 12, 8–21. [Google Scholar] [CrossRef]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H.J. Understanding Nucleotide Excision Repair and Its Roles in Cancer and Ageing. Nat. Rev. Mol. Cell. Biol 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Gillet, L.C.; Scharer, O.D. Molecular Mechanisms of Mammalian Global Genome Nucleotide Excision Repair. Chem. Rev. 2006, 106, 253–276. [Google Scholar] [CrossRef] [PubMed]
- Durik, M.; Kavousi, M.; van der Pluijm, I.; Isaacs, A.; Cheng, C.; Verdonk, K.; Loot, A.E.; Oeseburg, H.; Bhaggoe, U.M.; Leijten, F. Nucleotide Excision DNA Repair Is Associated with Age-Related Vascular Dysfunction. Circulation 2012, 126, 468–478. [Google Scholar] [CrossRef]
- Weeda, G.; Donker, I.; de Wit, J.; Morreau, H.; Janssens, R.; Vissers, C.J.; Nigg, A.; van Steeg, H.; Bootsma, D.; Hoeijmakers, J.H.J. Disruption of Mouse ERCC1 Results in a Novel Repair Syndrome with Growth Failure, Nuclear Abnormalities and Senescence. Curr. Biol. 1997, 7, 427–439. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Zhao, J.; Bukata, C.; Wade, E.A.; McGowan, S.J.; Angelini, L.A.; Bank, M.P.; Gurkar, A.U.; McGuckian, C.A.; Calubag, M.F.; et al. Tissue Specificity of Senescent Cell Accumulation during Physiologic and Accelerated Aging of Mice. Aging Cell 2020, 19, e13094. [Google Scholar] [CrossRef]
- Dollé, M.E.T.; Kuiper, R.V.; Roodbergen, M.; Robinson, J.; de Vlugt, S.; Wijnhoven, S.W.P.; Beems, R.B.; de la Fonteyne, L.; de With, P.; van der Pluijm, I.; et al. Broad Segmental Progeroid Changes in Short-Lived Ercc1 −/Δ7 Mice. Pathobiol. Aging Age-Relat. Dis. 2011, 1, 7219. [Google Scholar] [CrossRef]
- Bautista-Niño, P.K.; Portilla-Fernandez, E.; Rubio-Beltrán, E.; van der Linden, J.J.; de Vries, R.; van Veghel, R.; de Boer, M.; Durik, M.; Ridwan, Y.; Brandt, R.; et al. Local Endothelial DNA Repair Deficiency Causes Aging-Resembling Endothelial-Specific Dysfunction. Clin. Sci. 2020, 134, 727–746. [Google Scholar] [CrossRef] [PubMed]
- Ataei Ataabadi, E.; Golshiri, K.; Van Der Linden, J.; De Boer, M.; Duncker, D.J.; Jüttner, A.; De Vries, R.; Van Veghel, R.; Van Der Pluijm, I.; Dutheil, S.; et al. Vascular Ageing Features Caused by Selective DNA Damage in Smooth Muscle Cell. Oxid. Med. Cell. Longev. 2021, 2021, 2308317. [Google Scholar] [CrossRef] [PubMed]
- Kashiyama, K.; Nakazawa, Y.; Pilz, D.T.; Guo, C.; Shimada, M.; Sasaki, K.; Fawcett, H.; Wing, J.F.; Lewin, S.O.; Carr, L.; et al. Malfunction of Nuclease ERCC1-XPF Results in Diverse Clinical Manifestations and Causes Cockayne Syndrome, Xeroderma Pigmentosum, and Fanconi Anemia. Am. J. Hum. Genet. 2013, 92, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, N.G.J.; Raams, A.; Silengo, M.C.; Wijgers, N.; Niedernhofer, L.J.; Robinson, A.R.; Giglia-Mari, G.; Hoogstraten, D.; Kleijer, W.J.; Hoeijmakers, J.H.J.; et al. First Reported Patient with Human ERCC1 Deficiency Has Cerebro-Oculo-Facio-Skeletal Syndrome with a Mild Defect in Nucleotide Excision Repair and Severe Developmental Failure. Am. J. Hum. Genet. 2007, 80, 457–466. [Google Scholar] [CrossRef]
- Baer, S.; Obringer, C.; Julia, S.; Chelly, J.; Capri, Y.; Gras, D.; Baujat, G.; Felix, T.M.; Doray, B.; Sanchez del Pozo, J.; et al. Early-onset Nucleotide Excision Repair Disorders with Neurological Impairment: Clues for Early Diagnosis and Prognostic Counseling. Clin. Genet. 2020, 98, 251–260. [Google Scholar] [CrossRef]
