
Loss of Key EMT-Regulating miRNAs Highlight the Role of ZEB1 in EGFR Tyrosine Kinase Inhibitor-Resistant NSCLC

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
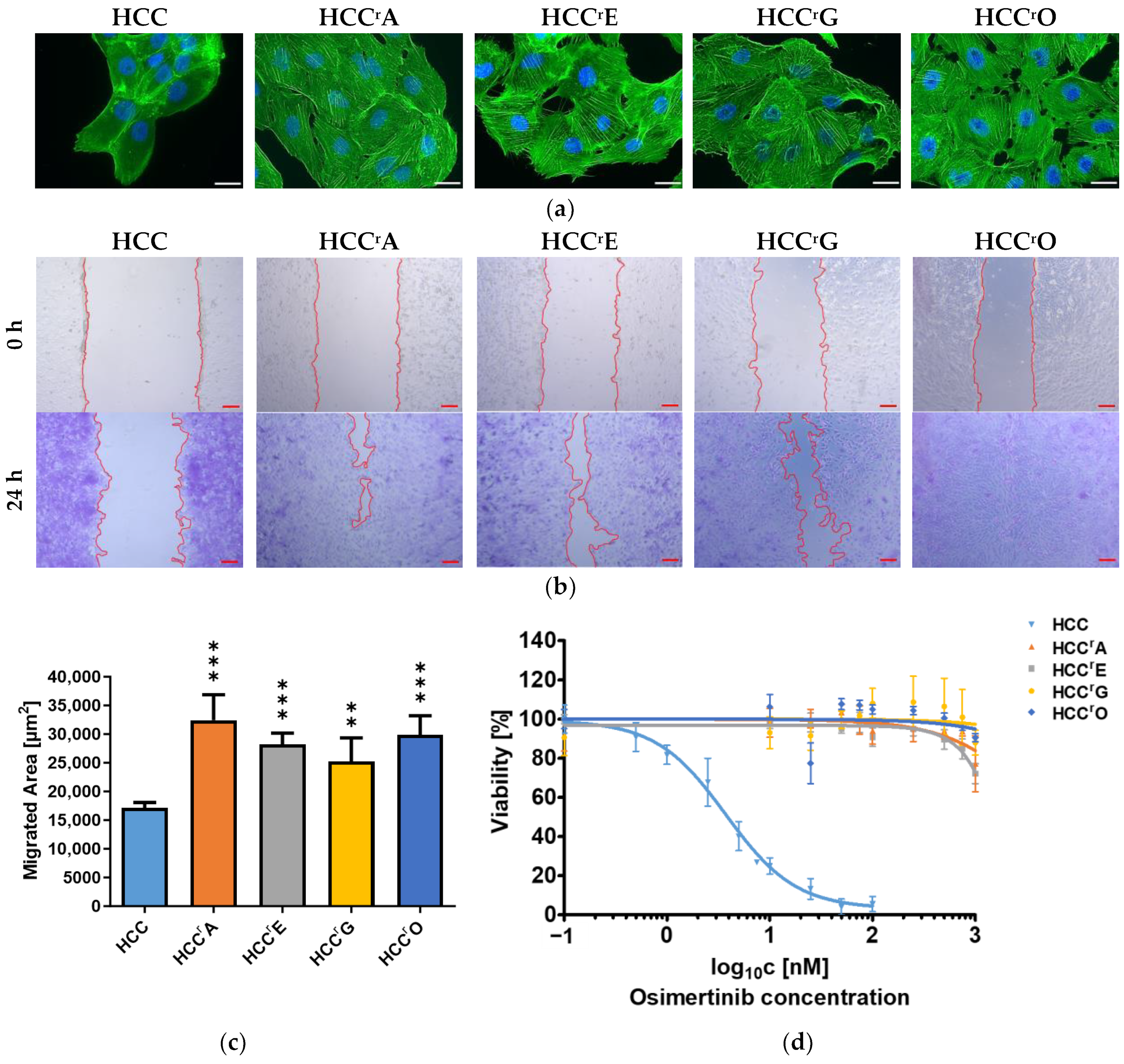
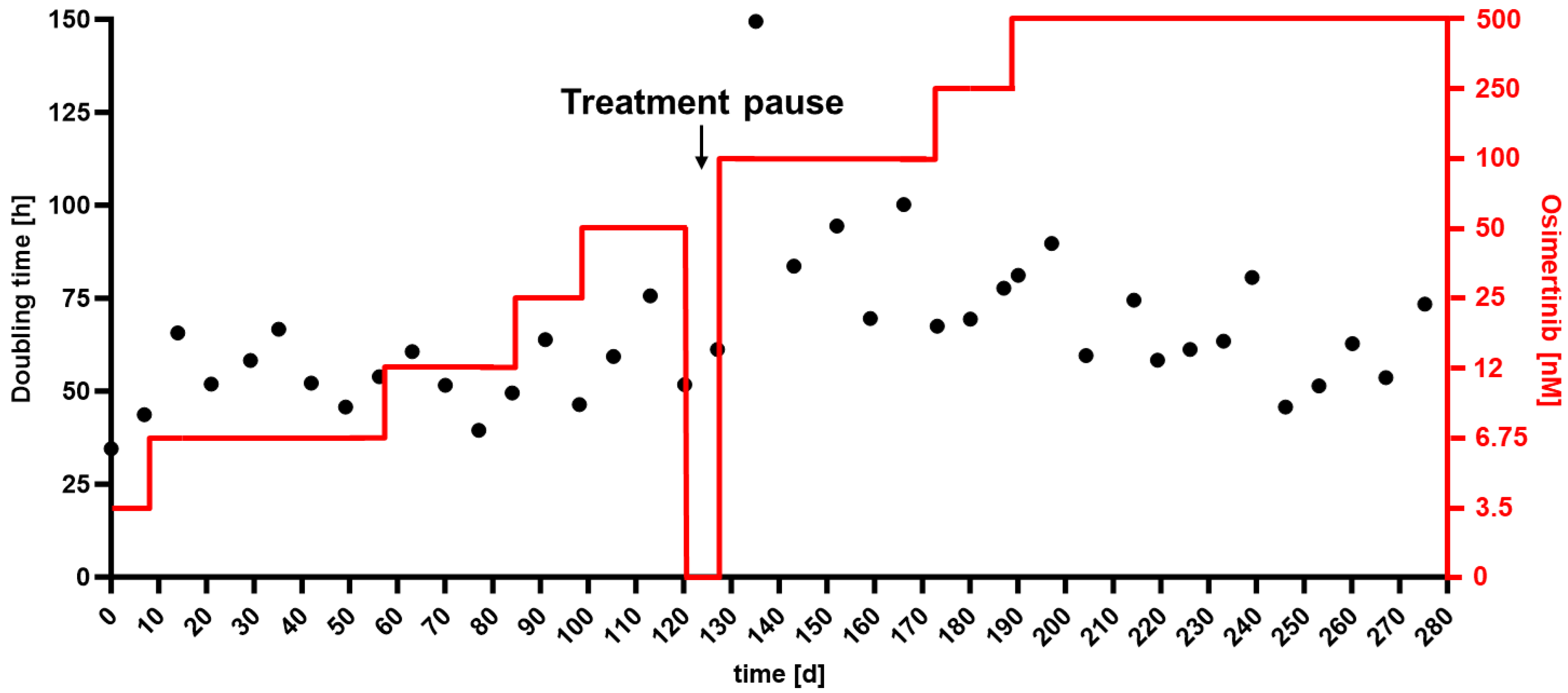
2.1. Drug Resistance Was Accompanied by EMT-Induced Phenotypic Changes and Increased Migration
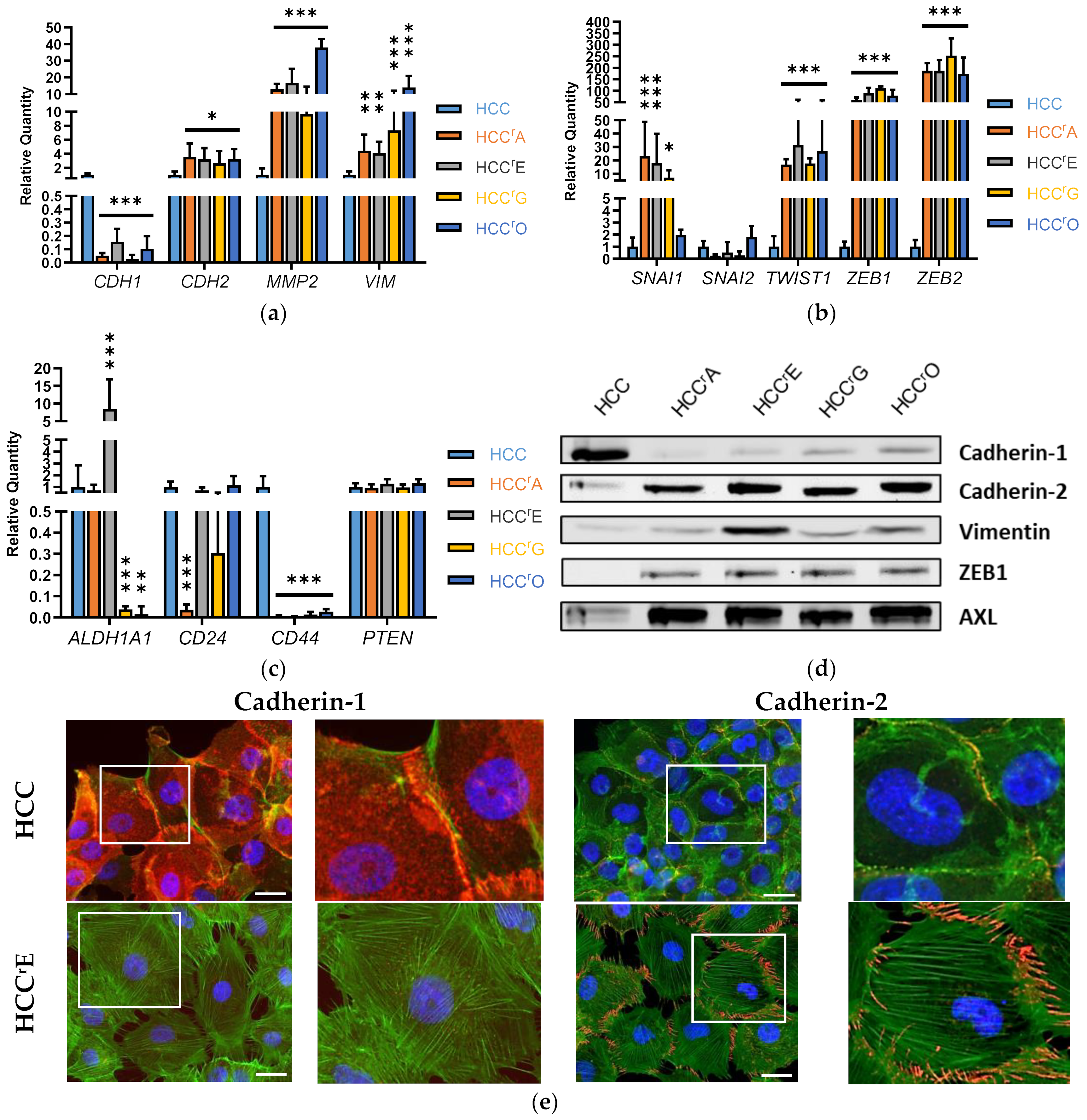
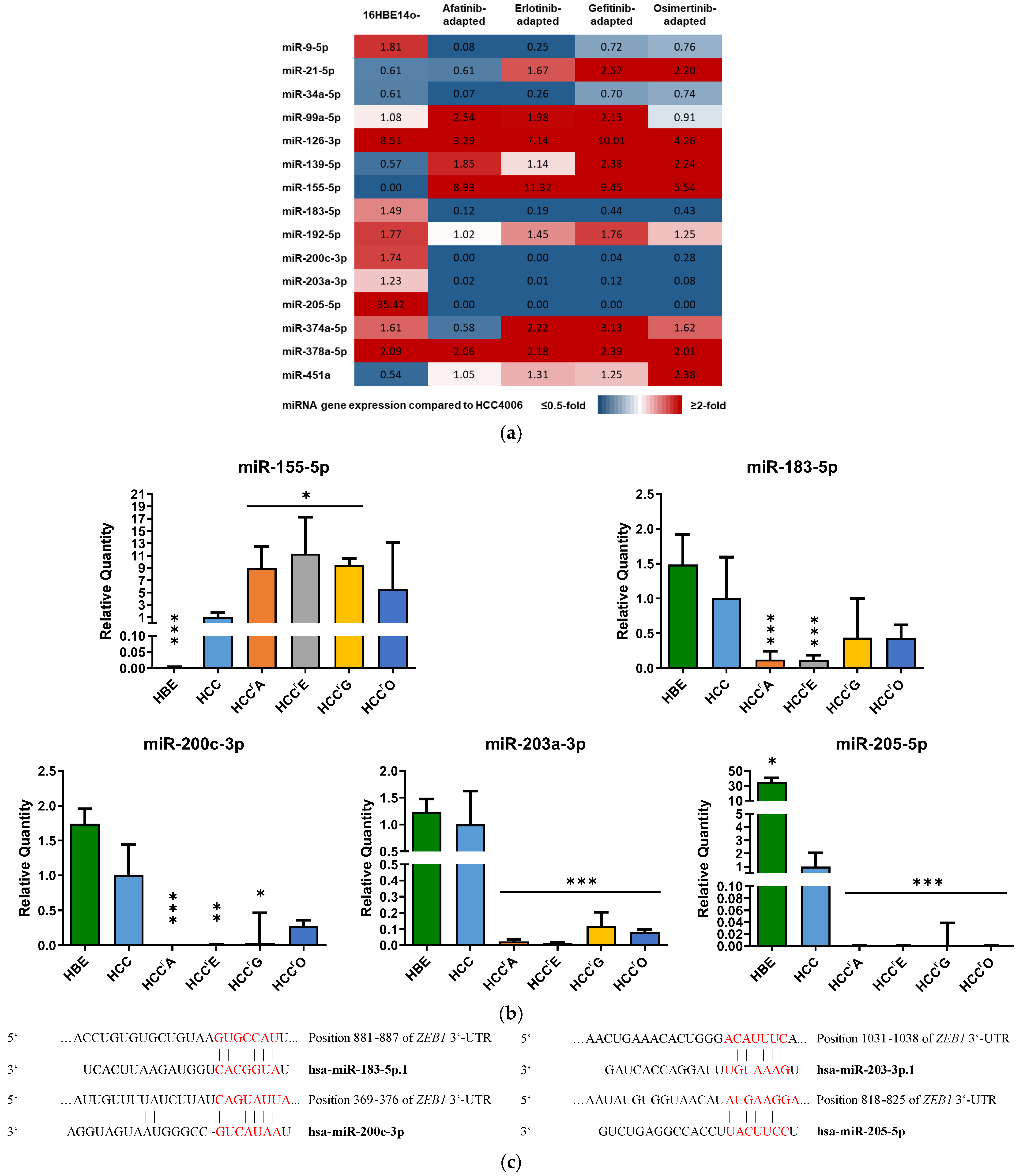
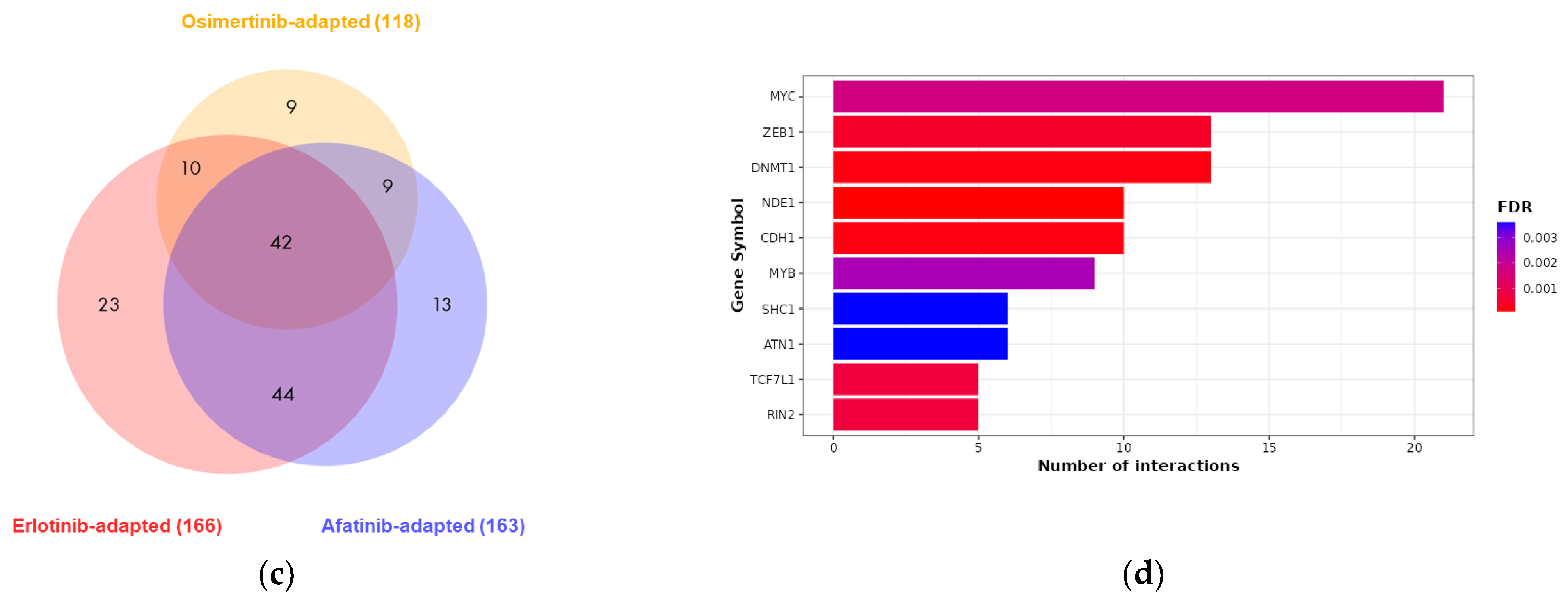
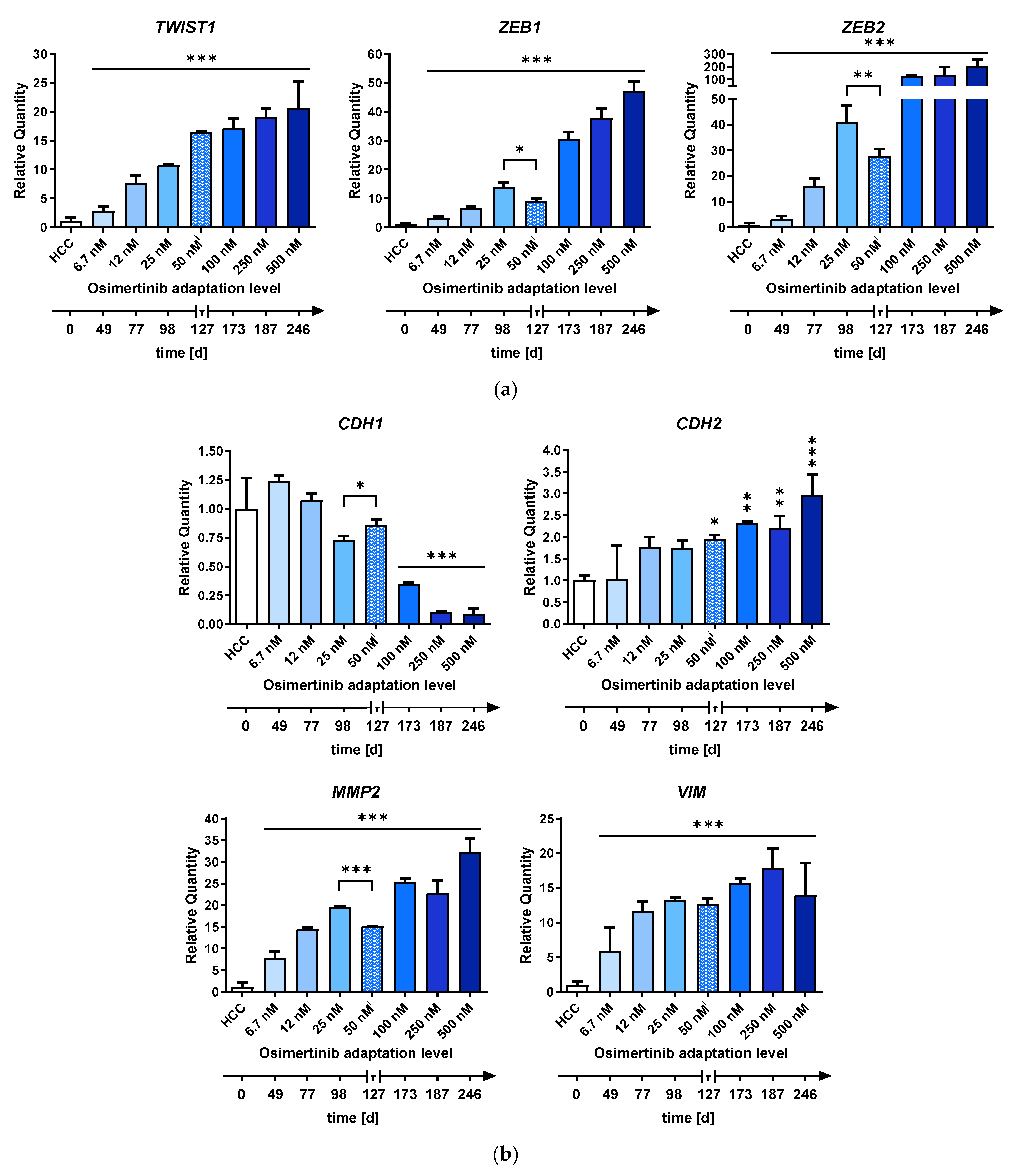
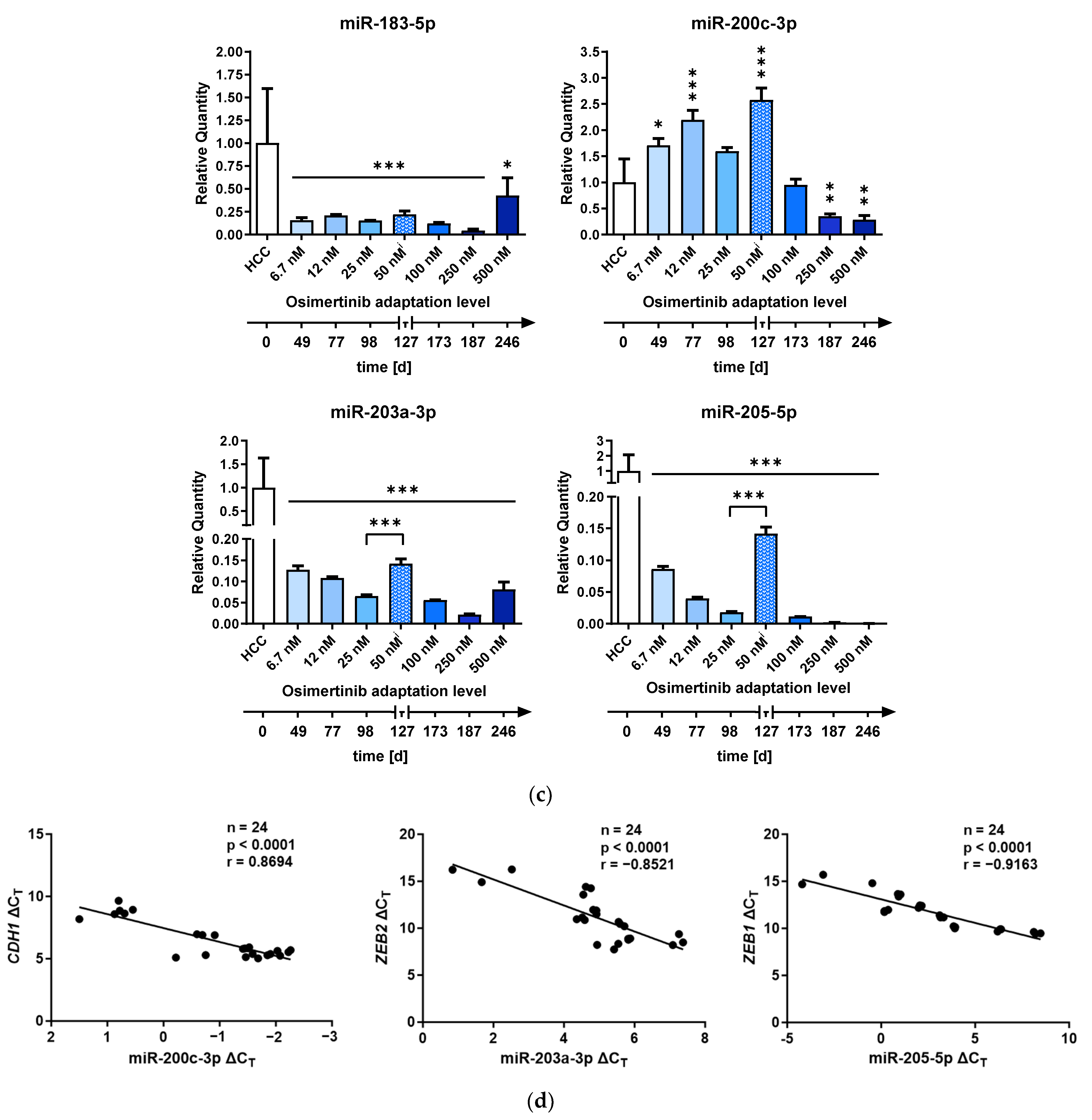
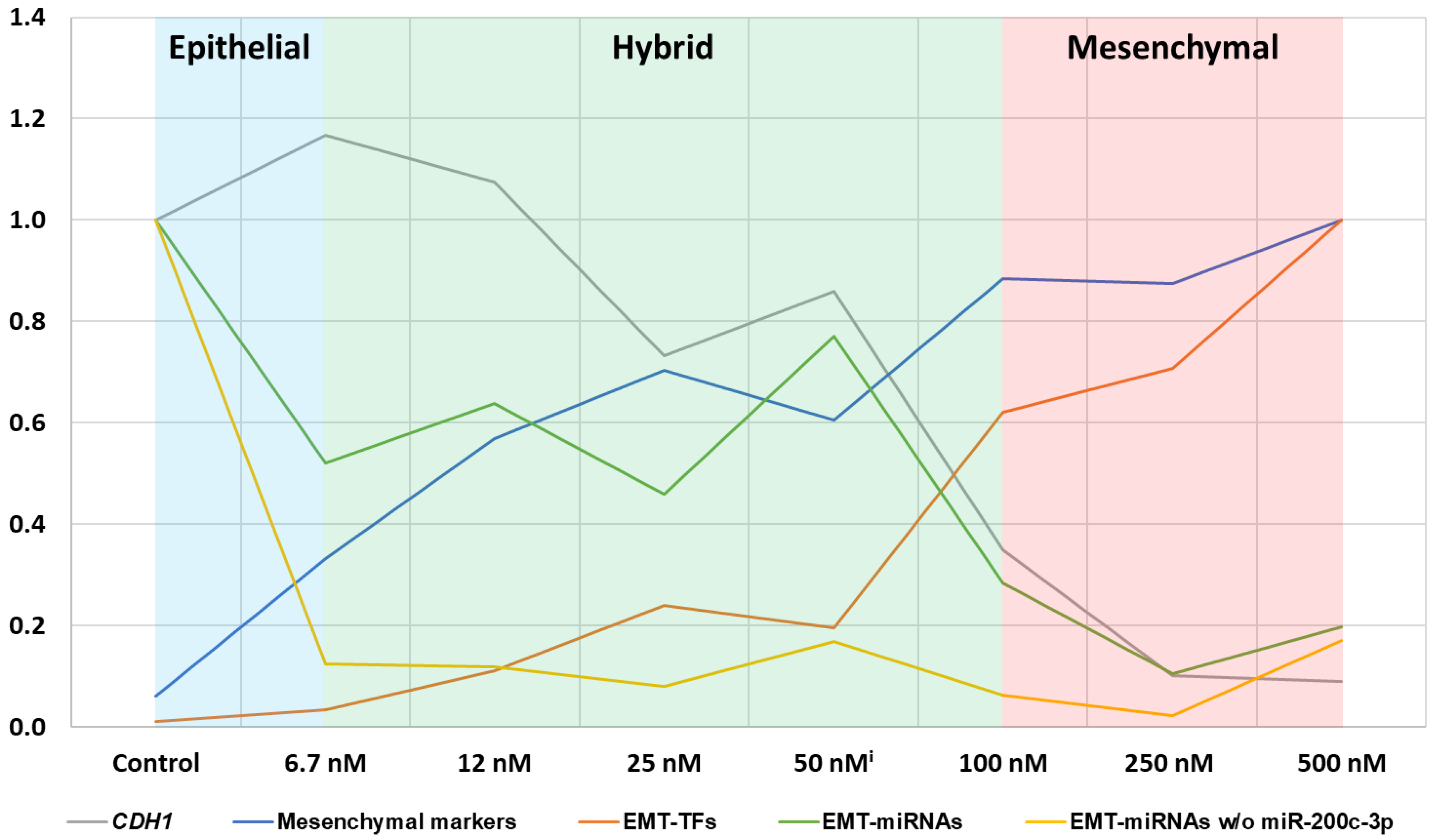
2.2. Differential Expression of EMT-Associated miRNAs Indicates ZEB1 as the Main Driver of EMT-Linked Drug Resistance
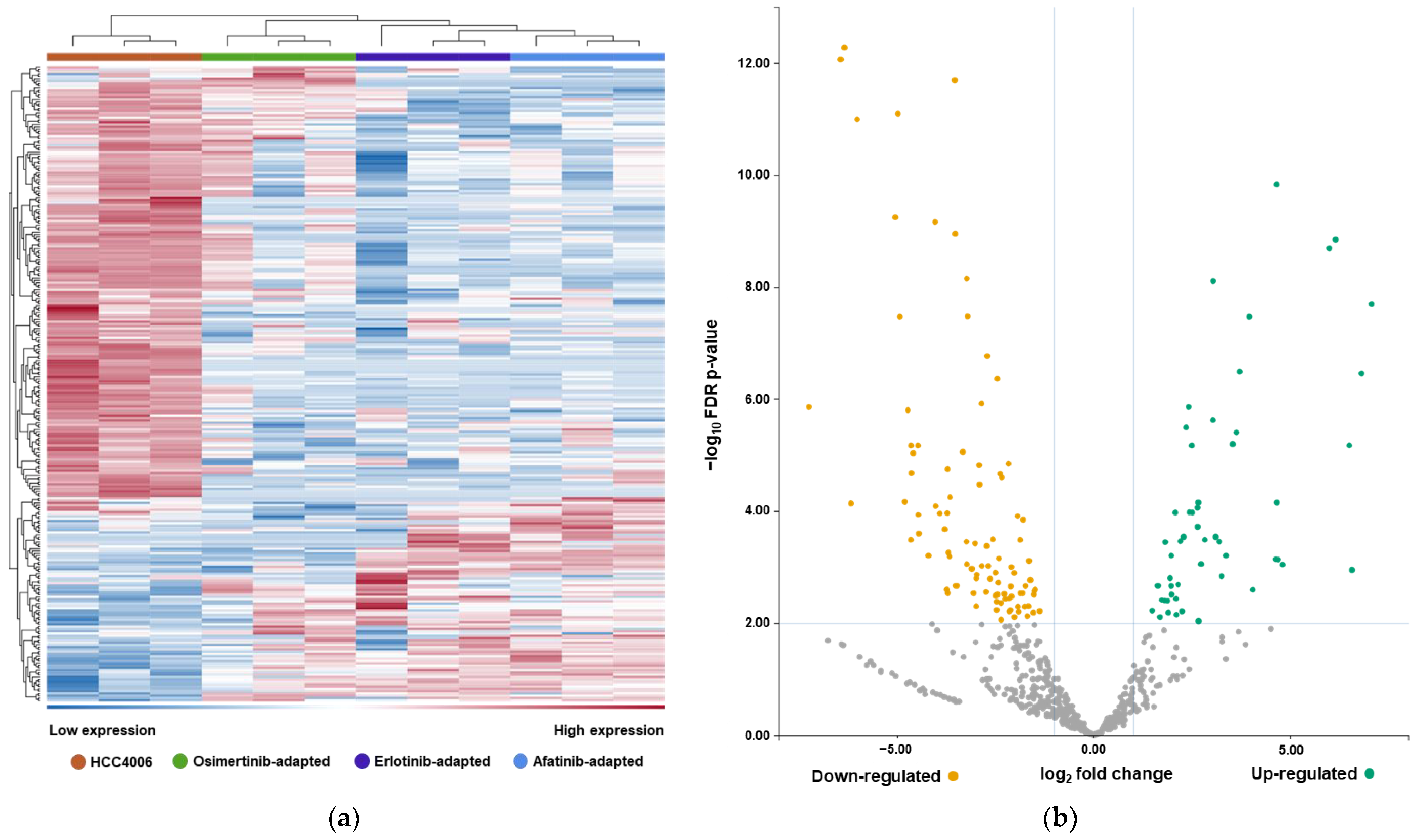
2.3. EMT-Associated miRNAs Were among the Highest-Ranked EV-Derived miRNAs Revealed by RNA-Sequencing
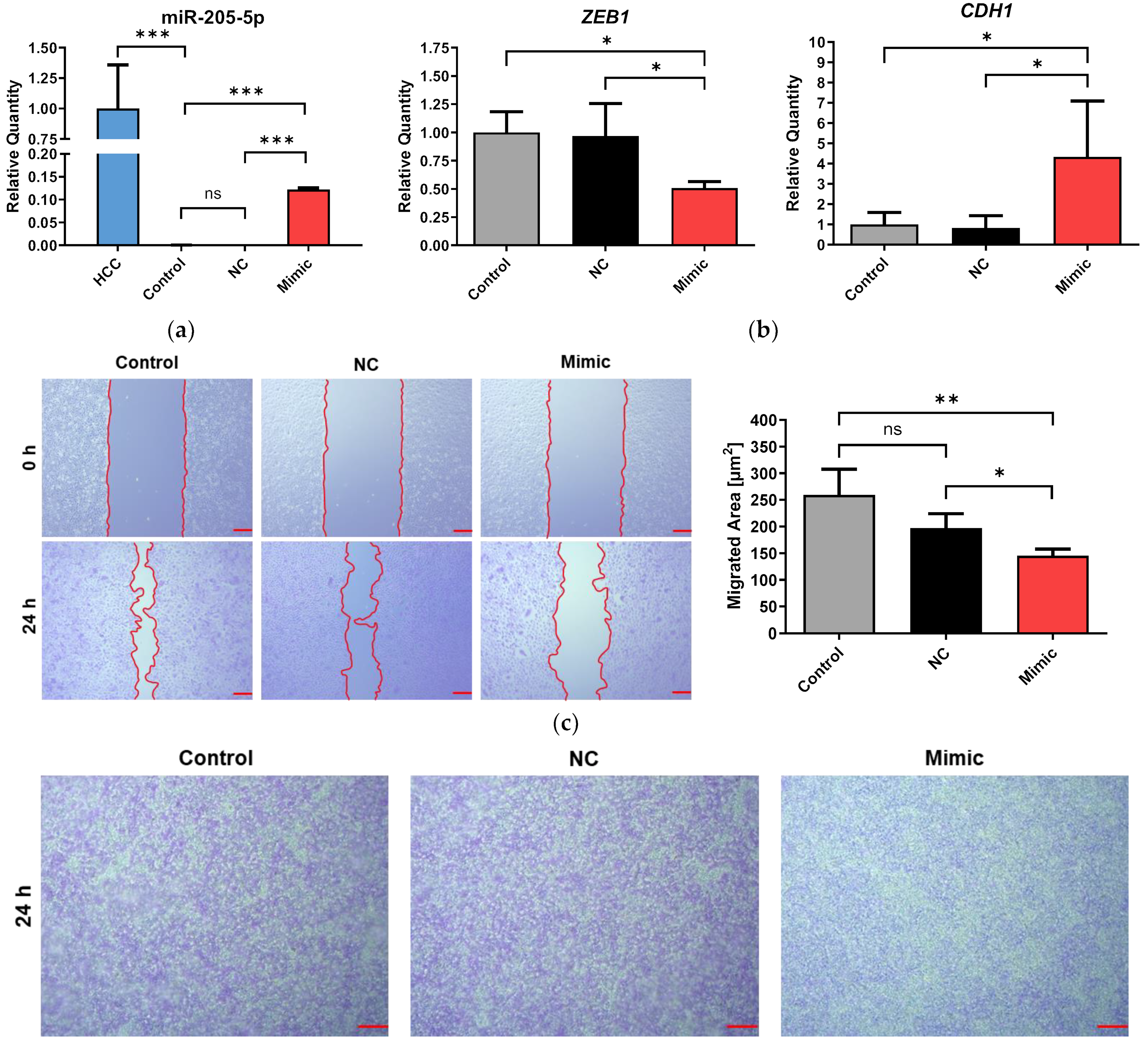
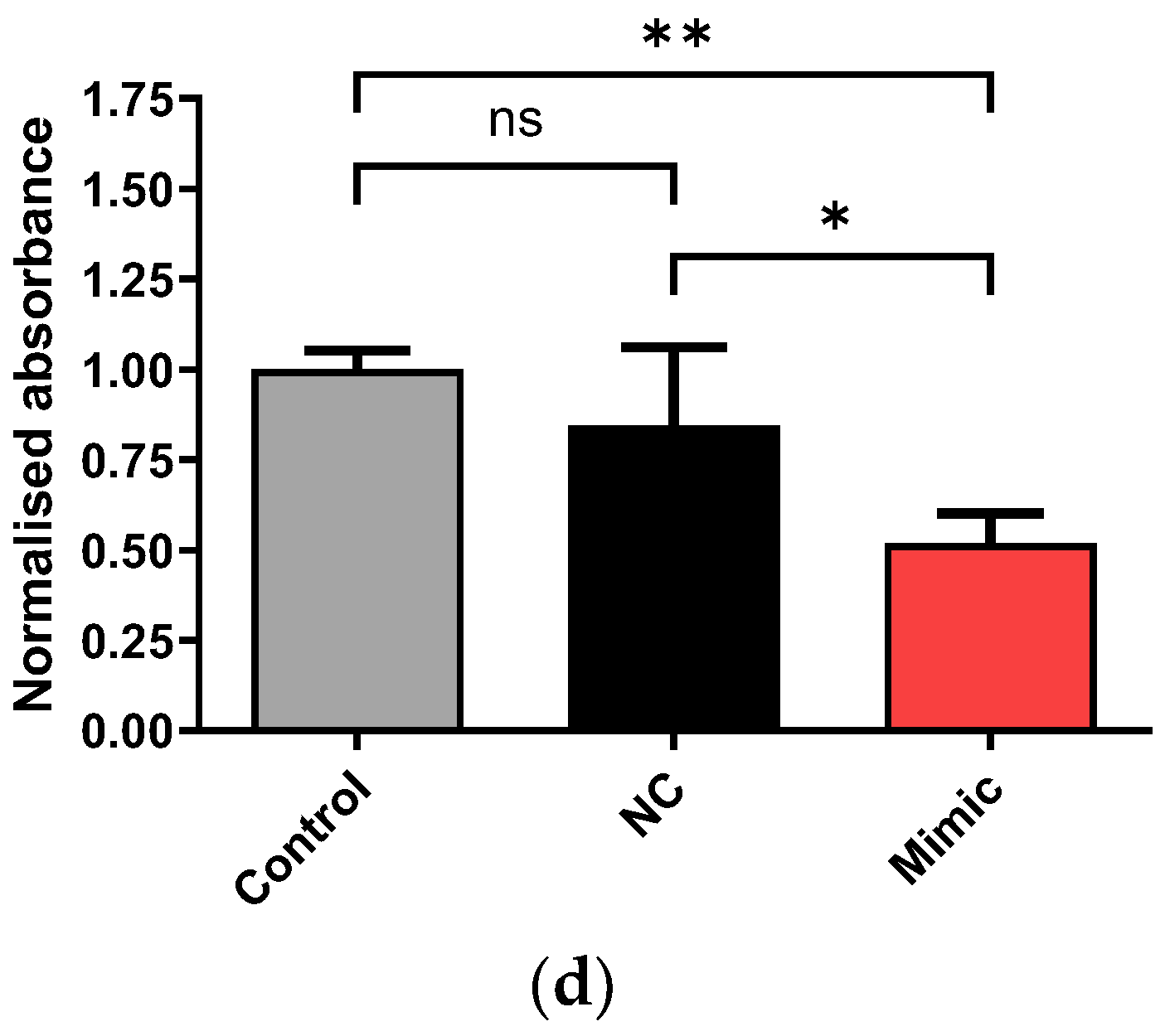
2.4. Transfection of Erlotinib-Adapted HCC4006 (HCCrE) Cells with a miR-205-5p Mimic Partly Reversed EMT
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Cell Proliferation Analysis
4.4. RNA Extraction and cDNA Synthesis
4.5. Quantitative Real-Time PCR
4.6. Western Immunoblotting
4.7. Immunofluorescent Microscopy
4.8. RNA Interference
4.9. Wound Healing Migration Assay
4.10. Transwell-Migration Assay
4.11. miRNA Isolation from Extracellular Vesicles
4.12. Size and Size Distribution of Extracellular Vesicles
4.13. RNA-Sequencing and Analysis
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Liao, B.-C.; Lin, C.-C.; Yang, J.C.-H. Second and third-generation epidermal growth factor receptor tyrosine kinase inhibitors in advanced nonsmall cell lung cancer. Curr. Opin. Oncol. 2015, 27, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Gazdar, A.F. Activating and resistance mutations of EGFR in non-small-cell lung cancer: Role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009, 28 (Suppl. 1), S24–S31. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Yang, J.C.-H.; Lee, C.K.; Kurata, T.; Kim, D.-W.; John, T.; Nogami, N.; Ohe, Y.; Mann, H.; Rukazenkov, Y.; et al. Osimertinib As First-Line Treatment of EGFR Mutation–Positive Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed]
- Tomasello, C.; Baldessari, C.; Napolitano, M.; Orsi, G.; Grizzi, G.; Bertolini, F.; Barbieri, F.; Cascinu, S. Resistance to EGFR inhibitors in non-small cell lung cancer: Clinical management and future perspectives. Crit. Rev. Oncol. /Hematol. 2018, 123, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Cuaran, S.; Scheffler, M.; Plenker, D.; Dahmen, L.; Scheel, A.H.; Fernandez-Cuesta, L.; Meder, L.; Lovly, C.M.; Persigehl, T.; Merkelbach-Bruse, S.; et al. Heterogeneous Mechanisms of Primary and Acquired Resistance to Third-Generation EGFR Inhibitors. Clin. Cancer Res. 2016, 22, 4837–4847. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yang, N.; Ou, Q.; Xiang, Y.; Jiang, T.; Wu, X.; Bao, H.; Tong, X.; Wang, X.; Shao, Y.W.; et al. Investigating Novel Resistance Mechanisms to Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib in Non–Small Cell Lung Cancer Patients. Clin. Cancer Res. 2018, 24, 3097–3107. [Google Scholar] [CrossRef]
