

Insulin Receptor Isoforms and Insulin Growth Factor-like Receptors: Implications in Cell Signaling, Carcinogenesis, and Chemoresistance

Abstract
:1. Introduction
2. IRand IGFR Genes and Structures
2.1. IR
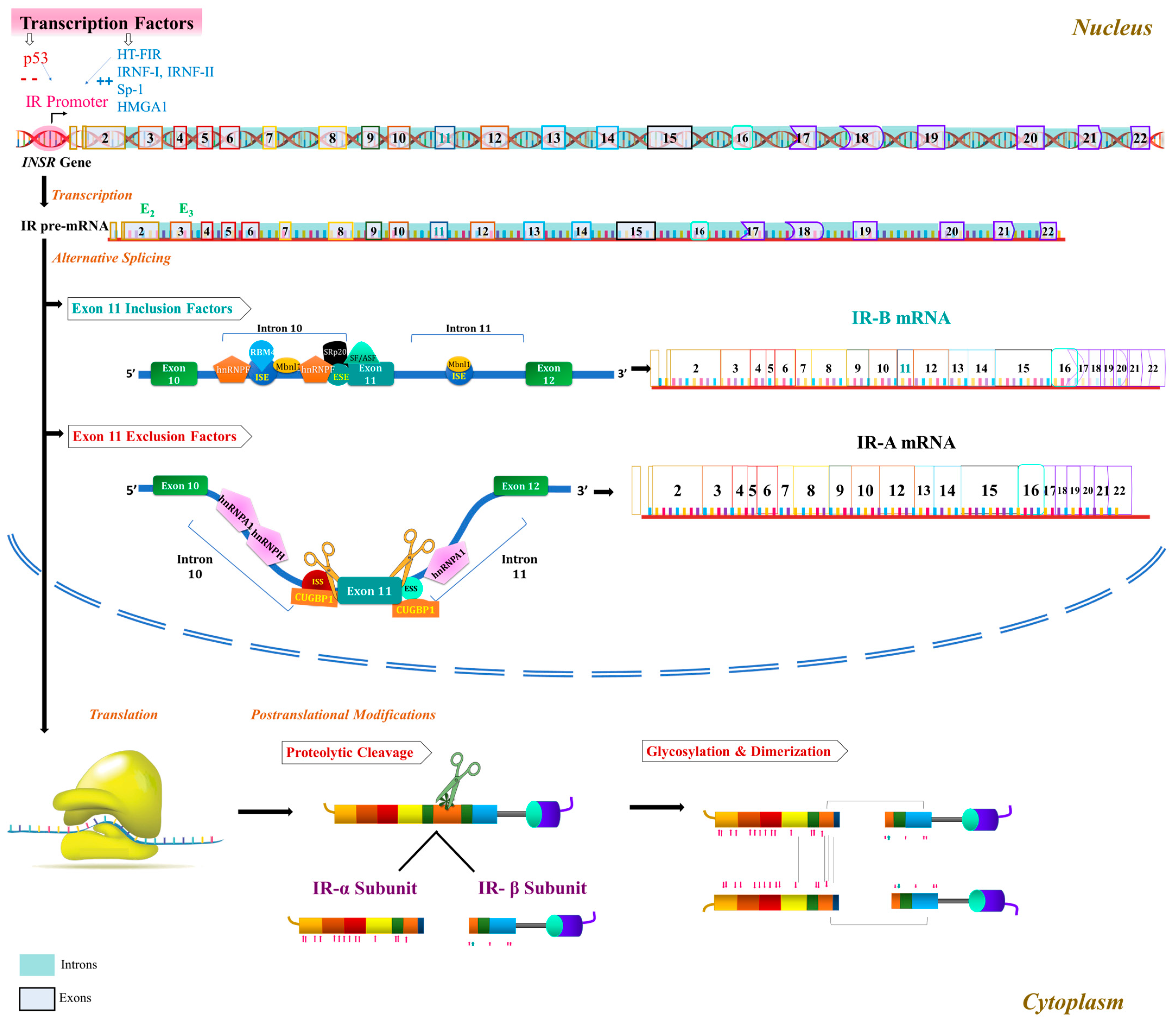
2.1.1. INSR Gene and IR Isoform Formation
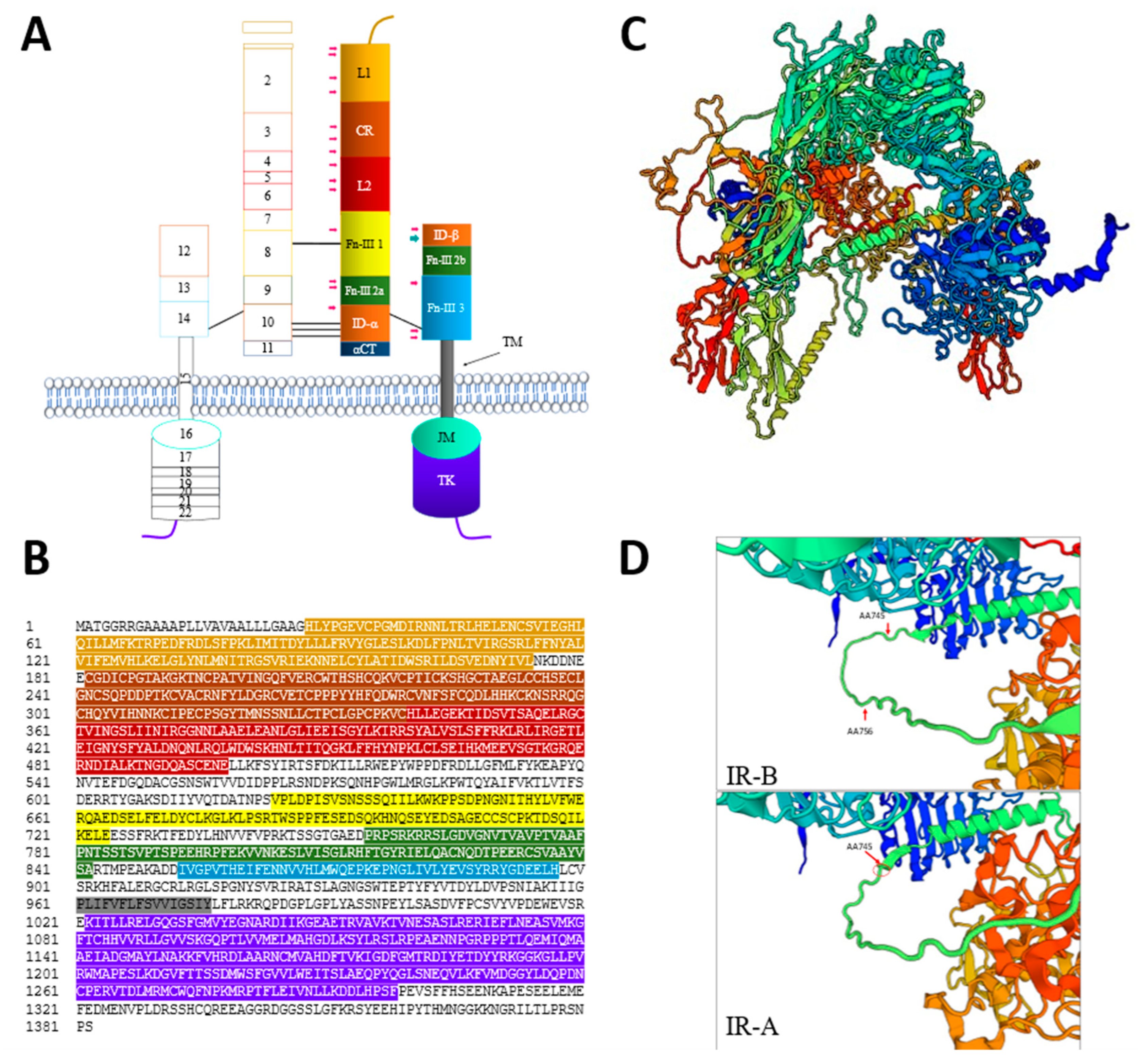
2.1.2. IR Structure
2.1.3. Characteristics of IR Isoforms
2.2. IGFR
2.2.1. Type 1 Insulin-Like Growth Factor Receptor (IGF-1R)
2.2.2. Type 2 Insulin-Like Growth Factor Receptor (IGF-2R)
2.3. Hybrid Receptor (HR)
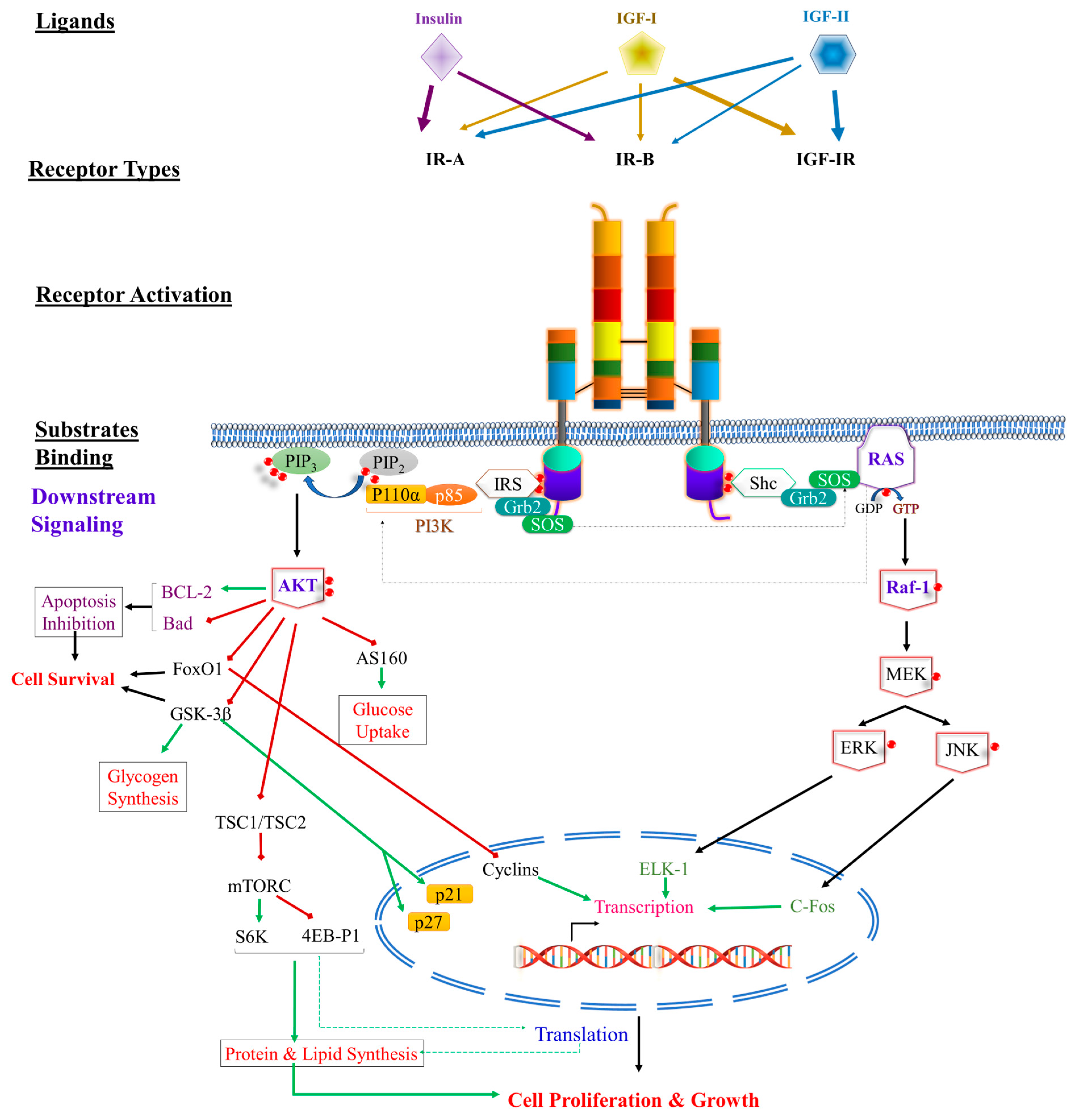
3. Receptor Activation and Signaling Pathway
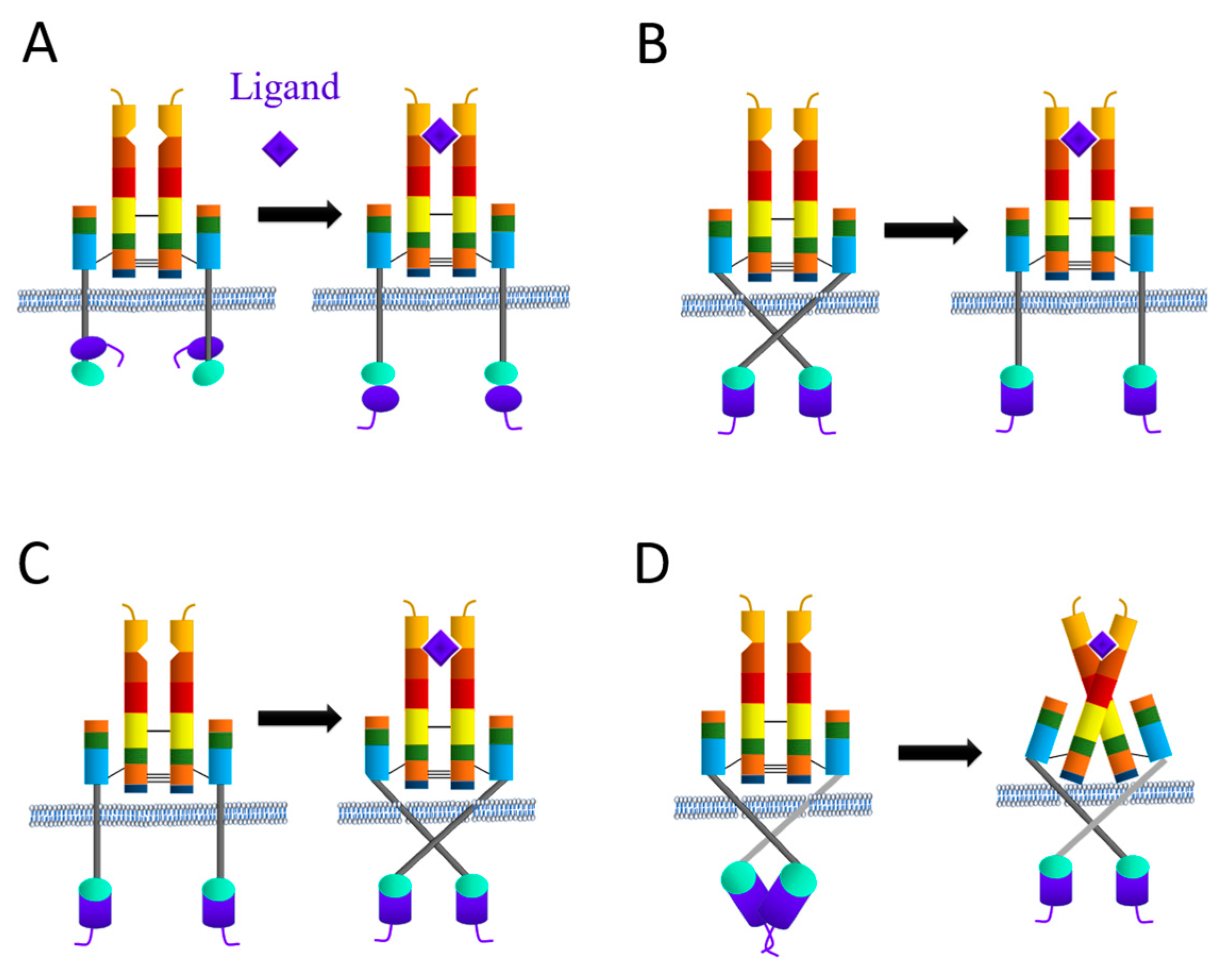
3.1. Activation of Receptor Tyrosine Kinase (RTK) Superfamily
3.1.1. Proposed IR Activation Mechanisms
3.1.2. Intracellular IR Signal Transduction Network
3.1.3. PI3K Signaling Pathway
3.1.4. The MAPK-ERK Signaling Pathway
3.1.5. Intracellular Domain Pathway (LYKi)
3.1.6. Activation and Signaling Pathway Differences between IR Isoforms
3.1.7. Intracellular IGFR Signal Transduction Network
4. Role of IR and IGFR in Carcinogenesis
4.1. Role of IR Isoforms in Carcinogenesis
4.2. Role of IGFRs in Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Tissue Types/Cell Lines | Quantification Method | Causes of the Overexpression | Prognostic Implications | References |
|---|---|---|---|---|---|
| Breast Cancer | Breast tissue subtypes: Luminal type a and type b | Gene expression profiling (microarray)/Molecular profiling | Overexpression was hormonally driven | IGF-1R overexpression was predominantly seen in ER-positive (+) tumors contributing to chemoresistance, and reducing IGF-1R levels significantly reduced ER+ tumor size | [258,259,260] |
| Triple-negative breast cancer | N/A | IGF-1R expression was found to be associated with a lower disease-free survival rate (p = 0.031). | [261] | ||
| Colorectal Cancer | Electrophoresis | IGF-1R has a relation to SNP implication in CRC | There was a significant association between IGF-1R rs2229765 polymorphism and advanced CRC (AA/AG vs. GG: OR = 3.06, p = 0.004) | [262] | |
| Esophageal Cancer | Esophageal cells CE48T/VGH cell line | Northern blot analysis and ligand-binding assay | N/A | Overexpression of IGF-1R and autocrine growth regulation may concertedly control the proliferation of esophageal carcinoma | [263] |
| Gastric Carcinoma (GC) | N/A | Immunohistochemistry | N/A | IGF-1R overexpression positively correlated with MRP-1 overexpression (rp = 0.39, p < 0.01). IGF-1R and MPR-1 overexpression was correlated with poor prognosis of GC (p < 0.01) | [264] |
| Head and Neck Squamous Cell Carcinomas (HNSCCs) | Squamous cell | IHC | N/A | OS and DSS were reduced in patients whose tumors contained high membrane IGF-1R | [234] |
| Lung Cancer | Small-cell lung cancer (SCLC) | NA (serum and lung cancer histological tissues from small-cell lung carcinoma (SCLC)) | N/A | IGF-1R overexpression leads to increased cell survival and suppressed cell apoptosis, whereas IGF1R silencing mediated by RNAi abrogates this response of NCI-H446 | [265] |
| Osteosarcoma | Human primary and metastatic osteosarcomas | Reverse transcriptase polymerase chain reaction | N/A | _ | [266] |
| Prostate Cancer | Malignant epithelia | IHC | N/A | High IGF-1R was associated with high risk of metastasis | [233] |
| Cancer Type | Tissue Types/Cell Lines | Quantification Method | Expression | Causes of Altered Expression | Prognostic Implications | References |
|---|---|---|---|---|---|---|
| Bladder Cancer | Bladder carcinoma | qPCR and IHC | ↓ in 70% vs. ↑ in 30% | N/A | Low expression is associated with poor prognosis.