- Osorio, F.G.; Navarro, C.L.; Cadiñanos, J.; López-Mejía, I.C.; Quirós, P.M.; Bartoli, C.; Rivera, J.; Tazi, J.; Guzmán, G.; Varela, I.; et al. Splicing-Directed Therapy in a New Mouse Model of Human Accelerated Aging. Sci. Transl. Med. 2011, 3, 106–107. [Google Scholar] [CrossRef]
- Cabral, W.A.; Tavarez, U.L.; Beeram, I.; Yeritsyan, D.; Boku, Y.D.; Eckhaus, M.A.; Nazarian, A.; Erdos, M.R.; Collins, F.S. Genetic Reduction of MTOR Extends Lifespan in a Mouse Model of Hutchinson-Gilford Progeria Syndrome. Aging. Cell 2021, 20, e13457. [Google Scholar] [CrossRef]
- del Campo, L.; Sánchez-López, A.; Salaices, M.; von Kleeck, R.A.; Expósito, E.; González-Gómez, C.; Cussó, L.; Guzmán-Martínez, G.; Ruiz-Cabello, J.; Desco, M.; et al. Vascular Smooth Muscle Cell-specific Progerin Expression in a Mouse Model of Hutchinson–Gilford Progeria Syndrome Promotes Arterial Stiffness: Therapeutic Effect of Dietary Nitrite. Aging. Cell 2019, 18, e12936. [Google Scholar] [CrossRef]
- Nevado, R.M.; Hamczyk, M.R.; Gonzalo, P.; Andrés-Manzano, M.J.; Andrés, V. Premature Vascular Aging with Features of Plaque Vulnerability in an Atheroprone Mouse Model of Hutchinson–Gilford Progeria Syndrome with Ldlr Deficiency. Cells 2020, 9, 2252. [Google Scholar] [CrossRef]
- Coll-Bonfill, N.; Mahajan, U.; Shashkova, E.V.; Lin, C.-J.; Mecham, R.P.; Gonzalo, S. Progerin Induces a Phenotypic Switch in Vascular Smooth Muscle Cells and Triggers Replication Stress and an Aging-Associated Secretory Signature. Geroscience 2023, 45, 965–982. [Google Scholar] [CrossRef]
- SH, Y.; MO, B.; JI, T.; Qiao, X.; Hu, Y.; Sandoval, S.; Meta, M.; Bendale, P.; MH, G.; SG, Y.; et al. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc. Natl. Acad. Sci. USA 2005, 102, 10291–10296. [Google Scholar] [CrossRef]
- Benedicto, I.; Dorado, B.; Andrés, V. Molecular and Cellular Mechanisms Driving Cardiovascular Disease in Hutchinson-Gilford Progeria Syndrome: Lessons Learned from Animal Models. Cells 2021, 10, 1157. [Google Scholar] [CrossRef]
- Hamczyk, M.R.; Villa-Bellosta, R.; Gonzalo, P.; Andrés-Manzano, M.J.; Nogales, P.; Bentzon, J.F.; López-Otín, C.; Andrés, V. Vascular Smooth Muscle–Specific Progerin Expression Accelerates Atherosclerosis and Death in a Mouse Model of Hutchinson-Gilford Progeria Syndrome. Circulation 2018, 138, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Baker, D.J.; d’Uscio, L.V.; Mozammel, G.; Katusic, Z.S.; van Deursen, J.M. Aging-Associated Vascular Phenotype in Mutant Mice with Low Levels of BubR1. Stroke 2007, 38, 1050–1056. [Google Scholar] [CrossRef]
- Baker, D.J.; Weaver, R.L.; van Deursen, J.M. P21 Both Attenuates and Drives Senescence and Aging in BubR1 Progeroid Mice. Cell. Rep 2013, 3, 1164–1174. [Google Scholar] [CrossRef]
- Okadome, J.; Matsumoto, T.; Yoshiya, K.; Matsuda, D.; Tamada, K.; Onimaru, M.; Nakano, K.; Egashira, K.; Yonemitsu, Y.; Maehara, Y. BubR1 Insufficiency Impairs Angiogenesis in Aging and in Experimental Critical Limb Ischemic Mice. J. Vasc. Surg. 2018, 68, 576–586.e1. [Google Scholar] [CrossRef]