- Wang, S.; Song, Y.; Yan, F.; Liu, D. Mechanisms of resistance to third-generation EGFR tyrosine kinase inhibitors. Front. Med. 2016, 10, 383–388. [Google Scholar] [CrossRef]
- Ricordel, C.; Friboulet, L.; Facchinetti, F.; Soria, J.C. Molecular mechanisms of acquired resistance to third-generation EGFR-TKIs in EGFR T790M-mutant lung cancer. Ann. Oncol. 2018, 29 (Suppl. 1), i28–i37. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, S.; Quader, S.; Cabral, H.; Ono, R. Interplay of EMT and CSC in Cancer and the Potential Therapeutic Strategies. Front. Pharmacol. 2020, 11, 904. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.-H.; Chen, L.-Y.; Lin, Y.-C.; Shih, J.-Y.; Lin, Y.-C.; Tseng, R.-Y.; Chiu, A.-C.; Yeh, Y.-H.; Liu, C.; Lin, Y.-T.; et al. Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene 2018, 38, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Tulchinsky, E.; Demidov, O.; Kriajevska, M.; Barlev, N.A.; Imyanitov, E. EMT: A mechanism for escape from EGFR-targeted therapy in lung cancer. Biochim. Biophys. Acta (BBA) Rev. Cancer 2018, 1871, 29–39. [Google Scholar] [CrossRef]
- Na Kim, B.; Ahn, D.H.; Kang, N.; Yeo, C.D.; Kim, Y.K.; Lee, K.Y.; Kim, T.-J.; Lee, S.H.; Park, M.S.; Yim, H.W.; et al. TGF-β induced EMT and stemness characteristics are associated with epigenetic regulation in lung cancer. Sci. Rep. 2020, 10, 10597. [Google Scholar] [CrossRef]
- Strauss, R.; Li, Z.-Y.; Liu, Y.; Beyer, I.; Persson, J.; Sova, P.; Möller, T.; Pesonen, S.; Hemminki, A.; Hamerlik, P.; et al. Analysis of epithelial and mesenchymal markers in ovarian cancer reveals phenotypic heterogeneity and plasticity. PLoS ONE 2011, 6, e16186. [Google Scholar] [CrossRef]
- Liao, T.-T.; Yang, M.-H. Hybrid Epithelial/Mesenchymal State in Cancer Metastasis: Clinical Significance and Regulatory Mechanisms. Cells 2020, 9, 623. [Google Scholar] [CrossRef]
- Haerinck, J.; Goossens, S.; Berx, G. The epithelial–mesenchymal plasticity landscape: Principles of design and mechanisms of regulation. Nat. Rev. Genet. 2023, 24, 590–609. [Google Scholar] [CrossRef] [PubMed]
- Andriani, F.; Bertolini, G.; Facchinetti, F.; Baldoli, E.; Moro, M.; Casalini, P.; Caserini, R.; Milione, M.; Leone, G.; Pelosi, G.; et al. Conversion to stem-cell state in response to microenvironmental cues is regulated by balance between epithelial and mesenchymal features in lung cancer cells. Mol. Oncol. 2015, 10, 253–271. [Google Scholar] [CrossRef]
- Aktas, B.; Tewes, M.; Fehm, T.; Hauch, S.; Kimmig, R.; Kasimir-Bauer, S. Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res. 2009, 11, R46. [Google Scholar] [CrossRef] [PubMed]