| [267] |
| Bladder carcinoma | PCR, FISH | ↑ | Autophagy-associated circular RNA hsa_circ_0007813 upregulation sponge hsa-miR-361-3p to regulate IGF-2R expression | Unfavorable prognosis | [268] | |
| Breast Cancer | Triple-negative breast cancer | IHC | ↑ | N/A | Patients with IGF-2R-positive expression had lower OS (p < 0.001) and DFS rates than those with IGF-2R-negative expression (67.8% vs. 78.5%, p = 0.023) | [269] |
| Cervical Cancer | Cervical cancer tissue and cell lines | DNA microarray, IHC | ↑ | N/A | Poor prognosis Lower OS compared to cases with intermediate- and high-IGF-2R expression, p < 0.05 and p < 0.01, respectively, in Stage I | [251] |
| Colon Cancer | Adenocarcinoma malignant tissue | RNase protection assay | ↑ | N/A | _ | [270] |
| Liver Cancer | Primary HCC | PCR | ↓ | LOH of M6P/IGF-2R | Poor Prognosis
| [271] |
| Lung Cancer | NSCLC tissue and cell lines | IHC | ↓ in 56% vs. ↑ in 44% | N/A | Low IGF-2R expressions had a poorer prognosis than those with high IGF-2R expressions
| [249] |
| Oral Cancer | Squamous cell carcinoma tissues from patients | PCR | ↓ | Gly1619Arg polymorphism of M6P/IGF-2R domain 11 (rs62989) | Loss of IGF-2R function increased risk of advanced stage of OSCC by 3-fold | [253] |
| Osteosarcoma | Various cell lines | Flow cytometry analysis | ↑ | N/A | N/A | [272] |
| Pancreatic Cancer | Islet cells, acinar cells, and ductal cells | ISH | ↑ | N/A | _ | [273] |
5. Role of IR and Igfrs in Chemoresistance
5.1. Role of IR Isoforms in Chemoresistance
5.2. Role of IGFRs in Chemoresistance
6. Challenges of Quantification Methodologies and Future Insights
6.1. Quantification of IR Isoforms
6.2. Quantification of IGFR
6.3. Future Insights
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Broers, J.L.; Raymond, Y.; Rot, M.K.; Kuijpers, H.; Wagenaar, S.S.; Ramaekers, F.C. Nuclear A-type lamins are differentially expressed in human lung cancer subtypes. Am. J. Pathol. 1993, 143, 211–220. [Google Scholar] [PubMed]
- Rueff, J.; Rodrigues, A.S. Cancer Drug Resistance: A Brief Overview from a Genetic Viewpoint. Methods Mol. Biol. 2016, 1395, 1–18. [Google Scholar]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Lind, M. Principles of cytotoxic chemotherapy. Medicine 2008, 36, 19–23. [Google Scholar] [CrossRef]
- Ullrich, A.; Bell, J.R.; Chen, E.Y.; Herrera, R.; Petruzzelli, L.M.; Dull, T.J.; Gray, A.; Coussens, L.; Liao, Y.C.; Tsubokawa, M.; et al. Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 1985, 313, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Ebina, Y.; Ellis, L.; Jarnagin, K.; Edery, M.; Graf, L.; Clauser, E.; Ou, J.H.; Masiarz, F.; Kan, Y.W.; Goldfine, I.D.; et al. The human insulin receptor cDNA: The structural basis for hormone-activated transmembrane signalling. Cell 1985, 40, 747–758. [Google Scholar] [CrossRef]
- Seino, S.; Seino, M.; Nishi, S.; Bell, G.I. Structure of the human insulin receptor gene and characterization of its promoter. Proc. Natl. Acad. Sci. USA 1989, 86, 114–118. [Google Scholar] [CrossRef]
- Moller, D.E.; Yokota, A.; Caro, J.F.; Flier, J.S. Tissue-specific expression of two alternatively spliced insulin receptor mRNAs in man. Mol. Endocrinol. 1989, 3, 1263–1269. [Google Scholar] [CrossRef]
- Mosthaf, L.; Grako, K.; Dull, T.; Coussens, L.; Ullrich, A.; McClain, D. Functionally distinct insulin receptors generated by tissue-specific alternative splicing. EMBO J. 1990, 9, 2409–2413. [Google Scholar] [CrossRef]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin Receptor Isoforms and Insulin Receptor/Insulin-Like Growth Factor Receptor Hybrids in Physiology and Disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef]
- Yoshizato, K.; Shirotani, T.; Furukawa, N.; Taguchi, T.; Motoshima, H.; Toyonaga, T.; Hirashima, Y.; Kawashima, J.; Ebina, Y.; Shichiri, M.; et al. Identification of a cis-acting element and a novel trans-acting factor of the human insulin receptor gene in HepG2 and rat liver cells. Biochem. Biophys. Res. Commun. 2001, 280, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Tam, J.W.; Tsai, M.J.; Tsai, S.Y. Identification of cis- and trans-acting factors regulating the expression of the human insulin receptor gene. J. Biol. Chem. 1992, 267, 4638–4645. [Google Scholar] [CrossRef] [PubMed]
- Barthel, A.; Schmoll, D.; Unterman, T.G. FoxO proteins in insulin action and metabolism. Trends Endocrinol. Metab. TEM 2005, 16, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Foti, D.; Iuliano, R.; Chiefari, E.; Brunetti, A. A nucleoprotein complex containing Sp1, C/EBP beta, and HMGI-Y controls human insulin receptor gene transcription. Mol. Cell. Biol. 2003, 23, 2720–2732. [Google Scholar] [CrossRef] [PubMed]
- Webster, N.J.; Resnik, J.L.; Reichart, D.B.; Strauss, B.; Haas, M.; Seely, B.L. Repression of the insulin receptor promoter by the tumor suppressor gene product p53: A possible mechanism for receptor overexpression in breast cancer. Cancer Res. 1996, 56, 2781–2788. [Google Scholar]
- Pillai, R.S.; Bhattacharyya, S.N.; Filipowicz, W. Repression of protein synthesis by miRNAs: How many mechanisms? Trends Cell Biol. 2007, 17, 118–126. [Google Scholar] [CrossRef]
- Yang, W.M.; Jeong, H.J.; Park, S.W.; Lee, W. Obesity-induced miR-15b is linked causally to the development of insulin resistance through the repression of the insulin receptor in hepatocytes. Mol. Nutr. Food Res. 2015, 59, 2303–2314. [Google Scholar] [CrossRef]
- Yang, W.M.; Jeong, H.J.; Park, S.Y.; Lee, W. Saturated fatty acid-induced miR-195 impairs insulin signaling and glycogen metabolism in HepG2 cells. FEBS Lett. 2014, 588, 3939–3946. [Google Scholar] [CrossRef]
- Wang, X.; Wang, M.; Li, H.; Lan, X.; Liu, L.; Li, J.; Li, Y.; Li, J.; Yi, J.; Du, X.; et al. Upregulation of miR-497 induces hepatic insulin resistance in E3 rats with HFD-MetS by targeting insulin receptor. Mol. Cell. Endocrinol. 2015, 416, 57–69. [Google Scholar] [CrossRef]
- Trajkovski, M.; Hausser, J.; Soutschek, J.; Bhat, B.; Akin, A.; Zavolan, M.; Heim, M.H.; Stoffel, M. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 2011, 474, 649–653. [Google Scholar] [CrossRef]
- Mayr, C.; Hemann, M.T.; Bartel, D.P. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 2007, 315, 1576–1579. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Talukdar, I.; Liu, Y.; Tam, J.; Reddy, S.; Webster, N.J. Muscleblind-like 1 (Mbnl1) promotes insulin receptor exon 11 inclusion via binding to a downstream evolutionarily conserved intronic enhancer. J. Biol. Chem. 2010, 285, 25426–25437. [Google Scholar] [CrossRef] [PubMed]
- Geuens, T.; Bouhy, D.; Timmerman, V. The hnRNP family: Insights into their role in health and disease. Hum. Genet. 2016, 135, 851–867. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, I.; Sen, S.; Urbano, R.; Thompson, J.; Yates, J.R., 3rd; Webster, N.J. hnRNP A1 and hnRNP F modulate the alternative splicing of exon 11 of the insulin receptor gene. PLoS ONE 2011, 6, e27869. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Dansithong, W.; Kim, D.; Rossi, J.; Webster, N.J.; Comai, L.; Reddy, S. Interaction of muscleblind, CUG-BP1 and hnRNP H proteins in DM1-associated aberrant IR splicing. EMBO J. 2006, 25, 4271–4283. [Google Scholar] [CrossRef] [PubMed]
- Boukis, L.A.; Liu, N.; Furuyama, S.; Bruzik, J.P. Ser/Arg-rich protein-mediated communication between U1 and U2 small nuclear ribonucleoprotein particles. J. Biol. Chem. 2004, 279, 29647–29653. [Google Scholar] [CrossRef]
- Lu, C.C.; Chen, T.H.; Wu, J.R.; Chen, H.H.; Yu, H.Y.; Tarn, W.Y. Phylogenetic and molecular characterization of the splicing factor RBM4. PLoS ONE 2013, 8, e59092. [Google Scholar] [CrossRef]
- Malakar, P.; Chartarifsky, L.; Hija, A.; Leibowitz, G.; Glaser, B.; Dor, Y.; Karni, R. Insulin receptor alternative splicing is regulated by insulin signaling and modulates beta cell survival. Sci. Rep. 2016, 6, 31222. [Google Scholar] [CrossRef]
- Tatulian, S.A. Structural Dynamics of Insulin Receptor and Transmembrane Signaling. Biochemistry 2015, 54, 5523–5532. [Google Scholar] [CrossRef]
- Cuatrecasas, P. Insulin--receptor interactions in adipose tissue cells: Direct measurement and properties. Proc. Natl. Acad. Sci. USA 1971, 68, 1264–1268. [Google Scholar] [CrossRef]
- Jacobs, S.; Hazum, E.; Shechter, Y.; Cuatrecasas, P. Insulin receptor: Covalent labeling and identification of subunits. Proc. Natl. Acad. Sci. USA 1979, 76, 4918–4921. [Google Scholar] [CrossRef] [PubMed]
- Yip, C.C.; Yeung, C.W.; Moule, M.L. Photoaffinity labeling of insulin receptor proteins of liver plasma membrane preparations. Biochemistry 1980, 19, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Pilch, P.F.; Czech, M.P. The subunit structure of the high affinity insulin receptor. Evidence for a disulfide-linked receptor complex in fat cell and liver plasma membranes. J. Biol. Chem. 1980, 255, 1722–1731. [Google Scholar] [CrossRef]
- McKern, N.M.; Lawrence, M.C.; Streltsov, V.A.; Lou, M.Z.; Adams, T.E.; Lovrecz, G.O.; Elleman, T.C.; Richards, K.M.; Bentley, J.D.; Pilling, P.A.; et al. Structure of the insulin receptor ectodomain reveals a folded-over conformation. Nature 2006, 443, 218–221. [Google Scholar] [CrossRef]
- Lou, M.; Garrett, T.P.; McKern, N.M.; Hoyne, P.A.; Epa, V.C.; Bentley, J.D.; Lovrecz, G.O.; Cosgrove, L.J.; Frenkel, M.J.; Ward, C.W. The first three domains of the insulin receptor differ structurally from the insulin-like growth factor 1 receptor in the regions governing ligand specificity. Proc. Natl. Acad. Sci. USA 2006, 103, 12429–12434. [Google Scholar] [CrossRef] [PubMed]
- Frasca, F.; Pandini, G.; Sciacca, L.; Pezzino, V.; Squatrito, S.; Belfiore, A.; Vigneri, R. The role of insulin receptors and IGF-I receptors in cancer and other diseases. Arch. Physiol. Biochem. 2008, 114, 23–37. [Google Scholar] [CrossRef]
- Andersen, A.S.; Wiberg, F.C.; Kjeldsen, T. Localization of Specific Amino Acids Contributing to Insulin Specificity of the Insulin Receptor. Ann. N. Y. Acad. Sci. 1995, 766, 466–468. [Google Scholar] [CrossRef]
- Whittaker, J.; Whittaker, L. Characterization of the functional insulin binding epitopes of the full-length insulin receptor. J. Biol. Chem. 2005, 280, 20932–20936. [Google Scholar] [CrossRef]
- De Meyts, P. Receptor Tyrosine Kinase Signal Transduction and the Molecular Basis of Signalling Specificity. In Receptor Tyrosine Kinases: Structure, Functions and Role in Human Disease; Wheeler, D.L., Yarden, Y., Eds.; Springer: New York, NY, USA, 2015; pp. 51–76. [Google Scholar]
- Bajaj, M.; Waterfield, M.D.; Schlessinger, J.; Taylor, W.R.; Blundell, T. On the tertiary structure of the extracellular domains of the epidermal growth factor and insulin receptors. Biochim. Biophys. Acta 1987, 916, 220–226. [Google Scholar] [CrossRef]
- Sparrow, L.G.; Lawrence, M.C.; Gorman, J.J.; Strike, P.M.; Robinson, C.P.; McKern, N.M.; Ward, C.W. N-linked glycans of the human insulin receptor and their distribution over the crystal structure. Proteins 2008, 71, 426–439. [Google Scholar] [CrossRef]
- Rentería, M.E.; Gandhi, N.S.; Vinuesa, P.; Helmerhorst, E.; Mancera, R.L. A comparative structural bioinformatics analysis of the insulin receptor family ectodomain based on phylogenetic information. PLoS ONE 2008, 3, e3667. [Google Scholar] [CrossRef] [PubMed]
- Faria, T.N.; Blakesley, V.A.; Kato, H.; Stannard, B.; LeRoith, D.; Roberts, C.T., Jr. Role of the carboxyl-terminal domains of the insulin and insulin-like growth factor I receptors in receptor function. J. Biol. Chem. 1994, 269, 13922–13928. [Google Scholar] [CrossRef] [PubMed]
- Vigneri, R.; Goldfine, I.D.; Frittitta, L. Insulin, insulin receptors, and cancer. J. Endocrinol. Investig. 2016, 39, 1365–1376. [Google Scholar]