- Wong, L.S.M.; Oeseburg, H.; de Boer, R.A.; van Gilst, W.H.; van Veldhuisen, D.J.; van der Harst, P. Telomere Biology in Cardiovascular Disease: The TERC-/- Mouse as a Model for Heart Failure and Ageing. Cardiovasc. Res. 2008, 81, 244–252. [Google Scholar] [CrossRef]
- Blasco, M.A.; Lee, H.-W.; Hande, M.P.; Samper, E.; Lansdorp, P.M.; DePinho, R.A.; Greider, C.W. Telomere Shortening and Tumor Formation by Mouse Cells Lacking Telomerase RNA. Cell 1997, 91, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Bhayadia, R.; Schmidt, B.M.W.; Melk, A.; Hömme, M. Senescence-Induced Oxidative Stress Causes Endothelial Dysfunction. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Rivero, G.; Ruiz-Torres, M.P.; Rivas-Elena, J.V.; Jerkic, M.; Díez-Marques, M.L.; Lopez-Novoa, J.M.; Blasco, M.A.; Rodríguez-Puyol, D. Mice Deficient in Telomerase Activity Develop Hypertension Because of an Excess of Endothelin Production. Circulation 2006, 114, 309–317. [Google Scholar] [CrossRef]
- Klein, M.A.; Denu, J.M. Biological and Catalytic Functions of Sirtuin 6 as Targets for Small-Molecule Modulators. J. Biol. Chem. 2020, 295, 11021–11041. [Google Scholar] [CrossRef] [PubMed]
- Labinskyy, N.; Csiszar, A.; Veress, G.; Stef, G.; Pacher, P.; Oroszi, G.; Wu, J.; Ungvari, Z. Vascular Dysfunction in Aging: Potential Effects of Resveratrol, an Anti- Inflammatory Phytoestrogen. Curr. Med. Chem. 2006, 13, 989–996. [Google Scholar] [CrossRef]
- Greiten, L.E.; Zhang, B.; Roos, C.M.; Hagler, M.; Jahns, F.-P.; Miller, J.D. Sirtuin 6 Protects Against Oxidative Stress and Vascular Dysfunction in Mice. Front. Physiol. 2021, 12, 753501. [Google Scholar] [CrossRef]
- Lee, O.-H.; Woo, Y.M.; Moon, S.; Lee, J.; Park, H.; Jang, H.; Park, Y.-Y.; Bae, S.-K.; Park, K.-H.; Heo, J.H.; et al. Sirtuin 6 Deficiency Induces Endothelial Cell Senescence via Downregulation of Forkhead Box M1 Expression. Aging 2020, 12, 20946–20967. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Feng, W.; Su, X.; Luo, D.; Li, Z.; Zhou, Y.; Zhu, Y.; Zhang, M.; Chen, J.; Liu, B.; et al. SIRT6 Protects Vascular Smooth Muscle Cells from Osteogenic Transdifferentiation via Runx2 in Chronic Kidney Disease. J. Clin. Investig. 2022, 132, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Yin, M.; Koroleva, M.; Mastrangelo, M.A.; Zhang, W.; Bai, P.; Little, P.J.; Jin, Z.G. SIRT6 Protects against Endothelial Dysfunction and Atherosclerosis in Mice. Aging 2016, 8, 1064–1082. [Google Scholar] [CrossRef] [PubMed]
- Peshti, V.; Obolensky, A.; Nahum, L.; Kanfi, Y.; Rathaus, M.; Avraham, M.; Tinman, S.; Alt, F.W.; Banin, E.; Cohen, H.Y. Characterization of Physiological Defects in Adult SIRT6-/- Mice. PLoS ONE 2017, 12, e0176371. [Google Scholar] [CrossRef]
- Moskalev, A.A.; Shaposhnikov, M.V.; Plyusnina, E.N.; Zhavoronkov, A.; Budovsky, A.; Yanai, H.; Fraifeld, V.E. The Role of DNA Damage and Repair in Aging through the Prism of Koch-like Criteria. Ageing Res. Rev. 2013, 12, 661–684. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Niño, P.K.; Portilla-Fernandez, E.; Vaughan, D.E.; Danser, A.H.; Roks, A.J. DNA Damage: A Main Determinant of Vascular Aging. Int. J. Mol. Sci. 2016, 17, 748. [Google Scholar] [CrossRef]
- Lin, Z.; Ding, Q.; Li, X.; Feng, Y.; He, H.; Huang, C.; Zhu, Y. Targeting Epigenetic Mechanisms in Vascular Aging. Front. Cardiovasc. Med. 2022, 8, 806988. [Google Scholar] [CrossRef]