- Ntzifa, A.; Strati, A.; Kallergi, G.; Kotsakis, A.; Georgoulias, V.; Lianidou, E. Gene expression in circulating tumor cells reveals a dynamic role of EMT and PD-L1 during osimertinib treatment in NSCLC patients. Sci. Rep. 2021, 11, 2313. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Wu, K.-L.; Tsai, Y.-M.; Lien, C.-T.; Kuo, P.-L.; Hung, J.-Y. The Roles of MicroRNA in Lung Cancer. Int. J. Mol. Sci. 2019, 20, 1611. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.A.; Arora, S.; Prakasam, G.; Calin, G.A.; Syed, M.A. MicroRNA in lung cancer: Role, mechanisms, pathways and therapeutic relevance. Mol. Asp. Med. 2018, 70, 3–20. [Google Scholar] [CrossRef]
- Chen, Y.; Lu, L.; Feng, B.; Han, S.; Cui, S.; Chu, X.; Chen, L.; Wang, R. Non-coding RNAs as emerging regulators of epithelial to mesenchymal transition in non-small cell lung cancer. Oncotarget 2017, 8, 36787–36799. [Google Scholar] [CrossRef]
- Legras, A.; Pécuchet, N.; Imbeaud, S.; Pallier, K.; Didelot, A.; Roussel, H.; Gibault, L.; Fabre, E.; Le Pimpec-Barthes, F.; Laurent-Puig, P.; et al. Epithelial-to-Mesenchymal Transition and MicroRNAs in Lung Cancer. Cancers 2017, 9, 101. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Jian, Y. MiR-203a-3p regulates TGF-β1-induced epithelial–mesenchymal transition (EMT) in asthma by regulating Smad3 pathway through SIX1. Biosci. Rep. 2020, 40, BSR20192645. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Liu, A.Y.; Fan, C.; Zheng, H.; Li, Y.; Zhang, C.; Wu, S.; Yu, D.; Huang, Z.; Liu, F.; et al. MicroRNA-33b Inhibits Breast Cancer Metastasis by Targeting HMGA2, SALL4 and Twist1. Sci. Rep. 2015, 5, 9995. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Zhang, Y.; Xu, Q.; Guo, W.; Guo, A. miR-126 suppresses epithelial-to-mesenchymal transition and metastasis by targeting PI3K/AKT/Snail signaling of lung cancer cells. Oncol. Lett. 2018, 15, 7369–7375. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Ansieau, S.; Morel, A.-P.; Hinkal, G.; Bastid, J.; Puisieux, A. TWISTing an embryonic transcription factor into an oncoprotein. Oncogene 2010, 29, 3173–3184. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.E.; Nathan, V.; Osborne, J.K.; Farrow, R.K.; Deb, D.; Sullivan, J.P.; Dospoy, P.D.; Augustyn, A.; Hight, S.K.; Sato, M.; et al. ZEB1 drives epithelial-to-mesenchymal transition in lung cancer. J. Clin. Investig. 2016, 126, 3219–3235. [Google Scholar] [CrossRef]
- Jing, C.; Cao, H.; Qin, X.; Yu, S.; Wu, J.; Wang, Z.; Ma, R.; Feng, J. Exosome-mediated gefitinib resistance in lung cancer HCC827 cells via delivery of miR-21. Oncol. Lett. 2018, 15, 9811–9817. [Google Scholar] [CrossRef]
- Li, X.; Chen, C.; Wang, Z.; Liu, J.; Sun, W.; Shen, K.; Lv, Y.; Zhu, S.; Zhan, P.; Lv, T.; et al. Elevated exosome-derived miRNAs predict osimertinib resistance in non-small cell lung cancer. Cancer Cell Int. 2021, 21, 428. [Google Scholar] [CrossRef]