- Kosaki, A.; Webster, N. Effect of dexamethasone on the alternative splicing of the insulin receptor mRNA and insulin action in HepG2 hepatoma cells. J. Biol. Chem. 1993, 268, 21990–21996. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Flier, J.S.; Yokota, A.; Benecke, H.; Backer, J.M.; Moller, D.E. Functional properties of two naturally occurring isoforms of the human insulin receptor in Chinese hamster ovary cells. Endocrinology 1991, 129, 2058–2066. [Google Scholar] [CrossRef]
- Frasca, F.; Pandini, G.; Scalia, P.; Sciacca, L.; Mineo, R.; Costantino, A.; Goldfine, I.D.; Belfiore, A.; Vigneri, R. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol. Cell. Biol. 1999, 19, 3278–3288. [Google Scholar] [CrossRef]
- Vogt, B.; Carrascosa, J.M.; Ermel, B.; Ullrich, A.; Häring, H.-U. The two isotypes of the human insulin receptor (HIR-A and HIR-B) follow different internalization kinetics. Biochem. Biophys. Res. Commun. 1991, 177, 1013–1018. [Google Scholar] [CrossRef]
- Sacco, A.; Morcavallo, A.; Pandini, G.; Vigneri, R.; Belfiore, A. Differential Signaling Activation by Insulin and Insulin-Like Growth Factors I and II upon Binding to Insulin Receptor Isoform A. Endocrinology 2009, 150, 3594–3602. [Google Scholar] [CrossRef]
- Sciacca, L.; Cassarino, M.F.; Genua, M.; Pandini, G.; Le Moli, R.; Squatrito, S.; Vigneri, R. Insulin analogues differently activate insulin receptor isoforms and post-receptor signalling. Diabetologia 2010, 53, 1743–1753. [Google Scholar] [CrossRef]
- Pierre-Eugene, C.; Pagesy, P.; Nguyen, T.T.; Neuillé, M.; Tschank, G.; Tennagels, N.; Hampe, C.; Issad, T. Effect of Insulin Analogues on Insulin/IGF1 Hybrid Receptors: Increased Activation by Glargine but Not by Its Metabolites M1 and M2. PLoS ONE 2012, 7, e41992. [Google Scholar]
- Pandini, G.; Frasca, F.; Mineo, R.; Sciacca, L.; Vigneri, R.; Belfiore, A. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J. Biol. Chem. 2002, 277, 39684–39695. [Google Scholar] [CrossRef] [PubMed]
- Slaaby, R.; Schäffer, L.; Lautrup-Larsen, I.; Andersen, A.S.; Shaw, A.C.; Mathiasen, I.S.; Brandt, J. Hybrid receptors formed by insulin receptor (IR) and insulin-like growth factor I receptor (IGF-IR) have low insulin and high IGF-1 affinity irrespective of the IR splice variant. J. Biol. Chem. 2006, 281, 25869–25874. [Google Scholar] [CrossRef] [PubMed]
- Slaaby, R. Specific insulin/IGF1 hybrid receptor activation assay reveals IGF1 as a more potent ligand than insulin. Sci. Rep. 2015, 5, 7911. [Google Scholar] [CrossRef] [PubMed]
- Versteyhe, S.; Klaproth, B.; Borup, R.; Palsgaard, J.; Jensen, M.; Gray, S.G.; De Meyts, P. IGF-I, IGF-II, and Insulin Stimulate Different Gene Expression Responses through Binding to the IGF-I Receptor. Front. Endocrinol. 2013, 4, 98. [Google Scholar] [CrossRef]
- Rajapaksha, H.; Forbes, B.E. Ligand-Binding Affinity at the Insulin Receptor Isoform-A and Subsequent IR-A Tyrosine Phosphorylation Kinetics are Important Determinants of Mitogenic Biological Outcomes. Front. Endocrinol. 2015, 6, 107. [Google Scholar] [CrossRef]
- Benyoucef, S.; Surinya, K.H.; Hadaschik, D.; Siddle, K. Characterization of insulin/IGF hybrid receptors: Contributions of the insulin receptor L2 and Fn1 domains and the alternatively spliced exon 11 sequence to ligand binding and receptor activation. Biochem. J. 2007, 403, 603–613. [Google Scholar] [CrossRef]
- Ziegler, A.N.; Chidambaram, S.; Forbes, B.E.; Wood, T.L.; Levison, S.W. Insulin-like Growth Factor-II (IGF-II) and IGF-II Analogs with Enhanced Insulin Receptor-a Binding Affinity Promote Neural Stem Cell Expansion*. J. Biol. Chem. 2014, 289, 4626–4633. [Google Scholar] [CrossRef]
- Beck-Nielsen, H.; Kühl, C.; Pedersen, O.; Bjerre-Christensen, C.; Nielsen, T.T.; Klebe, J.G. Decreased insulin binding to monocytes from normal pregnant women. J. Clin. Endocrinol. Metab. 1979, 49, 810–814. [Google Scholar] [CrossRef]
- Goldstein, B.J.; Dudley, A.L. The rat insulin receptor: Primary structure and conservation of tissue-specific alternative messenger RNA splicing. Mol. Endocrinol. 1990, 4, 235–244. [Google Scholar] [CrossRef]
- Kaminska, D.; Hämäläinen, M.; Cederberg, H.; Käkelä, P.; Venesmaa, S.; Miettinen, P.; Ilves, I.; Herzig, K.H.; Kolehmainen, M.; Karhunen, L.; et al. Adipose tissue INSR splicing in humans associates with fasting insulin level and is regulated by weight loss. Diabetologia 2014, 57, 347–351. [Google Scholar] [CrossRef]
- Muller, D.; Huang, G.C.; Amiel, S.; Jones, P.M.; Persaud, S.J. Identification of insulin signaling elements in human beta-cells: Autocrine regulation of insulin gene expression. Diabetes 2006, 55, 2835–2842. [Google Scholar] [CrossRef] [PubMed]
- Moruzzi, N.; Lazzeri-Barcelo, F.; Valladolid-Acebes, I.; Moede, T.; Paschen, M.; Leibiger, B.; Berggren, P.O.; Leibiger, I.B. Tissue-specific expression of insulin receptor isoforms in obesity/type 2 diabetes mouse models. J. Cell. Mol. Med. 2021, 25, 4800–4813. [Google Scholar] [CrossRef] [PubMed]
- Westermeier, F.; Salomón, C.; Farías, M.; Arroyo, P.; Fuenzalida, B.; Sáez, T.; Salsoso, R.; Sanhueza, C.; Guzmán-Gutiérrez, E.; Pardo, F.; et al. Insulin requires normal expression and signaling of insulin receptor A to reverse gestational diabetes-reduced adenosine transport in human umbilical vein endothelium. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Garwood, C.J.; Ratcliffe, L.E.; Morgan, S.V.; Simpson, J.E.; Owens, H.; Vazquez-Villaseñor, I.; Heath, P.R.; Romero, I.A.; Ince, P.G.; Wharton, S.B. Insulin and IGF1 signalling pathways in human astrocytes in vitro and in vivo; characterisation, subcellular localisation and modulation of the receptors. Mol. Brain 2015, 8, 51. [Google Scholar] [CrossRef]
- Payankaulam, S.; Raicu, A.M.; Arnosti, D.N. Transcriptional Regulation of INSR, the Insulin Receptor Gene. Genes 2019, 10, 984. [Google Scholar] [CrossRef]
- Diaz-Castroverde, S.; Baos, S.; Luque, M.; Di Scala, M.; González-Aseguinolaza, G.; Gómez-Hernández, A.; Beneit, N.; Escribano, O.; Benito, M. Prevalent role of the insulin receptor isoform A in the regulation of hepatic glycogen metabolism in hepatocytes and in mice. Diabetologia 2016, 59, 2702–2710. [Google Scholar] [CrossRef]
- Sciacca, L.; Prisco, M.; Wu, A.; Belfiore, A.; Vigneri, R.; Baserga, R. Signaling Differences from the A and B Isoforms of the Insulin Receptor (IR) in 32D Cells in the Presence or Absence of IR Substrate-1. Endocrinology 2003, 144, 2650–2658. [Google Scholar] [CrossRef]
- Escribano, O.; Gómez-Hernández, A.; Díaz-Castroverde, S.; Nevado, C.; García, G.; Otero, Y.F.; Perdomo, L.; Beneit, N.; Benito, M. Insulin receptor isoform A confers a higher proliferative capability to pancreatic beta cells enabling glucose availability and IGF-I signaling. Mol. Cell. Endocrinol. 2015, 409, 82–91. [Google Scholar] [CrossRef]
- Khatib, A.-M.; Siegfried, G.; Prat, A.; Luis, J.; Chrétien, M.; Metrakos, P.; Seidah, N.G. Inhibition of proprotein convertases is associated with loss of growth and tumorigenicity of HT-29 human colon carcinoma cells: Importance of insulin-like growth factor-1 (IGF-1) receptor processing in IGF-1-mediated functions. J. Biol. Chem. 2001, 276, 30686–30693. [Google Scholar] [CrossRef]
- LeRoith, D.; Werner, H.; Beitner-Johnson, D.; Roberts, C.T., Jr. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr. Rev. 1995, 16, 143–163. [Google Scholar] [CrossRef]
- Ullrich, A.; Gray, A.; Tam, A.W.; Yang-Feng, T.; Tsubokawa, M.; Collins, C.; Henzel, W.; Le Bon, T.; Kathuria, S.; Chen, E.; et al. Insulin-like growth factor I receptor primary structure: Comparison with insulin receptor suggests structural determinants that define functional specificity. Embo J. 1986, 5, 2503–2512. [Google Scholar] [CrossRef] [PubMed]
- Adams, T.E.; Epa, V.C.; Garrett, T.P.; Ward, C.W. Structure and function of the type 1 insulin-like growth factor receptor. Cell. Mol. Life Sci. CMLS 2000, 57, 1050–1093. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, J.; Groth, A.V.; Mynarcik, D.C.; Pluzek, L.; Gadsbøll, V.L.; Whittaker, L.J. Alanine scanning mutagenesis of a type 1 insulin-like growth factor receptor ligand binding site. J. Biol. Chem. 2001, 276, 43980–43986. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, M.M.; Yuen, J.S.; Protheroe, A.S.; Pollak, M.; Macaulay, V.M. The type 1 insulin-like growth factor receptor pathway. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 6364–6370. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.B. Keys to the hidden treasures of the mannose 6-phosphate/insulin-like growth factor 2 receptor. Am. J. Pathol. 2003, 162, 3–6. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, R.G.; Pfeffer, S.R.; Coussens, L.; Tepper, M.A.; Brocklebank, C.M.; Mole, J.E.; Anderson, J.K.; Chen, E.; Czech, M.P.; Ullrich, A. A single receptor binds both insulin-like growth factor II and mannose-6-phosphate. Science 1988, 239, 1134–1137. [Google Scholar] [CrossRef]
- Kornfeld, S. Structure and function of the mannose 6-phosphate/insulinlike growth factor II receptors. Annu. Rev. Biochem. 1992, 61, 307–330. [Google Scholar] [CrossRef]
- Morgan, D.O.; Edman, J.C.; Standring, D.N.; Fried, V.A.; Smith, M.C.; Roth, R.A.; Rutter, W.J. Insulin-like growth factor II receptor as a multifunctional binding protein. Nature 1987, 329, 301–307. [Google Scholar] [CrossRef]
- Oka, Y.; Rozek, L.M.; Czech, M.P. Direct demonstration of rapid insulin-like growth factor II Receptor internalization and recycling in rat adipocytes. Insulin stimulates 125I-insulin-like growth factor II degradation by modulating the IGF-II receptor recycling process. J. Biol. Chem. 1985, 260, 9435–9442. [Google Scholar] [CrossRef]
- Dennis, P.A.; Rifkin, D.B. Cellular activation of latent transforming growth factor beta requires binding to the cation-independent mannose 6-phosphate/insulin-like growth factor type II receptor. Proc. Natl. Acad. Sci. USA 1991, 88, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Nissley, P.; Kiess, W.; Sklar, M. Developmental expression of the IGF-II/mannose 6-phosphate receptor. Mol. Reprod. Dev. 1993, 35, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Sinha, M.K.; Buchanan, C.; Raineri-Maldonado, C.; Khazanie, P.; Atkinson, S.; DiMarchi, R.; Caro, J.F. IGF-II receptors and IGF-II-stimulated glucose transport in human fat cells. Am. J. Physiol.-Endocrinol. Metab. 1990, 258, E534–E542. [Google Scholar] [CrossRef]
- Ohradanova-Repic, A.; Machacek, C.; Donner, C.; Mühlgrabner, V.; Petrovčíková, E.; Zahradníková, A.; Vičíková, K.; Hořejší, V.; Stockinger, H.; Leksa, V. The mannose 6-phosphate/insulin-like growth factor 2 receptor mediates plasminogen-induced efferocytosis. J. Leukoc. Biol. 2019, 105, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Leksa, V.; Godar, S.; Schiller, H.B.; Fuertbauer, E.; Muhammad, A.; Slezakova, K.; Horejsi, V.; Steinlein, P.; Weidle, U.H.; Binder, B.R. TGF-β-induced apoptosis in endothelial cells mediated by M6P/IGFII-R and mini-plasminogen. J. Cell Sci. 2005, 118, 4577–4586. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. The transforming growth factor-beta family. Annu. Rev. Cell Biol. 1990, 6, 597–641. [Google Scholar] [CrossRef]
- Blanquart, C.; Achi, J.; Issad, T. Characterization of IRA/IRB hybrid insulin receptors using bioluminescence resonance energy transfer. Biochem. Pharmacol. 2008, 76, 873–883. [Google Scholar] [CrossRef]
- Bailyes, E.M.; Navé, B.T.; Soos, M.A.; Orr, S.R.; Hayward, A.C.; Siddle, K. Insulin receptor/IGF-I receptor hybrids are widely distributed in mammalian tissues: Quantification of individual receptor species by selective immunoprecipitation and immunoblotting. Biochem. J. 1997, 327, 209–215. [Google Scholar] [CrossRef]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef]
- Langlois, W.J.; Sasaoka, T.; Yip, C.C.; Olefsky, J.M. Functional characterization of hybrid receptors composed of a truncated insulin receptor and wild type insulin-like growth factor 1 or insulin receptors. Endocrinology 1995, 136, 1978–1986. [Google Scholar] [CrossRef]