- Farina, F.M.; Hall, I.F.; Serio, S.; Zani, S.; Climent, M.; Salvarani, N.; Carullo, P.; Civilini, E.; Condorelli, G.; Elia, L. MiR-128-3p Is a Novel Regulator of Vascular Smooth Muscle Cell Phenotypic Switch and Vascular Diseases. Circ. Res. 2020, 126, e120–e135. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.E.; Chen, T.M.; Wallingford, M.C.; Nguyen, N.B.; Yamada, S.; Sawangmake, C.; Zhang, J.; Speer, M.Y.; Giachelli, C.M. Runx2 deletion in smooth muscle cells inhibits vascular osteochondrogenesis and calcification but not atherosclerotic lesion formation. Cardiovasc. Res. 2016, 112, 606–616. [Google Scholar] [CrossRef]
- Cobb, A.M.; Yusoff, S.; Hayward, R.; Ahmad, S.; Sun, M.; Verhulst, A.; D’Haese, P.C.; Shanahan, C.M. Runx2 (Runt-Related Transcription Factor 2) Links the DNA Damage Response to Osteogenic Reprogramming and Apoptosis of Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1339–1357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Guo, X.; Xia, Y.; Mao, L. An Update on the Phenotypic Switching of Vascular Smooth Muscle Cells in the Pathogenesis of Atherosclerosis. Cell. Mol. Life Sci. 2022, 79, 6. [Google Scholar] [CrossRef]
- Heerkens, E.H.J.; Izzard, A.S.; Heagerty, A.M. Integrins, Vascular Remodeling, and Hypertension. Hypertension 2007, 49, 1–4. [Google Scholar] [CrossRef]
- Hartmann, C.; Herling, L.; Hartmann, A.; Köckritz, V.; Fuellen, G.; Walter, M.; Hermann, A. Systematic Estimation of Biological Age of in Vitro Cell Culture Systems by an Age-Associated Marker Panel. Front. Aging 2023, 4, 1129107. [Google Scholar] [CrossRef] [PubMed]
- Jimenez Trinidad, F.R.; Arrieta Ruiz, M.; Solanes Batlló, N.; Vea Badenes, À.; Bobi Gibert, J.; Valera Cañellas, A.; Roqué Moreno, M.; Freixa Rofastes, X.; Sabaté Tenas, M.; Dantas, A.P.; et al. Linking In Vitro Models of Endothelial Dysfunction with Cell Senescence. Life 2021, 11, 1323. [Google Scholar] [CrossRef]
- DeCicco-Skinner, K.L.; Henry, G.H.; Cataisson, C.; Tabib, T.; Gwilliam, J.C.; Watson, N.J.; Bullwinkle, E.M.; Falkenburg, L.; O’Neill, R.C.; Morin, A.; et al. Endothelial Cell Tube Formation Assay for the In Vitro Study of Angiogenesis. J. Vis. Exp. 2014, 91, e51312. [Google Scholar] [CrossRef]
- Borenstein, J.T.; Tupper, M.M.; MacK, P.J.; Weinberg, E.J.; Khalil, A.S.; Hsiao, J.; García-Cardeña, G. Functional Endothelialized Microvascular Networks with Circular Cross-Sections in a Tissue Culture Substrate. Biomed. Microdevices 2010, 12, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, I.; Spicar-Mihalic, P.; Kuyper, C.L.; Fiorini, G.S.; Chiu, D.T. Rapid Prototyping of Glass Microchannels. Anal. Chim. Acta 2003, 496, 205–215. [Google Scholar] [CrossRef]
- Pollet, A.M.A.O.; Den Toonder, J.M.J. Recapitulating the Vasculature Using Organ-on-Chip Technology. Bioengineering 2020, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Mandrycky, C.J.; Howard, C.C.; Rayner, S.G.; Shin, Y.J.; Zheng, Y. Organ-on-a-Chip Systems for Vascular Biology. J. Mol. Cell. Cardiol. 2021, 159, 1–13. [Google Scholar] [CrossRef]