- Liu, C.; Kannisto, E.; Yu, G.; Yang, Y.; Reid, M.E.; Patnaik, S.K.; Wu, Y. Non-invasive Detection of Exosomal MicroRNAs via Tethered Cationic Lipoplex Nanoparticles (tCLN) Biochip for Lung Cancer Early Detection. Front. Genet. 2020, 11, 258. [Google Scholar] [CrossRef]
- Roa, W.H.; Kim, J.O.; Razzak, R.; Du, H.; Guo, L.; Singh, R.; Gazala, S.; Ghosh, S.; Wong, E.; Joy, A.A.; et al. Sputum microRNA profiling: A novel approach for the early detection of non-small cell lung cancer. Clin. Investig. Med. 2012, 35, 271–E281. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhu, J.; Lin, Y.; Zhang, Z.; Liu, J.; Wang, C.; Wu, H.; Zou, T. The potential diagnostic value of extracellular vesicle miRNA for human non-small cell lung cancer: A systematic review and meta-analysis. Expert Rev. Mol. Diagn. 2021, 21, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Liu, Z.; Todd, N.W.; Zhang, H.; Liao, J.; Yu, L.; Guarnera, M.A.; Li, R.; Cai, L.; Zhan, M.; et al. Diagnosis of lung cancer in individuals with solitary pulmonary nodules by plasma microRNA biomarkers. BMC Cancer 2011, 11, 374. [Google Scholar] [CrossRef] [PubMed]
- Smolarz, M.; Widlak, P. Serum Exosomes and Their miRNA Load—A Potential Biomarker of Lung Cancer. Cancers 2021, 13, 1373. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Yu, L.; Lin, X.; Zheng, Q.; Zhang, S.; Chen, D.; Pan, X.; Huang, Y. Combination of Serum miRNAs with Serum Exosomal miRNAs in Early Diagnosis for Non-Small-Cell Lung Cancer. Cancer Manag. Res. 2020, 12, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Zhou, K.; Zha, Y.; Chen, D.; He, J.; Ma, H.; Liu, X.; Le, H.; Zhang, Y. Diagnostic Value of Serum miR-182, miR-183, miR-210, and miR-126 Levels in Patients with Early-Stage Non-Small Cell Lung Cancer. PLoS ONE 2016, 11, e0153046. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Choe, G.; Jheon, S.; Sung, S.-W.; Lee, C.-T.; Chung, J.-H. CD24, a novel cancer biomarker, predicting disease-free survival of non-small cell lung carcinomas: A retrospective study of prognostic factor analysis from the viewpoint of forthcoming (seventh) new TNM classification. J. Thorac. Oncol. 2010, 5, 649–657. [Google Scholar] [CrossRef]
- Li, W.; Ma, H.; Zhang, J.; Zhu, L.; Wang, C.; Yang, Y. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci. Rep. 2017, 7, 13856. [Google Scholar] [CrossRef]
- Wang, J.; Nikhil, K.; Viccaro, K.; Chang, L.; White, J.; Shah, K. Phosphorylation-dependent regulation of ALDH1A1 by Aurora kinase A: Insights on their synergistic relationship in pancreatic cancer. BMC Biol. 2017, 15, 10. [Google Scholar] [CrossRef]
- Yin, J.; Zhang, H.; Wu, X.; Zhang, Y.; Li, J.; Shen, J.; Zhao, Y.; Xiao, Z.; Lu, L.; Huang, C.; et al. CD44 inhibition attenuates EGFR signaling and enhances cisplatin sensitivity in human EGFR wild-type non-small-cell lung cancer cells. Int. J. Mol. Med. 2020, 45, 1783–1792. [Google Scholar] [CrossRef]