- Soos, M.A.; Field, C.E.; Siddle, K. Purified hybrid insulin/insulin-like growth factor-I receptors bind insulin-like growth factor-I, but not insulin, with high affinity. Biochem. J. 1993, 290, 419–426. [Google Scholar] [CrossRef]
- Myers, M.G., Jr.; White, M.F. Insulin signal transduction and the IRS proteins. Annu. Rev. Pharmacol. Toxicol. 1996, 36, 615–658. [Google Scholar] [CrossRef] [PubMed]
- White, M.F. The IRS-signalling system: A network of docking proteins that mediate insulin action. Insul. Action 1998, 182, 3–11. [Google Scholar]
- Avruch, J. Insulin signal transduction through protein kinase cascades. Insul. Action 1998, 182, 31–48. [Google Scholar]
- Lawrence, M.C.; Ward, C.W. Structural Features of the Receptor Tyrosine Kinase Ectodomains. In Receptor Tyrosine Kinases: Structure, Functions and Role in Human Disease; Wheeler, D.L., Yarden, Y., Eds.; Springer: New York, NY, USA, 2015; pp. 163–193. [Google Scholar]
- Maruyama, I.N. Activation of transmembrane cell-surface receptors via a common mechanism? The “rotation model”. BioEssays News Rev. Mol. Cell. Dev. Biol. 2015, 37, 959–967. [Google Scholar] [CrossRef]
- Li, S.; Covino, N.D.; Stein, E.G.; Till, J.H.; Hubbard, S.R. Structural and biochemical evidence for an autoinhibitory role for tyrosine 984 in the juxtamembrane region of the insulin receptor. J. Biol. Chem. 2003, 278, 26007–26014. [Google Scholar] [CrossRef]
- Lee, J.; Miyazaki, M.; Romeo, G.R.; Shoelson, S.E. Insulin receptor activation with transmembrane domain ligands. J. Biol. Chem. 2014, 289, 19769–19777. [Google Scholar] [CrossRef]
- Kavran, J.M.; McCabe, J.M.; Byrne, P.O.; Connacher, M.K.; Wang, Z.; Ramek, A.; Sarabipour, S.; Shan, Y.; Shaw, D.E.; Hristova, K.; et al. How IGF-1 activates its receptor. Elife 2014, 3, e03772. [Google Scholar] [CrossRef]
- Pawson, T. Specificity in signal transduction: From phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell 2004, 116, 191–203. [Google Scholar] [CrossRef]
- Sun, X.J.; Rothenberg, P.; Kahn, C.R.; Backer, J.M.; Araki, E.; Wilden, P.A.; Cahill, D.A.; Goldstein, B.J.; White, M.F. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature 1991, 352, 73–77. [Google Scholar] [CrossRef]
- Ravichandran, K.S. Signaling via Shc family adapter proteins. Oncogene 2001, 20, 6322–6330. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.J.; Wang, L.M.; Zhang, Y.; Yenush, L.; Myers, M.G., Jr.; Glasheen, E.; Lane, W.S.; Pierce, J.H.; White, M.F. Role of IRS-2 in insulin and cytokine signalling. Nature 1995, 377, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Lavan, B.E.; Fantin, V.R.; Chang, E.T.; Lane, W.S.; Keller, S.R.; Lienhard, G.E. A novel 160-kDa phosphotyrosine protein in insulin-treated embryonic kidney cells is a new member of the insulin receptor substrate family. J. Biol. Chem. 1997, 272, 21403–21407. [Google Scholar] [CrossRef] [PubMed]
- Lavan, B.E.; Lane, W.S.; Lienhard, G.E. The 60-kDa phosphotyrosine protein in insulin-treated adipocytes is a new member of the insulin receptor substrate family. J. Biol. Chem. 1997, 272, 11439–11443. [Google Scholar] [CrossRef]
- Huang, C.; Thirone, A.C.; Huang, X.; Klip, A. Differential contribution of insulin receptor substrates 1 versus 2 to insulin signaling and glucose uptake in l6 myotubes. J. Biol. Chem. 2005, 280, 19426–19435. [Google Scholar] [CrossRef]
- Bouzakri, K.; Zachrisson, A.; Al-Khalili, L.; Zhang, B.B.; Koistinen, H.A.; Krook, A.; Zierath, J.R. siRNA-based gene silencing reveals specialized roles of IRS-1/Akt2 and IRS-2/Akt1 in glucose and lipid metabolism in human skeletal muscle. Cell Metab. 2006, 4, 89–96. [Google Scholar] [CrossRef]
- Araki, E.; Lipes, M.A.; Patti, M.E.; Brüning, J.C.; Haag, B., 3rd; Johnson, R.S.; Kahn, C.R. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature 1994, 372, 186–190. [Google Scholar] [CrossRef]
- Miki, H.; Yamauchi, T.; Suzuki, R.; Komeda, K.; Tsuchida, A.; Kubota, N.; Terauchi, Y.; Kamon, J.; Kaburagi, Y.; Matsui, J.; et al. Essential role of insulin receptor substrate 1 (IRS-1) and IRS-2 in adipocyte differentiation. Mol. Cell. Biol. 2001, 21, 2521–2532. [Google Scholar] [CrossRef]
- Tseng, Y.H.; Butte, A.J.; Kokkotou, E.; Yechoor, V.K.; Taniguchi, C.M.; Kriauciunas, K.M.; Cypess, A.M.; Niinobe, M.; Yoshikawa, K.; Patti, M.E.; et al. Prediction of preadipocyte differentiation by gene expression reveals role of insulin receptor substrates and necdin. Nat. Cell Biol. 2005, 7, 601–611. [Google Scholar] [CrossRef]
- Withers, D.J.; Gutierrez, J.S.; Towery, H.; Burks, D.J.; Ren, J.M.; Previs, S.; Zhang, Y.; Bernal, D.; Pons, S.; Shulman, G.I.; et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature 1998, 391, 900–904. [Google Scholar] [CrossRef]
- Björnholm, M.; He, A.R.; Attersand, A.; Lake, S.; Liu, S.C.; Lienhard, G.E.; Taylor, S.; Arner, P.; Zierath, J.R. Absence of functional insulin receptor substrate-3 (IRS-3) gene in humans. Diabetologia 2002, 45, 1697–1702. [Google Scholar] [PubMed]
- Fantin, V.R.; Lavan, B.E.; Wang, Q.; Jenkins, N.A.; Gilbert, D.J.; Copeland, N.G.; Keller, S.R.; Lienhard, G.E. Cloning, tissue expression, and chromosomal location of the mouse insulin receptor substrate 4 gene. Endocrinology 1999, 140, 1329–1337. [Google Scholar] [CrossRef] [PubMed]
- Fantin, V.R.; Wang, Q.; Lienhard, G.E.; Keller, S.R. Mice lacking insulin receptor substrate 4 exhibit mild defects in growth, reproduction, and glucose homeostasis. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E127–E133. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef]
- White, M.F. Insulin signaling in health and disease. Science 2003, 302, 1710–1711. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Versteyhe, S.; Blanquart, C.; Hampe, C.; Mahmood, S.; Christeff, N.; De Meyts, P.; Gray, S.G.; Issad, T. Insulin receptor substrates-5 and -6 are poor substrates for the insulin receptor. Mol. Med. Rep. 2010, 3, 189–193. [Google Scholar] [CrossRef]
- Shepherd, P.R.; Withers, D.J.; Siddle, K. Phosphoinositide 3-kinase: The key switch mechanism in insulin signalling. Biochem. J. 1998, 333, 471–490. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Harris, T.E.; Lawrence, J.C., Jr. TOR signaling. Sci. STKE Signal Transduct. Knowl. Environ. 2003, 2003, re15. [Google Scholar] [CrossRef]
- Cohen, P.; Frame, S. The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2001, 2, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Accili, D.; Arden, K.C. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 2004, 117, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Sano, H.; Kane, S.; Sano, E.; Mîinea, C.P.; Asara, J.M.; Lane, W.S.; Garner, C.W.; Lienhard, G.E. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J. Biol. Chem. 2003, 278, 14599–14602. [Google Scholar] [CrossRef] [PubMed]
- Skolnik, E.Y.; Lee, C.H.; Batzer, A.; Vicentini, L.M.; Zhou, M.; Daly, R.; Myers, M.J., Jr.; Backer, J.M.; Ullrich, A.; White, M.F.; et al. The SH2/SH3 domain-containing protein GRB2 interacts with tyrosine-phosphorylated IRS1 and Shc: Implications for insulin control of ras signalling. EMBO J. 1993, 12, 1929–1936. [Google Scholar] [CrossRef] [PubMed]
- White, M.F. Mechanisms of insulin action. In Atlas of Diabetes, 4th ed.; Skyler, J.S., Ed.; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Avruch, J. MAP kinase pathways: The first twenty years. Biochim. Biophys. Acta 2007, 1773, 1150–1160. [Google Scholar] [CrossRef]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef]
- Nagao, H.; Jayavelu, A.K.; Cai, W.; Pan, H.; Dreyfuss, J.M.; Batista, T.M.; Brandão, B.B.; Mann, M.; Kahn, C.R. Unique ligand and kinase-independent roles of the insulin receptor in regulation of cell cycle, senescence and apoptosis. Nat. Commun. 2023, 14, 57. [Google Scholar] [CrossRef]
- Ramirez, A.K.; Dankel, S.; Cai, W.; Sakaguchi, M.; Kasif, S.; Kahn, C.R. Membrane metallo-endopeptidase (Neprilysin) regulates inflammatory response and insulin signaling in white preadipocytes. Mol. Metab. 2019, 22, 21–36. [Google Scholar] [CrossRef]
- Batista, T.M.; Dagdeviren, S.; Carroll, S.H.; Cai, W.; Melnik, V.Y.; Noh, H.L.; Saengnipanthkul, S.; Kim, J.K.; Kahn, C.R.; Lee, R.T. Arrestin domain-containing 3 (Arrdc3) modulates insulin action and glucose metabolism in liver. Proc. Natl. Acad. Sci. USA 2020, 117, 6733–6740. [Google Scholar] [CrossRef]
- Hancock, M.L.; Meyer, R.C.; Mistry, M.; Khetani, R.S.; Wagschal, A.; Shin, T.; Sui, S.J.H.; Näär, A.M.; Flanagan, J.G. Insulin receptor associates with promoters genome-wide and regulates gene expression. Cell 2019, 177, 722–736.e22. [Google Scholar] [CrossRef]
- Leibiger, B.; Leibiger, I.B.; Moede, T.; Kemper, S.; Kulkarni, R.N.; Kahn, C.R.; de Vargas, L.M.; Berggren, P.O. Selective insulin signaling through A and B insulin receptors regulates transcription of insulin and glucokinase genes in pancreatic beta cells. Mol. Cell 2001, 7, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Nevado, C.; Benito, M.; Valverde, A.M. Role of insulin receptor and balance in insulin receptor isoforms A and B in regulation of apoptosis in simian virus 40-immortalized neonatal hepatocytes. Mol. Biol. Cell 2008, 19, 1185–1198. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cui, K.; Miyoshi, K.; Hennighausen, L.; Green, J.E.; Setser, J.; LeRoith, D.; Yakar, S. Reduced circulating insulin-like growth factor I levels delay the onset of chemically and genetically induced mammary tumors. Cancer Res. 2003, 63, 4384–4388. [Google Scholar] [PubMed]
- Giudice, J.; Barcos, L.S.; Guaimas, F.F.; Penas-Steinhardt, A.; Giordano, L.; Jares-Erijman, E.A.; Coluccio Leskow, F. Insulin and insulin like growth factor II endocytosis and signaling via insulin receptor B. Cell Commun. Signal. 2013, 11, 18. [Google Scholar] [CrossRef]
- Giudice, J.; Leskow, F.C.; Arndt-Jovin, D.J.; Jovin, T.M.; Jares-Erijman, E.A. Differential endocytosis and signaling dynamics of insulin receptor variants IR-A and IR-B. J. Cell Sci. 2011, 124, 801–811. [Google Scholar] [CrossRef]
- Sciacca, L.; Mineo, R.; Pandini, G.; Murabito, A.; Vigneri, R.; Belfiore, A. In IGF-I receptor-deficient leiomyosarcoma cells autocrine IGF-II induces cell invasion and protection from apoptosis via the insulin receptor isoform A. Oncogene 2002, 21, 8240–8250. [Google Scholar] [CrossRef]
- Hanks, S.K.; Quinn, A.M.; Hunter, T. The protein kinase family: Conserved features and deduced phylogeny of the catalytic domains. Science 1988, 241, 42–52. [Google Scholar] [CrossRef]
- Craparo, A.; O’Neill, T.J.; Gustafson, T.A. Non-SH2 domains within insulin receptor substrate-1 and SHC mediate their phosphotyrosine-dependent interaction with the NPEY motif of the insulin-like growth factor I receptor. J. Biol. Chem. 1995, 270, 15639–15643. [Google Scholar] [CrossRef]
- Tartare-Deckert, S.; Sawka-Verhelle, D.; Murdaca, J.; Van Obberghen, E. Evidence for a differential interaction of SHC and the insulin receptor substrate-1 (IRS-1) with the insulin-like growth factor-I (IGF-I) receptor in the yeast two-hybrid system. J. Biol. Chem. 1995, 270, 23456–23460. [Google Scholar] [CrossRef]
- Dey, B.R.; Frick, K.; Lopaczynski, W.; Nissley, S.P.; Furlanetto, R.W. Evidence for the direct interaction of the insulin-like growth factor I receptor with IRS-1, Shc, and Grb10. Mol. Endocrinol. 1996, 10, 631–641. [Google Scholar]
- Xu, P.; Jacobs, A.R.; Taylor, S.I. Interaction of insulin receptor substrate 3 with insulin receptor, insulin receptor-related receptor, insulin-like growth factor-1 receptor, and downstream signaling proteins. J. Biol. Chem. 1999, 274, 15262–15270. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ferber, A.; Miura, M.; Baserga, R. Mitogenicity and transforming activity of the insulin-like growth factor-I receptor with mutations in the tyrosine kinase domain. J. Biol. Chem. 1994, 269, 32558–32564. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Sánchez, C.; Blakesley, V.; Kalebic, T.; Helman, L.; LeRoith, D. The role of the tyrosine kinase domain of the insulin-like growth factor-I receptor in intracellular signaling, cellular proliferation, and tumorigenesis. J. Biol. Chem. 1995, 270, 29176–29181. [Google Scholar] [CrossRef] [PubMed]