- van Dijk, C.G.M.; Brandt, M.M.; Poulis, N.; Anten, J.; van der Moolen, M.; Kramer, L.; Homburg, E.; Louzao-Martinez, L.; Pei, J.; Krebber, M.M.; et al. A New Microfluidic Model That Allows Monitoring of Complex Vascular Structures and Cell Interactions in a 3D Biological Matrix. Lab. Chip. 2020, 20, 1827–1844. [Google Scholar] [CrossRef]
- Polacheck, W.J.; Kutys, M.L.; Tefft, J.B.; Chen, C.S. Microfabricated Blood Vessels for Modeling the Vascular Transport Barrier. Nat. Protoc 2019, 14, 1425–1454. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Phan, D.T.T.; Sobrino, A.; George, S.C.; Hughes, C.C.W.; Lee, A.P. Engineering Anastomosis between Living Capillary Networks and Endothelial Cell-Lined Microfluidic Channels. Lab. Chip. 2016, 16, 282–290. [Google Scholar] [CrossRef]
- Vila Cuenca, M.; Cochrane, A.; van den Hil, F.E.; de Vries, A.A.F.; Lesnik Oberstein, S.A.J.; Mummery, C.L.; Orlova, V.V. Engineered 3D Vessel-on-Chip Using HiPSC-Derived Endothelial- and Vascular Smooth Muscle Cells. Stem. Cell Rep. 2021, 16, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.; Kita, A.; Leach, J.; Rounsevell, R.; Huang, J.N.; Moake, J.; Ware, R.E.; Fletcher, D.A.; Lam, W.A. In Vitro Modeling of the Microvascular Occlusion and Thrombosis That Occur in Hematologic Diseases Using Microfluidic Technology. J. Clin. Investig. 2012, 122, 408–418. [Google Scholar] [CrossRef]
- Garvin, K.A.; Dalecki, D.; Yousefhussien, M.; Helguera, M.; Hocking, D.C. Spatial Patterning of Endothelial Cells and Vascular Network Formation Using Ultrasound Standing Wave Fields. J. Acoust. Soc. Am 2013, 134, 1483–1490. [Google Scholar] [CrossRef]
- Wu, C.-C.; Li, Y.-S.; Haga, J.H.; Kaunas, R.; Chiu, J.-J.; Su, F.-C.; Usami, S.; Chien, S. Directional Shear Flow and Rho Activation Prevent the Endothelial Cell Apoptosis Induced by Micropatterned Anisotropic Geometry. Proc. Natl. Acad. Sci. USA 2007, 104, 1254–1259. [Google Scholar] [CrossRef]
- Thomas, A.; Daniel Ou-Yang, H.; Lowe-Krentz, L.; Muzykantov, V.R.; Liu, Y. Biomimetic Channel Modeling Local Vascular Dynamics of Pro-Inflammatory Endothelial Changes. Biomicrofluidics 2016, 10, 014101. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lobatto, M.E.; Kawahara, T.; Lee Chung, B.; Mieszawska, A.J.; Sanchez-Gaytan, B.L.; Fay, F.; Senders, M.L.; Calcagno, C.; Becraft, J.; et al. Probing Nanoparticle Translocation across the Permeable Endothelium in Experimental Atherosclerosis. Proc. Natl. Acad. Sci. USA 2014, 111, 1078–1083. [Google Scholar] [CrossRef]
- Chen, M.B.; Whisler, J.A.; Jeon, J.S.; Kamm, R.D. Mechanisms of Tumor Cell Extravasation in an in Vitro Microvascular Network Platform. Integr. Biol. 2013, 5, 1262–1271. [Google Scholar] [CrossRef]
- Bray, L.J.; Binner, M.; Holzheu, A.; Friedrichs, J.; Freudenberg, U.; Hutmacher, D.W.; Werner, C. Multi-Parametric Hydrogels Support 3D in Vitro Bioengineered Microenvironment Models of Tumour Angiogenesis. Biomaterials 2015, 53, 609–620. [Google Scholar] [CrossRef]
- Stroock, A.D.; Fischbach, C. Microfluidic Culture Models of Tumor Angiogenesis. Tissue Eng. Part A 2010, 16, 2143–2146. [Google Scholar] [CrossRef]