- Hu, B.; Ma, Y.; Yang, Y.; Zhang, L.; Han, H.; Chen, J. CD44 promotes cell proliferation in non-small cell lung cancer. Oncol. Lett. 2018, 15, 5627–5633. [Google Scholar] [CrossRef] [PubMed]
- Perumal, E.; Youn, K.S.; Sun, S.; Seung-Hyun, J.; Suji, M.; Jieying, L.; Yeun-Jun, C. PTEN inactivation induces epithelial-mesenchymal transition and metastasis by intranuclear translocation of β-catenin and snail/slug in non-small cell lung carcinoma cells. Lung Cancer 2019, 130, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Persad, A.; Venkateswaran, G.; Hao, L.; Garcia, M.E.; Yoon, J.; Sidhu, J.; Persad, S. Active β-catenin is regulated by the PTEN/PI3 kinase pathway: A role for protein phosphatase PP2A. Genes Cancer 2016, 7, 368–382. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Huang, Y.; Gong, W. miR-205 promotes the growth, metastasis and chemoresistance of NSCLC cells by targeting PTEN. Oncol. Rep. 2013, 30, 2897–2902. [Google Scholar] [CrossRef] [PubMed]
- Asiedu, M.K.; Beauchamp-Perez, F.D.; Ingle, J.N.; Behrens, M.D.; Radisky, D.C.; Knutson, K.L. AXL induces epithelial-to-mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene 2013, 33, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Yamada, T.; Wang, R.; Tanimura, K.; Adachi, Y.; Nishiyama, A.; Tanimoto, A.; Takeuchi, S.; Araujo, L.H.; Boroni, M.; et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat. Commun. 2019, 10, 259. [Google Scholar] [CrossRef] [PubMed]
- Antony, J.; Huang, R.Y.-J. AXL-Driven EMT State as a Targetable Conduit in Cancer. Cancer Res 2017, 77, 3725–3732. [Google Scholar] [CrossRef]
- Tang, J.; Li, Y.; Wang, J.; Wen, Z.; Lai, M.; Zhang, H. Molecular mechanisms of microRNAs in regulating epithelial–mesenchymal transitions in human cancers. Cancer Lett. 2016, 371, 301–313. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, Z.; Liang, X. Regulatory Network Analysis to Reveal Important miRNAs and Genes in Non-Small Cell Lung Cancer. Cell J. 2019, 21, 459–466. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef]
- Liu, W.; Wang, X. Prediction of functional microRNA targets by integrative modeling of microRNA binding and target expression data. Genome Biol. 2019, 20, 18. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2019, 48, D127–D131. [Google Scholar] [CrossRef] [PubMed]
- Licursi, V.; Conte, F.; Fiscon, G.; Paci, P. MIENTURNET: An interactive web tool for microRNA-target enrichment and network-based analysis. BMC Bioinform. 2019, 20, 545. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-Y.; Lin, Y.-C.-D.; Cui, S.; Huang, Y.; Tang, Y.; Xu, J.; Bao, J.; Li, Y.; Wen, J.; Zuo, H.; et al. miRTarBase update 2022: An informative resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2022, 50, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Chmielecki, J.; Foo, J.; Oxnard, G.R.; Hutchinson, K.; Ohashi, K.; Somwar, R.; Wang, L.; Amato, K.R.; Arcila, M.; Sos, M.L.; et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci. Transl. Med. 2011, 3, 90ra59. [Google Scholar] [CrossRef] [PubMed]