- Stannard, B.; Blakesley, V.; Kato, H.; Roberts, C.T., Jr.; LeRoith, D. Single tyrosine substitution in the insulin-like growth factor I receptor inhibits ligand-induced receptor autophosphorylation and internalization, but not mitogenesis. Endocrinology 1995, 136, 4918–4924. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Chan, J.L.; Zong, C.S.; Wang, L.H. Effect of tyrosine mutations on the kinase activity and transforming potential of an oncogenic human insulin-like growth factor I receptor. J. Biol. Chem. 1996, 271, 160–167. [Google Scholar] [CrossRef]
- O’Connor, R.; Kauffmann-Zeh, A.; Liu, Y.; Lehar, S.; Evan, G.I.; Baserga, R.; Blättler, W.A. Identification of domains of the insulin-like growth factor I receptor that are required for protection from apoptosis. Mol. Cell. Biol. 1997, 17, 427–435. [Google Scholar] [CrossRef]
- Miura, M.; Surmacz, E.; Burgaud, J.L.; Baserga, R. Different effects on mitogenesis and transformation of a mutation at tyrosine 1251 of the insulin-like growth factor I receptor. J. Biol. Chem. 1995, 270, 22639–22644. [Google Scholar] [CrossRef]
- Girnita, L.; Worrall, C.; Takahashi, S.; Seregard, S.; Girnita, A. Something old, something new and something borrowed: Emerging paradigm of insulin-like growth factor type 1 receptor (IGF-1R) signaling regulation. Cell. Mol. Life Sci. CMLS 2014, 71, 2403–2427. [Google Scholar] [CrossRef]
- Koval, A.P.; Blakesley, V.A.; Roberts, C.T., Jr.; Zick, Y.; Leroith, D. Interaction in vitro of the product of the c-Crk-II proto-oncogene with the insulin-like growth factor I receptor. Biochem. J. 1998, 330, 923–932. [Google Scholar] [CrossRef]
- Dearth, R.K.; Cui, X.; Kim, H.J.; Hadsell, D.L.; Lee, A.V. Oncogenic transformation by the signaling adaptor proteins insulin receptor substrate (IRS)-1 and IRS-2. Cell Cycle 2007, 6, 705–713. [Google Scholar] [CrossRef]
- Yamauchi, K.; Pessin, J.E. Insulin receptor substrate-1 (IRS1) and Shc compete for a limited pool of Grb2 in mediating insulin downstream signaling. J. Biol. Chem. 1994, 269, 31107–31114. [Google Scholar] [CrossRef] [PubMed]
- Peruzzi, F.; Prisco, M.; Dews, M.; Salomoni, P.; Grassilli, E.; Romano, G.; Calabretta, B.; Baserga, R. Multiple signaling pathways of the insulin-like growth factor 1 receptor in protection from apoptosis. Mol. Cell. Biol. 1999, 19, 7203–7215. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.J.; Johnson, M.D.; May, F.E.; Westley, B.R. Role of insulin-like growth factors and the type I insulin-like growth factor receptor in the estrogen-stimulated proliferation of human breast cancer cells. J. Biol. Chem. 1990, 265, 21172–21178. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.V.; Jackson, J.G.; Gooch, J.L.; Hilsenbeck, S.G.; Coronado-Heinsohn, E.; Osborne, C.K.; Yee, D. Enhancement of insulin-like growth factor signaling in human breast cancer: Estrogen regulation of insulin receptor substrate-1 expression in vitro and in vivo. Mol. Endocrinol. 1999, 13, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Rowinsky, E.K.; Youssoufian, H.; Tonra, J.R.; Solomon, P.; Burtrum, D.; Ludwig, D.L. IMC-A12, a human IgG1 monoclonal antibody to the insulin-like growth factor I receptor. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 5549s–5555s. [Google Scholar] [CrossRef] [PubMed]
- Macaulay, V.M.; Middleton, M.R.; Protheroe, A.S.; Tolcher, A.; Dieras, V.; Sessa, C.; Bahleda, R.; Blay, J.Y.; LoRusso, P.; Mery-Mignard, D.; et al. Phase I study of humanized monoclonal antibody AVE1642 directed against the type 1 insulin-like growth factor receptor (IGF-1R), administered in combination with anticancer therapies to patients with advanced solid tumors. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2013, 24, 784–791. [Google Scholar] [CrossRef]
- Naing, A.; Kurzrock, R.; Burger, A.; Gupta, S.; Lei, X.; Busaidy, N.; Hong, D.; Chen, H.X.; Doyle, L.A.; Heilbrun, L.K.; et al. Phase I trial of cixutumumab combined with temsirolimus in patients with advanced cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 6052–6060. [Google Scholar] [CrossRef]
- Chellappan, S.P.; Hiebert, S.; Mudryj, M.; Horowitz, J.M.; Nevins, J.R. The E2F transcription factor is a cellular target for the RB protein. Cell 1991, 65, 1053–1061. [Google Scholar] [CrossRef]
- Kato, J.-Y.; Matsushime, H.; Hiebert, S.; Ewen, M.; Sherr, C. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 1993, 7, 331–342. [Google Scholar]
- Ohtani, K.; Degregori, J.; Nevins, J.R. Regulation of the cyclin E gene by transcription factor E2F1. Proc. Natl. Acad. Sci. USA 1995, 92, 12146–12150. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Shin, I.; Yakes, F.M.; Rojo, F.; Shin, N.-Y.; Bakin, A.V.; Baselga, J.; Arteaga, C.L. PKB/Akt mediates cell-cycle progression by phosphorylation of p27Kip1 at threonine 157 and modulation of its cellular localization. Nat. Med. 2002, 8, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Liao, Y.; Xia, W.; Spohn, B.; Lee, M.-H.; Hung, M.-C. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat. Cell Biol. 2001, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, P.T.; Hay, N. The two TORCs and AKT. Dev. Cell 2007, 12, 487–502. [Google Scholar] [CrossRef]
- Averous, J.; Fonseca, B.; Proud, C. Regulation of cyclin D1 expression by mTORC1 signaling requires eukaryotic initiation factor 4E-binding protein 1. Oncogene 2008, 27, 1106–1113. [Google Scholar] [CrossRef]
- Vella, V.; Milluzzo, A.; Scalisi, N.M.; Vigneri, P.; Sciacca, L. Insulin Receptor Isoforms in Cancer. Int. J. Mol. Sci. 2018, 19, 3615. [Google Scholar] [CrossRef]
- Malaguarnera, R.; Frasca, F.; Garozzo, A.; Gianì, F.; Pandini, G.; Vella, V.; Vigneri, R.; Belfiore, A. Insulin receptor isoforms and insulin-like growth factor receptor in human follicular cell precursors from papillary thyroid cancer and normal thyroid. J. Clin. Endocrinol. Metab. 2011, 96, 766–774. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Zhao, T.; Xu, Y. p53 and stem cells: New developments and new concerns. Trends Cell Biol. 2010, 20, 170–175. [Google Scholar] [CrossRef]
- Loh, Y.H.; Wu, Q.; Chew, J.L.; Vega, V.B.; Zhang, W.; Chen, X.; Bourque, G.; George, J.; Leong, B.; Liu, J.; et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 2006, 38, 431–440. [Google Scholar] [CrossRef]
- Aljada, A.; Saleh, A.M.; Al-Aqeel, S.M.; Shamsa, H.B.; Al-Bawab, A.; Al Dubayee, M.; Ahmed, A.A. Quantification of insulin receptor mRNA splice variants as a diagnostic tumor marker in breast cancer. Cancer Biomark. Sect. A Dis. Markers 2015, 15, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Harrington, S.C.; Weroha, S.J.; Reynolds, C.; Suman, V.J.; Lingle, W.L.; Haluska, P. Quantifying insulin receptor isoform expression in FFPE breast tumors. Growth Horm. IGF Res. Off. J. Growth Horm. Res. Soc. Int. IGF Res. Soc. 2012, 22, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Law, J.H.; Habibi, G.; Hu, K.; Masoudi, H.; Wang, M.Y.; Stratford, A.L.; Park, E.; Gee, J.M.; Finlay, P.; Jones, H.E.; et al. Phosphorylated insulin-like growth factor-i/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res. 2008, 68, 10238–10246. [Google Scholar] [CrossRef] [PubMed]
- Flannery, C.A.; Saleh, F.L.; Choe, G.H.; Selen, D.J.; Kodaman, P.H.; Kliman, H.J.; Wood, T.L.; Taylor, H.S. Differential Expression of IR-A, IR-B and IGF-1R in Endometrial Physiology and Distinct Signature in Adenocarcinoma. J. Clin. Endocrinol. Metab. 2016, 101, 2883–2891. [Google Scholar] [CrossRef] [PubMed]
- Kalli, K.R.; Falowo, O.I.; Bale, L.K.; Zschunke, M.A.; Roche, P.C.; Conover, C.A. Functional insulin receptors on human epithelial ovarian carcinoma cells: Implications for IGF-II mitogenic signaling. Endocrinology 2002, 143, 3259–3267. [Google Scholar] [CrossRef]
- Breen, K.J.; O’Neill, A.; Murphy, L.; Fan, Y.; Boyce, S.; Fitzgerald, N.; Dorris, E.; Brady, L.; Finn, S.P.; Hayes, B.D.; et al. Investigating the role of the IGF axis as a predictor of biochemical recurrence in prostate cancer patients post-surgery. Prostate 2017, 77, 1288–1300. [Google Scholar] [CrossRef]
- Cox, M.E.; Gleave, M.E.; Zakikhani, M.; Bell, R.H.; Piura, E.; Vickers, E.; Cunningham, M.; Larsson, O.; Fazli, L.; Pollak, M. Insulin receptor expression by human prostate cancers. Prostate 2009, 69, 33–40. [Google Scholar] [CrossRef]
- Heni, M.; Hennenlotter, J.; Scharpf, M.; Lutz, S.Z.; Schwentner, C.; Todenhöfer, T.; Schilling, D.; Kühs, U.; Gerber, V.; Machicao, F.; et al. Insulin receptor isoforms A and B as well as insulin receptor substrates-1 and -2 are differentially expressed in prostate cancer. PLoS ONE 2012, 7, e50953. [Google Scholar] [CrossRef]
- Perks, C.M.; Zielinska, H.A.; Wang, J.; Jarrett, C.; Frankow, A.; Ladomery, M.R.; Bahl, A.; Rhodes, A.; Oxley, J.; Holly, J.M. Insulin Receptor Isoform Variations in Prostate Cancer Cells. Front. Endocrinol. 2016, 7, 132. [Google Scholar] [CrossRef]
- Roudnicky, F.; Dieterich, L.C.; Poyet, C.; Buser, L.; Wild, P.; Tang, D.; Camenzind, P.; Ho, C.H.; Otto, V.I.; Detmar, M. High expression of insulin receptor on tumour-associated blood vessels in invasive bladder cancer predicts poor overall and progression-free survival. J. Pathol. 2017, 242, 193–205. [Google Scholar] [CrossRef]
- Andres, S.F.; Simmons, J.G.; Mah, A.T.; Santoro, M.A.; Van Landeghem, L.; Lund, P.K. Insulin receptor isoform switching in intestinal stem cells, progenitors, differentiated lineages and tumors: Evidence that IR-B limits proliferation. J. Cell Sci. 2013, 126, 5645–5656. [Google Scholar] [CrossRef] [PubMed]
- Chettouh, H.; Fartoux, L.; Aoudjehane, L.; Wendum, D.; Clapéron, A.; Chrétien, Y.; Rey, C.; Scatton, O.; Soubrane, O.; Conti, F.; et al. Mitogenic insulin receptor-A is overexpressed in human hepatocellular carcinoma due to EGFR-mediated dysregulation of RNA splicing factors. Cancer Res. 2013, 73, 3974–3986. [Google Scholar] [CrossRef] [PubMed]
- Aleem, E.; Nehrbass, D.; Klimek, F.; Mayer, D.; Bannasch, P. Upregulation of the insulin receptor and type I insulin-like growth factor receptor are early events in hepatocarcinogenesis. Toxicol. Pathol. 2011, 39, 524–543. [Google Scholar] [CrossRef]
- Sakurai, Y.; Kubota, N.; Takamoto, I.; Obata, A.; Iwamoto, M.; Hayashi, T.; Aihara, M.; Kubota, T.; Nishihara, H.; Kadowaki, T. Role of insulin receptor substrates in the progression of hepatocellular carcinoma. Sci. Rep. 2017, 7, 5387. [Google Scholar] [CrossRef]
- Jiang, L.; Zhu, W.; Streicher, K.; Morehouse, C.; Brohawn, P.; Ge, X.; Dong, Z.; Yin, X.; Zhu, G.; Gu, Y.; et al. Increased IR-A/IR-B ratio in non-small cell lung cancers associates with lower epithelial-mesenchymal transition signature and longer survival in squamous cell lung carcinoma. BMC Cancer 2014, 14, 131. [Google Scholar] [CrossRef] [PubMed]
- Avnet, S.; Sciacca, L.; Salerno, M.; Gancitano, G.; Cassarino, M.F.; Longhi, A.; Zakikhani, M.; Carboni, J.M.; Gottardis, M.; Giunti, A.; et al. Insulin receptor isoform A and insulin-like growth factor II as additional treatment targets in human osteosarcoma. Cancer Res. 2009, 69, 2443–2452. [Google Scholar] [CrossRef]
- Nowak-Sliwinska, P.; van Beijnum, J.R.; Huijbers, E.J.M.; Gasull, P.C.; Mans, L.; Bex, A.; Griffioen, A.W. Oncofoetal insulin receptor isoform A marks the tumour endothelium; an underestimated pathway during tumour angiogenesis and angiostatic treatment. Br. J. Cancer 2019, 120, 218–228. [Google Scholar] [CrossRef]
- Benabou, E.; Salamé, Z.; Wendum, D.; Lequoy, M.; Tahraoui, S.; Merabtene, F.; Chrétien, Y.; Scatton, O.; Rosmorduc, O.; Fouassier, L.; et al. Insulin receptor isoform A favors tumor progression in human hepatocellular carcinoma by increasing stem/progenitor cell features. Cancer Lett. 2019, 450, 155–168. [Google Scholar] [CrossRef]