- Bersini, S.; Jeon, J.S.; Dubini, G.; Arrigoni, C.; Chung, S.; Charest, J.L.; Moretti, M.; Kamm, R.D. A Microfluidic 3D in Vitro Model for Specificity of Breast Cancer Metastasis to Bone. Biomaterials 2014, 35, 2454–2461. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.S.; Bersini, S.; Gilardi, M.; Dubini, G.; Charest, J.L.; Moretti, M.; Kamm, R.D. Human 3D Vascularized Organotypic Microfluidic Assays to Study Breast Cancer Cell Extravasation. Proc. Natl. Acad. Sci. USA 2015, 112, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Leslie, D.C.; Matthews, B.D.; Fraser, J.P.; Jurek, S.; Hamilton, G.A.; Thorneloe, K.S.; McAlexander, M.A.; Ingber, D.E. A Human Disease Model of Drug Toxicity-Induced Pulmonary Edema in a Lung-on-a-Chip Microdevice. Sci. Transl. Med. 2012, 4, 159ra147. [Google Scholar] [CrossRef] [PubMed]
- Namdee, K.; Thompson, A.J.; Charoenphol, P.; Eniola-Adefeso, O. Margination Propensity of Vascular-Targeted Spheres from Blood Flow in a Microfluidic Model of Human Microvessels. Langmuir 2013, 29, 2530–2535. [Google Scholar] [CrossRef]
- Kastrup, C.J.; Nahrendorf, M.; Figueiredo, J.L.; Lee, H.; Kambhampati, S.; Lee, T.; Cho, S.-W.; Gorbatov, R.; Iwamoto, Y.; Dang, T.T.; et al. Painting Blood Vessels and Atherosclerotic Plaques with an Adhesive Drug Depot. Proc. Natl. Acad. Sci. USA 2012, 109, 21444–21449. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lang, B.; Qu, Y.; Li, L.; Song, Z.; Wang, Z. Single-Cell Patterning Technology for Biological Applications. Biomicrofluidics 2019, 13, 61502. [Google Scholar] [CrossRef] [PubMed]
- Leopold, J.A.; Loscalzo, J. Emerging Role of Precision Medicine in Cardiovascular Disease. Circ. Res. 2018, 122, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Strianese, O.; Rizzo, F.; Ciccarelli, M.; Galasso, G.; D’Agostino, Y.; Salvati, A.; Del Giudice, C.; Tesorio, P.; Rusciano, M.R. Precision and Personalized Medicine: How Genomic Approach Improves the Management of Cardiovascular and Neurodegenerative Disease. Genes 2020, 11, 747. [Google Scholar] [CrossRef] [PubMed]
- Gray, K.M.; Stroka, K.M. Vascular Endothelial Cell Mechanosensing: New Insights Gained from Biomimetic Microfluidic Models. Semin. Cell. Dev. Biol. 2017, 71, 106–117. [Google Scholar] [CrossRef]
- Hesh, C.A.; Qiu, Y.; Lam, W.A. Vascularized Microfluidics and the Blood-Endothelium Interface. Micromachines 2019, 11, 18. [Google Scholar] [CrossRef] [PubMed]
- Luna, D.J.; Pandian, N.K.R.; Mathur, T.; Bui, J.; Gadangi, P.; Kostousov, V.V.; Hui, S.-K.R.; Teruya, J.; Jain, A. Tortuosity-Powered Microfluidic Device for Assessment of Thrombosis and Antithrombotic Therapy in Whole Blood. Sci. Rep. 2020, 10, 5742. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Linden, J.; Trap, L.; Scherer, C.V.; Roks, A.J.M.; Danser, A.H.J.; van der Pluijm, I.; Cheng, C. Model Systems to Study the Mechanism of Vascular Aging. Int. J. Mol. Sci. 2023, 24, 15379. https://doi.org/10.3390/ijms242015379
van der Linden J, Trap L, Scherer CV, Roks AJM, Danser AHJ, van der Pluijm I, Cheng C. Model Systems to Study the Mechanism of Vascular Aging. International Journal of Molecular Sciences. 2023; 24(20):15379. https://doi.org/10.3390/ijms242015379
Chicago/Turabian Stylevan der Linden, Janette, Lianne Trap, Caroline V. Scherer, Anton J. M. Roks, A. H. Jan Danser, Ingrid van der Pluijm, and Caroline Cheng. 2023. "Model Systems to Study the Mechanism of Vascular Aging" International Journal of Molecular Sciences 24, no. 20: 15379. https://doi.org/10.3390/ijms242015379