- Mazza, V.; Cappuzzo, F. Treating EGFR mutation resistance in non-small cell lung cancer—Role of osimertinib. Appl. Clin. Genet. 2017, 10, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 2019, 121, 725–737. [Google Scholar] [CrossRef]
- Shen, S.; Vagner, S.; Robert, C. Persistent Cancer Cells: The Deadly Survivors. Cell 2020, 183, 860–874. [Google Scholar] [CrossRef]
- Suda, K.; Tomizawa, K.; Fujii, M.; Murakami, H.; Osada, H.; Maehara, Y.; Yatabe, Y.; Sekido, Y.; Mitsudomi, T. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired re-sistance to erlotinib. J. Thorac. Oncol. 2011, 6, 1152–1161. [Google Scholar] [CrossRef]
- Qin, Q.; Li, X.; Liang, X.; Zeng, L.; Wang, J.; Sun, L.; Zhong, D. Targeting the EMT transcription factor Snail overcomes resistance to osimertinib in EGFR-mutant non-small cell lung cancer. Thorac. Cancer 2021, 12, 1708–1715. [Google Scholar] [CrossRef]
- Okabe, H.; Aoki, K.; Yogosawa, S.; Saito, M.; Marumo, K.; Yoshida, K. Downregulation of CD24 suppresses bone metastasis of lung cancer. Cancer Sci. 2017, 109, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Gao, Y.; Cui, Y.; Zhang, T.; Cui, R.; Jiang, Y.; Shi, J. Overexpression of CD44 is associated with the occurrence and migration of non-small cell lung cancer. Mol. Med. Rep. 2016, 14, 3159–3167. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Song, L.; Bai, Y.; Kinose, F.; Li, J.; Ohaegbulam, K.C.; Muñoz-Antonia, T.; Qu, X.; Eschrich, S.; Uramoto, H.; et al. ZEB1 Mediates Acquired Resistance to the Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors in Non-Small Cell Lung Cancer. PLoS ONE 2016, 11, e0147344. [Google Scholar] [CrossRef] [PubMed]
- Yochum, Z.A.; Cades, J.; Wang, H.; Chatterjee, S.; Simons, B.W.; O’brien, J.P.; Khetarpal, S.K.; Lemtiri-Chlieh, G.; Myers, K.V.; Huang, E.H.-B.; et al. Targeting the EMT transcription factor TWIST1 overcomes resistance to EGFR inhibitors in EGFR-mutant non-small-cell lung cancer. Oncogene 2018, 38, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Gao, W. Overcoming acquired resistance of gefitinib in lung cancer cells without T790M by AZD9291 or Twist1 knockdown in vitro and in vivo. Arch. Toxicol. 2019, 93, 1555–1571. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Liu, Y.; Wang, Y.; Meng, M.; Wang, K.; Zang, X.; Zhao, S.; Sun, X.; Cui, L.; Pan, L.; et al. MiR-21 and MiR-155 promote non-small cell lung cancer progression by downregulating SOCS1, SOCS6, and PTEN. Oncotarget 2016, 7, 84508–84519. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Zhu, X.; Ma, J.; Wang, W. miR-205 regulates A549 cells proliferation by targeting PTEN. Int. J. Clin. Exp. Pathol. 2015, 8, 1175–1183. [Google Scholar]
- Zhang, P.; Lu, X.; Shi, Z.; Li, X.; Zhang, Y.; Zhao, S.; Liu, H. miR-205-5p regulates epithelial-mesenchymal transition by targeting PTEN via PI3K/AKT signaling pathway in cisplatin-resistant nasopharyngeal carcinoma cells. Gene 2019, 710, 103–113. [Google Scholar] [CrossRef]