- Leitinger, B. Discoidin domain receptor functions in physiological and pathological conditions. Int. Rev. Cell Mol. Biol. 2014, 310, 39–87. [Google Scholar]
- Malaguarnera, R.; Nicolosi, M.L.; Sacco, A.; Morcavallo, A.; Vella, V.; Voci, C.; Spatuzza, M.; Xu, S.Q.; Iozzo, R.V.; Vigneri, R.; et al. Novel cross talk between IGF-IR and DDR1 regulates IGF-IR trafficking, signaling and biological responses. Oncotarget 2015, 6, 16084–16105. [Google Scholar] [CrossRef]
- Matà, R.; Palladino, C.; Nicolosi, M.L.; Lo Presti, A.R.; Malaguarnera, R.; Ragusa, M.; Sciortino, D.; Morrione, A.; Maggiolini, M.; Vella, V.; et al. IGF-I induces upregulation of DDR1 collagen receptor in breast cancer cells by suppressing MIR-199a-5p through the PI3K/AKT pathway. Oncotarget 2016, 7, 7683–7700. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, A.; Malaguarnera, R.; Nicolosi, M.L.; Lappano, R.; Ragusa, M.; Morrione, A.; Vella, V. A novel functional crosstalk between DDR1 and the IGF axis and its relevance for breast cancer. Cell Adhes. Migr. 2018, 12, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Vella, V.; Malaguarnera, R.; Nicolosi, M.L.; Palladino, C.; Spoleti, C.; Massimino, M.; Vigneri, P.; Purrello, M.; Ragusa, M.; Morrione, A.; et al. Discoidin domain receptor 1 modulates insulin receptor signaling and biological responses in breast cancer cells. Oncotarget 2017, 8, 43248–43270. [Google Scholar] [CrossRef] [PubMed]
- Vella, V.; Nicolosi, M.L.; Cantafio, P.; Massimino, M.; Lappano, R.; Vigneri, P.; Ciuni, R.; Gangemi, P.; Morrione, A.; Malaguarnera, R.; et al. DDR1 regulates thyroid cancer cell differentiation via IGF-2/IR-A autocrine signaling loop. Endocr.-Relat. Cancer 2019, 26, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Vellingiri, B.; Iyer, M.; Devi Subramaniam, M.; Jayaramayya, K.; Siama, Z.; Giridharan, B.; Narayanasamy, A.; Abdal Dayem, A.; Cho, S.G. Understanding the Role of the Transcription Factor Sp1 in Ovarian Cancer: From Theory to Practice. Int. J. Mol. Sci. 2020, 21, 1153. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, L.; Zheng, Y.; Guo, L. HMGA1 in cancer: Cancer classification by location. J. Cell. Mol. Med. 2019, 23, 2293–2302. [Google Scholar] [CrossRef]
- Chang, E.T.; Donahue, J.M.; Xiao, L.; Cui, Y.; Rao, J.N.; Turner, D.J.; Twaddell, W.S.; Wang, J.Y.; Battafarano, R.J. The RNA-binding protein CUG-BP1 increases survivin expression in oesophageal cancer cells through enhanced mRNA stability. Biochem. J. 2012, 446, 113–123. [Google Scholar] [CrossRef]
- Philips, A.V.; Timchenko, L.T.; Cooper, T.A. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 1998, 280, 737–741. [Google Scholar] [CrossRef]
- Huang, G.; Song, C.; Wang, N.; Qin, T.; Sui, S.; Obr, A.; Zeng, L.; Wood, T.L.; Leroith, D.; Li, M.; et al. RNA-binding protein CUGBP1 controls the differential INSR splicing in molecular subtypes of breast cancer cells and affects cell aggressiveness. Carcinogenesis 2020, 41, 1294–1305. [Google Scholar] [CrossRef]
- Qi, Z.-P.; Chen, Z.-H.; He, D.-L.; Cai, S.-L.; Li, B.; Sun, D.; Lv, Z.-T.; Xu, E.-P.; Shi, Q.; Zhong, Y.-S.; et al. RNA binding protein CUGBP1 mediates the liver metastasis of colorectal cancer by regulating the ErbB signal pathway. Transl. Cancer Res. 2021, 10, 3373–3388. [Google Scholar] [CrossRef]
- Lee, E.K.; Kim, H.H.; Kuwano, Y.; Abdelmohsen, K.; Srikantan, S.; Subaran, S.S.; Gleichmann, M.; Mughal, M.R.; Martindale, J.L.; Yang, X.; et al. hnRNP C promotes APP translation by competing with FMRP for APP mRNA recruitment to P bodies. Nat. Struct. Mol. Biol. 2010, 17, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S. Regulation of urokinase receptor mRNA stability by hnRNP C in lung epithelial cells. Mol. Cell. Biochem. 2005, 272, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Dansithong, W.; Jog, S.P.; Holt, I.; Mittal, S.; Brook, J.D.; Morris, G.E.; Comai, L.; Reddy, S. Expanded CUG repeats Dysregulate RNA splicing by altering the stoichiometry of the muscleblind 1 complex. J. Biol. Chem. 2011, 286, 38427–38438. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.G.; Jung, Y.; Lee, N.; Seo, J.Y.; Kim, S.W.; Lee, K.H.; Kim, D.Y.; Kim, K.T. HNRNP A1 Promotes Lung Cancer Cell Proliferation by Modulating VRK1 Translation. Int. J. Mol. Sci. 2021, 22, 5506. [Google Scholar] [CrossRef]
- Cui, H.; Wu, F.; Sun, Y.; Fan, G.; Wang, Q. Up-regulation and subcellular localization of hnRNP A2/B1 in the development of hepatocellular carcinoma. BMC Cancer 2010, 10, 356. [Google Scholar] [CrossRef]
- Kozu, T.; Henrich, B.; Schäfer, K.P. Structure and expression of the gene (HNRPA2B1) encoding the human hnRNP protein A2/B1. Genomics 1995, 25, 365–371. [Google Scholar] [CrossRef]
- Carpenter, B.; MacKay, C.; Alnabulsi, A.; MacKay, M.; Telfer, C.; Melvin, W.T.; Murray, G.I. The roles of heterogeneous nuclear ribonucleoproteins in tumour development and progression. Biochim. Biophys. Acta 2006, 1765, 85–100. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, X.; Gu, Q.; Wang, J.; Sui, Y.; Wu, J.; Feng, J. Heterogeneous nuclear ribonucleoprotein A/B: An emerging group of cancer biomarkers and therapeutic targets. Cell Death Discov. 2022, 8, 337. [Google Scholar] [CrossRef]
- Sen, S.; Langiewicz, M.; Jumaa, H.; Webster, N.J. Deletion of serine/arginine-rich splicing factor 3 in hepatocytes predisposes to hepatocellular carcinoma in mice. Hepatology 2015, 61, 171–183. [Google Scholar] [CrossRef]
- Shepard, P.J.; Hertel, K.J. The SR protein family. Genome Biol. 2009, 10, 242. [Google Scholar] [CrossRef]
- Sen, S.; Talukdar, I.; Webster, N.J. SRp20 and CUG-BP1 modulate insulin receptor exon 11 alternative splicing. Mol. Cell. Biol. 2009, 29, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Konieczny, P.; Stepniak-Konieczna, E.; Sobczak, K. MBNL proteins and their target RNAs, interaction and splicing regulation. Nucleic Acids Res. 2014, 42, 10873–10887. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Miller, J.W.; Mankodi, A.; Kanadia, R.N.; Yuan, Y.; Moxley, R.T.; Swanson, M.S.; Thornton, C.A. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 2006, 15, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Lou, C.; Xiao, H.; Yang, Y.; Cai, X.; Li, C.; Jia, S.; Huang, Y. MiR-128 regulation of glucose metabolism and cell proliferation in triple-negative breast cancer. Br. J. Surg. 2018, 105, 75–85. [Google Scholar] [CrossRef]
- Budi, H.S.; Younus, L.A.; Lafta, M.H.; Parveen, S.; Mohammad, H.J.; Al-Qaim, Z.H.; Jawad, M.A.; Parra, R.M.R.; Mustafa, Y.F.; Alhachami, F.R.; et al. The role of miR-128 in cancer development, prevention, drug resistance, and immunotherapy. Front. Oncol. 2022, 12, 1067974. [Google Scholar] [CrossRef]
- Lovat, F.; Fassan, M.; Gasparini, P.; Rizzotto, L.; Cascione, L.; Pizzi, M.; Vicentini, C.; Balatti, V.; Palmieri, D.; Costinean, S.; et al. miR-15b/16-2 deletion promotes B-cell malignancies. Proc. Natl. Acad. Sci. USA 2015, 112, 11636–11641. [Google Scholar] [CrossRef]
- Yoshino, H.; Enokida, H.; Chiyomaru, T.; Tatarano, S.; Hidaka, H.; Yamasaki, T.; Gotannda, T.; Tachiwada, T.; Nohata, N.; Yamane, T.; et al. Tumor suppressive microRNA-1 mediated novel apoptosis pathways through direct inhibition of splicing factor serine/arginine-rich 9 (SRSF9/SRp30c) in bladder cancer. Biochem. Biophys. Res. Commun. 2012, 417, 588–593. [Google Scholar] [CrossRef]
- Spriggs, K.A.; Cobbold, L.C.; Ridley, S.H.; Coldwell, M.; Bottley, A.; Bushell, M.; Willis, A.E.; Siddle, K. The human insulin receptor mRNA contains a functional internal ribosome entry segment. Nucleic Acids Res. 2009, 37, 5881–5893. [Google Scholar] [CrossRef]
- Schönherr, E.; Sunderkötter, C.; Iozzo, R.V.; Schaefer, L. Decorin, a novel player in the insulin-like growth factor system. J. Biol. Chem. 2005, 280, 15767–15772. [Google Scholar] [CrossRef]
- Morcavallo, A.; Buraschi, S.; Xu, S.Q.; Belfiore, A.; Schaefer, L.; Iozzo, R.V.; Morrione, A. Decorin differentially modulates the activity of insulin receptor isoform A ligands. Matrix Biol. J. Int. Soc. Matrix Biol. 2014, 35, 82–90. [Google Scholar] [CrossRef]
- Fujimoto, J.; Shiota, M.; Iwahara, T.; Seki, N.; Satoh, H.; Mori, S.; Yamamoto, T. Characterization of the transforming activity of p80, a hyperphosphorylated protein in a Ki-1 lymphoma cell line with chromosomal translocation t(2;5). Proc. Natl. Acad. Sci. USA 1996, 93, 4181–4186. [Google Scholar] [CrossRef] [PubMed]
- Lannon, C.L.; Sorensen, P.H. ETV6-NTRK3: A chimeric protein tyrosine kinase with transformation activity in multiple cell lineages. Semin. Cancer Biol. 2005, 15, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, E.; Braga-Basaria, M.; Hardy, E.; Vasko, V.; Burman, K.D.; Jhiang, S.; Saji, M.; Ringel, M.D. Chronic expression of RET/PTC 3 enhances basal and insulin-stimulated PI3 kinase/AKT signaling and increases IRS-2 expression in FRTL-5 thyroid cells. Mol. Carcinog. 2004, 41, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Stoica, G.E.; Kuo, A.; Aigner, A.; Sunitha, I.; Souttou, B.; Malerczyk, C.; Caughey, D.J.; Wen, D.; Karavanov, A.; Riegel, A.T.; et al. Identification of anaplastic lymphoma kinase as a receptor for the growth factor pleiotrophin. J. Biol. Chem. 2001, 276, 16772–16779. [Google Scholar] [CrossRef] [PubMed]
- Chaves, J.; Saif, M.W. IGF system in cancer: From bench to clinic. Anti-Cancer Drugs 2011, 22, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Buck, E.; Mulvihill, M.J. Modulation of insulin-like growth factor-1 receptor and its signaling network for the treatment of cancer: Current status and future perspectives. Oncol. Rev. 2013, 7, e3. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, D.; Yee, D. Disrupting insulin-like growth factor signaling as a potential cancer therapy. Mol. Cancer Ther. 2007, 6, 1–12. [Google Scholar] [CrossRef]
- van de Luijtgaarden, A.C.; Versleijen-Jonkers, Y.M.; Roeffen, M.H.; Schreuder, H.W.; Flucke, U.E.; van der Graaf, W.T. Prognostic and therapeutic relevance of the IGF pathway in Ewing’s sarcoma patients. Target. Oncol. 2013, 8, 253–260. [Google Scholar] [CrossRef]
- Sell, C.; Dumenil, G.; Deveaud, C.; Miura, M.; Coppola, D.; DeAngelis, T.; Rubin, R.; Efstratiadis, A.; Baserga, R. Effect of a null mutation of the insulin-like growth factor I receptor gene on growth and transformation of mouse embryo fibroblasts. Mol. Cell. Biol. 1994, 14, 3604–3612. [Google Scholar]
- Aleksic, T.; Verrill, C.; Bryant, R.J.; Han, C.; Worrall, A.R.; Brureau, L.; Larré, S.; Higgins, G.S.; Fazal, F.; Sabbagh, A.; et al. IGF-1R associates with adverse outcomes after radical radiotherapy for prostate cancer. Br. J. Cancer 2017, 117, 1600–1606. [Google Scholar] [CrossRef]
- Dale, O.T.; Aleksic, T.; Shah, K.A.; Han, C.; Mehanna, H.; Rapozo, D.C.; Sheard, J.D.; Goodyear, P.; Upile, N.S.; Robinson, M.; et al. IGF-1R expression is associated with HPV-negative status and adverse survival in head and neck squamous cell cancer. Carcinogenesis 2015, 36, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Farabaugh, S.M.; Boone, D.N.; Lee, A.V. Role of IGF1R in breast cancer subtypes, stemness, and lineage differentiation. Front. Endocrinol. 2015, 6, 59. [Google Scholar] [CrossRef] [PubMed]
- Riedemann, J.; Macaulay, V.M. IGF1R signalling and its inhibition. Endocr.-Relat. Cancer 2006, 13 (Suppl. 1), S33–S43. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; Roberts, C.T., Jr. The IGFI receptor gene: A molecular target for disrupted transcription factors. Genes Chromosomes Cancer 2003, 36, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; Roberts, C.T., Jr.; Rauscher, F.J., 3rd; LeRoith, D. Regulation of insulin-like growth factor I receptor gene expression by the Wilms’ tumor suppressor WT1. J. Mol. Neurosci. MN 1996, 7, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Aguirre, J.; Balasubramanian, P.; Guevara-Aguirre, M.; Wei, M.; Madia, F.; Cheng, C.W.; Hwang, D.; Martin-Montalvo, A.; Saavedra, J.; Ingles, S.; et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci. Transl. Med. 2011, 3, 70ra13. [Google Scholar] [CrossRef]
- Steuerman, R.; Shevah, O.; Laron, Z. Congenital IGF1 deficiency tends to confer protection against post-natal development of malignancies. Eur. J. Endocrinol. 2011, 164, 485–489. [Google Scholar] [CrossRef]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef]
- Grimberg, A.; Cohen, P. Role of insulin-like growth factors and their binding proteins in growth control and carcinogenesis. J. Cell. Physiol. 2000, 183, 1–9. [Google Scholar] [CrossRef]
- Helle, S.I.; Geisler, S.; Aas, T.; Paulsen, T.; Holly, J.M.; Lønning, P.E. Plasma insulin-like growth factor binding protein-3 proteolysis is increased in primary breast cancer. Br. J. Cancer 2001, 85, 74–77. [Google Scholar] [CrossRef]
- Cohen, P.; Graves, H.C.; Peehl, D.M.; Kamarei, M.; Giudice, L.C.; Rosenfeld, R.G. Prostate-specific antigen (PSA) is an insulin-like growth factor binding protein-3 protease found in seminal plasma. J. Clin. Endocrinol. Metab. 1992, 75, 1046–1053. [Google Scholar]
- Yang, F.; Li, J.; Deng, H.; Wang, Y.; Lei, C.; Wang, Q.; Xiang, J.; Liang, L.; Xia, J.; Pan, X.; et al. GSTZ1-1 Deficiency Activates NRF2/IGF1R Axis in HCC via Accumulation of Oncometabolite Succinylacetone. EMBO J. 2019, 38, e101964. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, S.; Lee, J.; Klijn, C.; Gnad, F.; Bagniewska, M.; Schaefer, G.; Zhang, D.; Tan, J.; Watson, S.A.; Liu, L.; et al. ERBB3 and IGF1R Signaling Are Required for Nrf2-Dependent Growth in KEAP1-Mutant Lung Cancer. Cancer Res. 2019, 79, 4828–4839. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining Glutaminolysis Bolsters Chemotherapy While NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res. 2020, 80, 1630–1643. [Google Scholar] [CrossRef] [PubMed]
- Vella, V.; Sciacca, L.; Pandini, G.; Mineo, R.; Squatrito, S.; Vigneri, R.; Belfiore, A. The IGF system in thyroid cancer: New concepts. Mol. Pathol. MP 2001, 54, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Yao, G.; Song, H.; Zhou, Y.; Geng, J. IGF2R Expression is Associated with the Chemotherapy Response and Prognosis of Patients with Advanced NSCLC. Cell. Physiol. Biochem. 2014, 34, 1578–1588. [Google Scholar] [CrossRef]
- Wang, Y.; MacDonald, R.G.; Thinakaran, G.; Kar, S. Insulin-Like Growth Factor-II/Cation-Independent Mannose 6-Phosphate Receptor in Neurodegenerative Diseases. Mol. Neurobiol. 2017, 54, 2636–2658. [Google Scholar] [CrossRef]
- Takeda, T.; Komatsu, M.; Chiwaki, F.; Komatsuzaki, R.; Nakamura, K.; Tsuji, K.; Kobayashi, Y.; Tominaga, E.; Ono, M.; Banno, K.; et al. Upregulation of IGF2R evades lysosomal dysfunction-induced apoptosis of cervical cancer cells via transport of cathepsins. Cell Death Dis. 2019, 10, 876. [Google Scholar] [CrossRef]
- De Souza, A.T.; Yamada, T.; Mills, J.J.; Jirtle, R.L. Imprinted genes in liver carcinogenesis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1997, 11, 60–67. [Google Scholar] [CrossRef]
- Yoon, A.J.; Zavras, A.I.; Chen, M.K.; Lin, C.W.; Yang, S.F. Association between Gly1619ARG polymorphism of IGF2R domain 11 (rs629849) and advanced stage of oral cancer. Med. Oncol. 2012, 29, 682–685. [Google Scholar] [CrossRef]
- Probst, O.C.; Puxbaum, V.; Svoboda, B.; Leksa, V.; Stockinger, H.; Mikula, M.; Mikulits, W.; Mach, L. The mannose 6-phosphate/insulin-like growth factor II receptor restricts the tumourigenicity and invasiveness of squamous cell carcinoma cells. Int. J. Cancer 2009, 124, 2559–2567. [Google Scholar] [CrossRef] [PubMed]
- Morcavallo, A.; Genua, M.; Palummo, A.; Kletvikova, E.; Jiracek, J.; Brzozowski, A.M.; Iozzo, R.V.; Belfiore, A.; Morrione, A. Insulin and insulin-like growth factor II differentially regulate endocytic sorting and stability of insulin receptor isoform A. J. Biol. Chem. 2012, 287, 11422–11436. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Weiss, J.; Martin, J.L.; Scott, C.D. Increased expression of the mannose 6-phosphate/insulin-like growth factor-II receptor in breast cancer cells alters tumorigenic properties in vitro and in vivo. Int. J. Cancer 2003, 107, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.M.; Lian, W.S.; Qiu, M.K.; Dai, Y.X.; Dong, Q.; Shen, J.; Dong, P.; Wang, X.F.; Liu, Y.B.; Quan, Z.W.; et al. Knockdown of IGF2R suppresses proliferation and induces apoptosis in hemangioma cells in vitro and in vivo. Int. J. Oncol. 2014, 45, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Kolacinska, A.; Chalubinska, J.; Zawlik, I.; Szymanska, B.; Borowska-Garganisz, E.; Nowik, M.; Fendler, W.; Kubiak, R.; Pawlowska, Z.; Morawiec, Z.; et al. Apoptosis-, proliferation, immune function-, and drug resistance- related genes in ER positive, HER2 positive and triple negative breast cancer. Neoplasma 2012, 59, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, R.; Beriwal, S.; McManus, K.; Dabbs, D.J. Insulin-like growth factor receptor-1 (IGF-1R) expression in normal breast, proliferative breast lesions, and breast carcinoma. Appl. Immunohistochem. Mol. Morphol. AIMM 2011, 19, 218–225. [Google Scholar] [CrossRef]
- Yerushalmi, R.; Gelmon, K.A.; Leung, S.; Gao, D.; Cheang, M.; Pollak, M.; Turashvili, G.; Gilks, B.C.; Kennecke, H. Insulin-like growth factor receptor (IGF-1R) in breast cancer subtypes. Breast Cancer Res. Treat. 2012, 132, 131–142. [Google Scholar] [CrossRef]
- Shin, S.J.; Gong, G.; Lee, H.J.; Kang, J.; Bae, Y.K.; Lee, A.; Cho, E.Y.; Lee, J.S.; Suh, K.S.; Lee, D.W.; et al. Positive expression of insulin-like growth factor-1 receptor is associated with a positive hormone receptor status and a favorable prognosis in breast cancer. J. Breast Cancer 2014, 17, 113–120. [Google Scholar] [CrossRef]
- Stanilov, N.S.; Karakolev, I.A.; Deliysky, T.S.; Jovchev, J.P.; Stanilova, S.A. Association of insulin-like growth factor-I receptor polymorphism with colorectal cancer development. Mol. Biol. Rep. 2014, 41, 8099–8106. [Google Scholar] [CrossRef]
- Chen, S.C.; Chou, C.K.; Wong, F.H.; Chang, C.M.; Hu, C.P. Overexpression of epidermal growth factor and insulin-like growth factor-I receptors and autocrine stimulation in human esophageal carcinoma cells. Cancer Res. 1991, 51, 1898–1903. [Google Scholar]
- Ge, J.; Chen, Z.; Wu, S.; Chen, J.; Li, X.; Li, J.; Yin, J.; Chen, Z. Expression levels of insulin-like growth factor-1 and multidrug resistance-associated protein-1 indicate poor prognosis in patients with gastric cancer. Digestion 2009, 80, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lu, P.; Liang, Z.; Zhang, Z.; Shi, W.; Cai, X.; Chen, C. Increased insulin-like growth factor 1 receptor (IGF1R) expression in small cell lung cancer and the effect of inhibition of IGF1R expression by RNAi on growth of human small cell lung cancer NCI-H446 cell. Growth Factors 2015, 33, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Burrow, S.; Andrulis, I.L.; Pollak, M.; Bell, R.S. Expression of insulin-like growth factor receptor, IGF-1, and IGF-2 in primary and metastatic osteosarcoma. J. Surg. Oncol. 1998, 69, 21–27. [Google Scholar] [CrossRef]
- Liu, S.B.; Zhou, L.B.; Wang, H.F.; Li, G.; Xie, Q.P.; Hu, B. Loss of IGF2R indicates a poor prognosis and promotes cell proliferation and tumorigenesis in bladder cancer via AKT signaling pathway. Neoplasma 2020, 67, 129–136. [Google Scholar] [CrossRef]
- Zhang, Z.; Mou, Z.; Xu, C.; Wu, S.; Dai, X.; Chen, X.; Ou, Y.; Chen, Y.; Yang, C.; Jiang, H. Autophagy-associated circular RNA hsa_circ_0007813 modulates human bladder cancer progression via hsa-miR-361-3p/IGF2R regulation. Cell Death Dis. 2021, 12, 778. [Google Scholar] [CrossRef]
- Zhong, Y.; Ren, X.; Cao, X.; Xu, Y.; Song, Y.; Zhou, Y.; Mao, F.; Shen, S.; Wang, Z.; Sun, Q. Insulin-like growth factor 2 receptor is a key immune-related gene that is correlated with a poor prognosis in patients with triple-negative breast cancer: A bioinformatics analysis. Front. Oncol. 2022, 12, 871786. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, H.N.; Galanopoulos, T.; Neville-Golden, J.; Maxwell, M. Expression of insulin-like growth factors I and II and their receptor mRNAs in primary human astrocytomas and meningiomas; in vivo studies using in situ hybridization and immunocytochemistry. Int. J. Cancer 1992, 50, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.S.; Kang, K.M.; Choi, B.O.; Chai, G.Y.; Hong, S.C.; Ha, W.S.; Jirtle, R.L. Clinical significance of loss of heterozygosity for M6P/IGF2R in patients with primary hepatocellular carcinoma. World J. Gastroenterol. 2008, 14, 1394–1398. [Google Scholar] [CrossRef]
- Hassan, S.E.; Bekarev, M.; Kim, M.Y.; Lin, J.; Piperdi, S.; Gorlick, R.; Geller, D.S. Cell surface receptor expression patterns in osteosarcoma. Cancer 2012, 118, 740–749. [Google Scholar] [CrossRef]
- Ishiwata, T.; Bergmann, U.; Kornmann, M.; Lopez, M.; Beger, H.G.; Korc, M. Altered expression of insulin-like growth factor II receptor in human pancreatic cancer. Pancreas 1997, 15, 367–373. [Google Scholar] [CrossRef]
- Heidegger, I.; Kern, J.; Ofer, P.; Klocker, H.; Massoner, P. Oncogenic functions of IGF1R and INSR in prostate cancer include enhanced tumor growth, cell migration and angiogenesis. Oncotarget 2014, 5, 2723–2735. [Google Scholar] [CrossRef]
- Vella, V.; Pandini, G.; Sciacca, L.; Mineo, R.; Vigneri, R.; Pezzino, V.; Belfiore, A. A novel autocrine loop involving IGF-II and the insulin receptor isoform-A stimulates growth of thyroid cancer. J. Clin. Endocrinol. Metab. 2002, 87, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Buck, E.; Gokhale, P.C.; Koujak, S.; Brown, E.; Eyzaguirre, A.; Tao, N.; Rosenfeld-Franklin, M.; Lerner, L.; Chiu, M.I.; Wild, R.; et al. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): Rationale for cotargeting IGF-1R and IR in cancer. Mol. Cancer Ther. 2010, 9, 2652–2664. [Google Scholar] [CrossRef] [PubMed]
- Ulanet, D.B.; Ludwig, D.L.; Kahn, C.R.; Hanahan, D. Insulin receptor functionally enhances multistage tumor progression and conveys intrinsic resistance to IGF-1R targeted therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 10791–10798. [Google Scholar] [CrossRef] [PubMed]
- Dinchuk, J.E.; Cao, C.; Huang, F.; Reeves, K.A.; Wang, J.; Myers, F.; Cantor, G.H.; Zhou, X.; Attar, R.M.; Gottardis, M.; et al. Insulin receptor (IR) pathway hyperactivity in IGF-IR null cells and suppression of downstream growth signaling using the dual IGF-IR/IR inhibitor, BMS-754807. Endocrinology 2010, 151, 4123–4132. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, C.; Manara, M.C.; Nicoletti, G.; Marino, M.T.; Lollini, P.L.; Astolfi, A.; Pandini, G.; López-Guerrero, J.A.; Schaefer, K.L.; Belfiore, A.; et al. Efficacy of and resistance to anti-IGF-1R therapies in Ewing’s sarcoma is dependent on insulin receptor signaling. Oncogene 2011, 30, 2730–2740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Pelzer, A.M.; Kiang, D.T.; Yee, D. Down-regulation of type I insulin-like growth factor receptor increases sensitivity of breast cancer cells to insulin. Cancer Res. 2007, 67, 391–397. [Google Scholar] [CrossRef]
- Mulvihill, M.J.; Cooke, A.; Rosenfeld-Franklin, M.; Buck, E.; Foreman, K.; Landfair, D.; O’Connor, M.; Pirritt, C.; Sun, Y.; Yao, Y.; et al. Discovery of OSI-906: A selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med. Chem. 2009, 1, 1153–1171. [Google Scholar] [CrossRef]
- Kheirandish, M.; Mahboobi, H.; Yazdanparast, M.; Kamal, W.; Kamal, M.A. Anti-cancer Effects of Metformin: Recent Evidences for its Role in Prevention and Treatment of Cancer. Curr. Drug Metab. 2018, 19, 793–797. [Google Scholar] [CrossRef]
- Vella, V.; Nicolosi, M.L.; Giuliano, S.; Bellomo, M.; Belfiore, A.; Malaguarnera, R. PPAR-γ Agonists As Antineoplastic Agents in Cancers with Dysregulated IGF Axis. Front. Endocrinol. 2017, 8, 31. [Google Scholar] [CrossRef]