- Namba, K.; Shien, K.; Takahashi, Y.; Torigoe, H.; Sato, H.; Yoshioka, T.; Takeda, T.; Kurihara, E.; Ogoshi, Y.; Yamamoto, H.; et al. Activation of AXL as a Preclinical Acquired Resistance Mechanism Against Osimertinib Treatment in EGFR-Mutant Non–Small Cell Lung Cancer Cells. Mol. Cancer Res. 2019, 17, 499–507. [Google Scholar] [CrossRef]
- Kim, D.; Bach, D.-H.; Fan, Y.-H.; Luu, T.-T.; Hong, J.-Y.; Park, H.J.; Lee, S.K. AXL degradation in combination with EGFR-TKI can delay and overcome acquired resistance in human non-small cell lung cancer cells. Cell Death Dis. 2019, 10, 361. [Google Scholar] [CrossRef]
- Lin, Q.; Zhou, C.R.; Bai, M.J.; Zhu, D.; Chen, J.W.; Wang, H.F.; Li, M.A.; Wu, C.; Li, Z.R.; Huang, M.S. Exosome-mediated miRNA delivery promotes liver cancer EMT and metastasis. Am. J. Transl. Res. 2020, 12, 1080–1095. [Google Scholar] [PubMed]
- Qin, X.; Yu, S.; Zhou, L.; Shi, M.; Hu, Y.; Xu, X.; Shen, B.; Liu, S.; Yan, D.; Feng, J. Cisplatin-resistant lung cancer cell–derived exosomes increase cisplatin resistance of recipient cells in exosomal miR-100–5p-dependent manner. Int. J. Nanomed. 2017, 12, 3721–3733. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Y.; Liu, Y.-N.; Wu, S.-G.; Hsu, C.-L.; Chang, T.-H.; Tsai, M.-F.; Lin, Y.-T.; Shih, J.-Y. MiR-200c-3p suppression is associated with development of acquired resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors in EGFR mutant non-small cell lung cancer via a mediating epithelial-to-mesenchymal transition (EMT) process. Cancer Biomark. 2020, 28, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Zhang, F.; Guo, Y.; Huang, J.; Xie, Y.; Yue, S.; Chen, M.; Jiang, H.; Li, M. miR-200c enhances sensitivity of drug-resistant non-small cell lung cancer to gefitinib by suppression of PI3K/Akt signaling pathway and inhibites cell migration via targeting ZEB1. Biomed. Pharmacother. 2017, 85, 113–119. [Google Scholar] [CrossRef]
- Jiang, Q.; Zhou, Y.; Yang, H.; Li, L.; Deng, X.; Cheng, C.; Xie, Y.; Luo, X.; Fang, W.; Liu, Z. A directly negative interaction of miR-203 and ZEB2 modulates tumor stemness and chemotherapy resistance in nasopharyngeal carcinoma. Oncotarget 2016, 7, 67288–67301. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Zhang, D.L.; Fu, C.; Wei, B.Z.; Li, G.J. MiR-183 modulates multi-drug resistance in hepatocellular cancer (HCC) cells via miR-183-IDH2/SOCS6-HIF-1α feedback loop. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2020–2027. [Google Scholar] [PubMed]
- Zarogoulidis, P.; Petanidis, S.; Kioseoglou, E.; Domvri, K.; Anestakis, D.; Zarogoulidis, K. MiR-205 and miR-218 expression is associated with carboplatin chemoresistance and regulation of apoptosis via Mcl-1 and Survivin in lung cancer cells. Cell. Signal. 2015, 27, 1576–1588. [Google Scholar] [CrossRef] [PubMed]