- Chitnis, M.M.; Lodhia, K.A.; Aleksic, T.; Gao, S.; Protheroe, A.S.; Macaulay, V.M. IGF-1R inhibition enhances radiosensitivity and delays double-strand break repair by both non-homologous end-joining and homologous recombination. Oncogene 2014, 33, 5262–5273. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Bianco, R.; Damiano, V.; Gelardi, T.; Daniele, G.; Ciardiello, F.; Tortora, G. Rational combination of targeted therapies as a strategy to overcome the mechanisms of resistance to inhibitors of EGFR signaling. Curr. Pharm. Des. 2007, 13, 3358–3367. [Google Scholar] [CrossRef]
- Ciardiello, F.; Tortora, G. EGFR antagonists in cancer treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, T.; Chang, F.; Lorimer, D.; Smeekens, S.P.; Sawyers, C.L.; Pollak, M. In vivo progression of LAPC-9 and LNCaP prostate cancer models to androgen independence is associated with increased expression of insulin-like growth factor I (IGF-I) and IGF-I receptor (IGF-IR). Cancer Res. 2001, 61, 6276–6280. [Google Scholar] [PubMed]
- Yuan, T.A.; Yourk, V.; Farhat, A.; Guo, K.L.; Garcia, A.; Meyskens, F.L.; Liu-Smith, F. A Possible Link of Genetic Variations in ER/IGF1R Pathway and Risk of Melanoma. Int. J. Mol. Sci. 2020, 21, 1776. [Google Scholar] [CrossRef]
- Sanabria-Figueroa, E.; Donnelly, S.M.; Foy, K.C.; Buss, M.C.; Castellino, R.C.; Paplomata, E.; Taliaferro-Smith, L.; Kaumaya, P.T.; Nahta, R. Insulin-like growth factor-1 receptor signaling increases the invasive potential of human epidermal growth factor receptor 2-overexpressing breast cancer cells via Src-focal adhesion kinase and forkhead box protein M1. Mol. Pharmacol. 2015, 87, 150–161. [Google Scholar] [CrossRef]
- Qian, F.; Huo, D. Circulating Insulin-Like Growth Factor-1 and Risk of Total and 19 Site-Specific Cancers: Cohort Study Analyses from the UK Biobank. Cancer Epidemiol. Biomark. Prev. A Publ. Am. Assoc. Cancer Res. Cosponsored By Am. Soc. Prev. Oncol. 2020, 29, 2332–2342. [Google Scholar] [CrossRef]
- Fang, K.; Sun, M.; Leng, Z.; Chu, Y.; Zhao, Z.; Li, Z.; Zhang, Y.; Xu, A.; Zhang, Z.; Zhang, L.; et al. Targeting IGF1R signaling enhances the sensitivity of cisplatin by inhibiting proline and arginine metabolism in oesophageal squamous cell carcinoma under hypoxia. J. Exp. Clin. Cancer Res. CR 2023, 42, 73. [Google Scholar] [CrossRef]
- Peng, S.-W.; Ngo, M.-H.T.; Kuo, Y.-C.; Teng, M.-H.; Guo, C.-L.; Lai, H.-C.; Chang, T.-S.; Huang, Y.-H. Niclosamide Revitalizes Sorafenib through Insulin-like Growth Factor 1 Receptor (IGF-1R)/Stemness and Metabolic Changes in Hepatocellular Carcinoma. Cancers 2023, 15, 931. [Google Scholar] [CrossRef]
- Geoerger, B.; Brasme, J.F.; Daudigeos-Dubus, E.; Opolon, P.; Venot, C.; Debussche, L.; Vrignaud, P.; Vassal, G. Anti-insulin-like growth factor 1 receptor antibody EM164 (murine AVE1642) exhibits anti-tumour activity alone and in combination with temozolomide against neuroblastoma. Eur. J. Cancer 2010, 46, 3251–3262. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Gaikwad, S.M.; Jinager, A.; Chaudhury, S.; Maheshwari, A.; Ray, P. IGF-1R inhibition potentiates cytotoxic effects of chemotherapeutic agents in early stages of chemoresistant ovarian cancer cells. Cancer Lett. 2014, 354, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Dhadve, A.C.; Hari, K.; Rekhi, B.; Jolly, M.K.; De, A.; Ray, P. Decoding molecular interplay between RUNX1 and FOXO3a underlying the pulsatile IGF1R expression during acquirement of chemoresistance. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165754. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Jia, L.; Wang, T.; Zhang, Y.; Zhao, W.; Wang, X.; Chen, H. Trop2 binding IGF2R induces gefitinib resistance in NSCLC by remodeling the tumor microenvironment. J. Cancer 2021, 12, 5310–5319. [Google Scholar] [CrossRef] [PubMed]
- Flannery, C.A.; Rowzee, A.M.; Choe, G.H.; Saleh, F.L.; Radford, C.C.; Taylor, H.S.; Wood, T.L. Development of a Quantitative PCR Assay for Detection of Human Insulin-Like Growth Factor Receptor and Insulin Receptor Isoforms. Endocrinology 2016, 157, 1702–1708. [Google Scholar] [CrossRef]
- Arzalluz-Luque, Á.; Conesa, A. Single-cell RNAseq for the study of isoforms—How is that possible? Genome Biol. 2018, 19, 110. [Google Scholar] [CrossRef]
- Moruzzi, N.; Lazzeri-Barcelo, F. Insulin Receptor Isoforms in Physiology and Metabolic Disease. In Evolving Concepts in Insulin Resistance; IntechOpen: Rijeka, Croatia, 2022. [Google Scholar]
- DuBois, S.G.; Epling, C.L.; Tan, A.; Matthay, K.K.; Sinclair, E. Flow cytometric quantification of surface IGF-1R and intracellular p-AKT, p-S6, and p-ERK in Ewing sarcoma (EWS) cells. J. Clin. Oncol. 2012, 30 (Suppl. 30), 23. [Google Scholar] [CrossRef]
- Cao, Y.; Roth, M.; Piperdi, S.; Montoya, K.; Sowers, R.; Rao, P.; Geller, D.; Houghton, P.; Kolb, E.A.; Gill, J.; et al. Insulin-like growth factor 1 receptor and response to anti-IGF1R antibody therapy in osteosarcoma. PLoS ONE 2014, 9, e106249. [Google Scholar] [CrossRef]
- Zhang, H.; Zeng, X.; Li, Q.; Gaillard-Kelly, M.; Wagner, C.R.; Yee, D. Fluorescent tumour imaging of type I IGF receptor in vivo: Comparison of antibody-conjugated quantum dots and small-molecule fluorophore. Br. J. Cancer 2009, 101, 71–79. [Google Scholar] [CrossRef]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef]
- Khater, I.M.; Nabi, I.R.; Hamarneh, G. A Review of Super-Resolution Single-Molecule Localization Microscopy Cluster Analysis and Quantification Methods. Patterns 2020, 1, 100038. [Google Scholar] [CrossRef] [PubMed]
- Leibiger, B.; Moede, T.; Paschen, M.; Yunn, N.O.; Lim, J.H.; Ryu, S.H.; Pereira, T.; Berggren, P.O.; Leibiger, I.B. PI3K-C2α Knockdown Results in Rerouting of Insulin Signaling and Pancreatic Beta Cell Proliferation. Cell Rep. 2015, 13, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Jayasena, T.; Poljak, A.; Braidy, N.; Zhong, L.; Rowlands, B.; Muenchhoff, J.; Grant, R.; Smythe, G.; Teo, C.; Raftery, M.; et al. Application of Targeted Mass Spectrometry for the Quantification of Sirtuins in the Central Nervous System. Sci. Rep. 2016, 6, 35391. [Google Scholar] [CrossRef] [PubMed]
- Galal, M.A.; Abdel Jabar, M.; Zhra, M.; Abdel Rahman, A.M.; Aljada, A. Absolute quantification of senescence mediators in cells using multiple reaction monitoring liquid chromatography-Tandem mass spectrometry. Anal. Chim. Acta 2021, 1184, 339009. [Google Scholar] [CrossRef]




| Characteristics | IR-A | IR-B | IGF-1R | IR-A/IGF-1R | IR-B/IGFR | IGF-2R |
|---|---|---|---|---|---|---|
| Ligand-binding affinity | ||||||
| Pro-insulin | 4.5 ± 0.6 [49] | 31.0 ± 6.3 [49] | >100 [49] | _ | _ | |
| Insulin | 0.4 [50] − 2.7 [51] | 0.49 [50] − 2.6 [51] | >30 [52] − >1000 [50] | 1.1 [53] − 342 [54] | 1.1 [53] − 325 [54] | |
| IGF-I | >30 [52] | >30 [52] | 0.2 [52] − 1.49 [55] | 0.01 [53] − 6 [54] | 0.01 [53] − 12 [54] | |
| IGF-II | 3.3 [47] − 15 [56] | 36.0 ± 3.8 [47] | 0.6 [52] − 13 [10] | 0.18 [53] − 0.7 [57] | 0.19 − 15 [53] | 1.87 [58] |
| Distinctive structure | Absence of 12 AA | Presence of 12 AA encoded by Exon 11 | Formed by 21 exons | _ | _ | Lacks tyrosine kinase |
| Function | Embryogenesis and fetal development | Regulates glucose production and renal function of glomeruli and tubules. Because of its higher tyrosine kinase activity (2-folds), it is more involved in metabolic signaling [28] | Mediates apoptosis-inhibiting signals, and enhances cell metabolism and protein synthesis as well as tumor transformation and malignant cell survival | IGF-II scavenging, as well as regulating and trafficking lysosomal enzymes from the Golgi apparatus to lysosomes | ||
| Distribution in cells | More in brain cells, cancer cells, human placenta, osteoblasts precursor, spleen cells, and skeletal muscle | More in liver cells, mature osteoblasts, epididymal adipose tissue, kidney cells, and thyroid | Ubiquitous so it can be found in the kidney, ovary, and prostate | Ubiquitous and thus can be found in the liver, kidney, lung, adipose tissues, skeletal muscle, and placenta | ||
| Hormone-dependent cancer | |||||||
| Cancer | Cell type | IR | IR-A | IR-B | IR-A:IR-B ratio | IGFR | References |
| Breast | h-BC | ↑ | ↑ | ↓ | ↑ | _ | [174,175,176] |
| h-BC | ↑ | ↑ | ↓ | ↑ | ↓ | [175] | |
| Endometrial Adenocarcinoma | _ | ↑ | ↑ | ↑ | ↑ | [177] | |
| Ovarian | h-OV cell lines | _ | ↑ | ↑ | _ | _ | [178] |
| Prostate (Androgen-Dependent) | h-PC | ↑ | _ | _ | _ | _ | [179] |
| h-PC | ↑ | ↑ | ↑ | _ | _ | [180] | |
| h-PC | ↑ | _ | _ | ↑ | ↓ | [181] | |
| LNCaP and VCaP | ↑ | ↑ | ↓ | _ | _ | [182] | |
| Hormone-independent cancer | |||||||
| Cancer | Cell type | IR | IR-A | IR-B | IR-A:IR-B ratio | IGFR | References |
| Bladder | h-BLC specimens | ↑ | ↑ | _ | _ | _ | [183] |
| Colon | m-PCA, h-CC | ↓ | _ | ↓ | _ | _ | [184] |
| Liver | h-HCC | ↑ | ↑ | ↓ | ↑ | _ | [185] |
| r-HCC | ↑ | _ | _ | _ | ↑ | [186] | |
| m-HCC | ↑ | ↑ | _ | ↑ | ↓ | [187] | |
| Lung | h-NSCLC | _ | ↑ | ↓ | ↑ | _ | [188] |
| Osteosarcoma | h-OS | _ | ↑ | _ | _ | ↑ | [189] |
| Prostate (Androgen-Independent) | DU145 | _ | ↓ | ↑ | ↓ | _ | [182] |
| PC3 | _ | ↑ | ↓ | ↑ | _ | [182] | |
| Thyroid | h-TC | _ | ↑ | _ | _ | ↑ | [170] |
| Alteration of Transcription | ||||
|---|---|---|---|---|
| IR Dysregulated Expression Molecular Pathways | Function | Dysregulation Consequences | Cancer Type | References |
| Sp1, HMGA1, FOXO1 | IR transcription gene | IR upregulation | Breast, gastric, ovarian, liver, colorectal, prostate | [169,198,199] |
| p53 inactivation | Tumor suppression gene | IR upregulation | [25] | |
| Post-Transcriptional Dysregulation Due to Alternative Splicing Regulatory Factor and Non-Coding RNAs | ||||
| (A) Alternative Splicing Regulatory factor | ||||
| I. CUG-BP1 increase |
| Increased IRA:IRB ratio since it favors IRA expression | Breast, lung, colorectal | [22,25,200,201,202,203] |
| II. hRNP H increase |
| Increased IRA:IRB ratio | [25,204,205,206] | |
|
| Increased IRA:IRB ratio | NSCLC | [24,207] |
|
| Increased IRA:IRB ratio | Multiple myeloma, NSCLC, PDAC, HCC, breast | [208,209,210,211] |
| III. Loss of SRSF3 (Serine/arginine-rich protein 3)and SRp20 |
| Loss of SRSF3 and SRp20 causes increased IRA:IRB ratio since favoring IRA expression | HCC | [212,213,214] |
| IV. Muscleblind-like (MBNL) protein downregulation |
| When downregulated, IR-B levels decrease, increasing IRA:IRB ratio | Breast, HCC, lung, renal | [25,215,216] |
| V. mir-128 downregulation |
| Increased IRA:IRB ratio | Breast, melanoma, ALL | [217,218] |
| VI. mir-15b/16-2 downregulation |
| Increased IRA:IRB ratio | Lymphocytic leukemia | [219] |
| VII. mir-1 downregulation | Its deletion can affect cyclin D2 and IGF-R; thus, it controls the apoptosis of β cells | Increased IRA:IRB ratio | Bladder cancer | [220] |
| (B) Non-Coding RNAs | ||||
| Enhanced IRES-mediated IR mRNA translocation to the ribosomes | Initiates translocation | IR upregulation | [221] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galal, M.A.; Alouch, S.S.; Alsultan, B.S.; Dahman, H.; Alyabis, N.A.; Alammar, S.A.; Aljada, A. Insulin Receptor Isoforms and Insulin Growth Factor-like Receptors: Implications in Cell Signaling, Carcinogenesis, and Chemoresistance. Int. J. Mol. Sci. 2023, 24, 15006. https://doi.org/10.3390/ijms241915006
Galal MA, Alouch SS, Alsultan BS, Dahman H, Alyabis NA, Alammar SA, Aljada A. Insulin Receptor Isoforms and Insulin Growth Factor-like Receptors: Implications in Cell Signaling, Carcinogenesis, and Chemoresistance. International Journal of Molecular Sciences. 2023; 24(19):15006. https://doi.org/10.3390/ijms241915006
Chicago/Turabian StyleGalal, Mariam Ahmed, Samhar Samer Alouch, Buthainah Saad Alsultan, Huda Dahman, Nouf Abdullah Alyabis, Sarah Ammar Alammar, and Ahmad Aljada. 2023. "Insulin Receptor Isoforms and Insulin Growth Factor-like Receptors: Implications in Cell Signaling, Carcinogenesis, and Chemoresistance" International Journal of Molecular Sciences 24, no. 19: 15006. https://doi.org/10.3390/ijms241915006