- Dinami, R.; Ercolani, C.; Petti, E.; Piazza, S.; Ciani, Y.; Sestito, R.; Sacconi, A.; Biagioni, F.; le Sage, C.; Agami, R.; et al. miR-155 drives telomere fragility in human breast cancer by targeting TRF1. Cancer Res 2014, 74, 4145–4156. [Google Scholar] [CrossRef]
- Lin, S.; Ruan, H.; Qin, L.; Zhao, C.; Gu, M.; Wang, Z.; Liu, B.; Wang, H.; Wang, J. Acquired resistance to EGFR-TKIs in NSCLC mediates epigenetic downregulation of MUC17 by facilitating NF-κB activity via UHRF1/DNMT1 complex. Int. J. Biol. Sci. 2023, 19, 832–851. [Google Scholar] [CrossRef]
- Liu, F.; Zhou, Y.; Zhou, D.; Kan, M.; Niu, X.; Zhang, Z.; Zhang, D.; Tao, L.; He, L.; Zhan, L.; et al. Whole DNA methylome profiling in lung cancer cells before and after epithelial-to-mesenchymal transition. Diagn. Pathol. 2014, 9, 66. [Google Scholar] [CrossRef]
- Sui, C.; Meng, F.; Li, Y.; Jiang, Y. miR-148b reverses cisplatin-resistance in non-small cell cancer cells via negatively regulating DNA (cytosine-5)-methyltransferase 1(DNMT1) expression. J. Transl. Med. 2015, 13, 132. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Dong, Z.; Gao, H.; Zhang, Y.-Y.; Shao, L.-H.; Jin, L.-L.; Lv, Y.-H.; Zhao, G.; Shen, Y.-N.; Jin, S.-Z. MicroRNA-9 enhanced radiosensitivity and its mechanism of DNA methylation in non-small cell lung cancer. Gene 2019, 710, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Sunaga, N.; Shames, D.S.; Toyooka, S.; Gazdar, A.F.; Minna, J.D. RNA interference-mediated knockdown of DNA methyltransferase 1 leads to promoter demethylation and gene re-expression in human lung and breast cancer cells. Cancer Res 2004, 64, 3137–3143. [Google Scholar] [CrossRef] [PubMed]
- Bu, X.; Zhang, X.; Xu, J.; Yang, H.; Zhou, X.; Wang, H.; Gong, L. Inhibition of DNA methyltransferase 1 by RNA interference reverses epithelial-mesenchymal transition in highly metastatic 95D lung cancer cells by inhibiting the Wnt signaling pathway. Oncol. Lett. 2018, 15, 9242–9250. [Google Scholar] [CrossRef] [PubMed]
- Khanna, A.; Pimanda, J.E.; Westermarck, J. Cancerous inhibitor of protein phosphatase 2A, an emerging human oncoprotein and a potential cancer therapy target. Cancer Res 2013, 73, 6548–6553. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-C.; Chen, H.-J.; Chang, Y.-L.; Liu, C.-Y.; Shiau, C.-W.; Cheng, A.-L.; Chen, K.-F. Inhibition of CIP2A determines erlotinib-induced apoptosis in hepatocellular carcinoma. Biochem. Pharmacol. 2013, 85, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Saafan, H.; Alahdab, A.; Michelet, R.; Gohlke, L.; Ziemann, J.; Holdenrieder, S.; McLaughlin, K.-M.; Wass, M.N.; Cinatl, J.; Michaelis, M.; et al. Constitutive Cell Proliferation Regulating Inhibitor of Protein Phosphatase 2A (CIP2A) Mediates Drug Resistance to Erlotinib in an EGFR Activating Mutated NSCLC Cell Line. Cells 2021, 10, 716. [Google Scholar] [CrossRef]
- Kaushik, V.; Yakisich, J.S.; Kulkarni, Y.; Azad, N.; Iyer, A.K.V. Chemoresistance of Lung Cancer Cells: 2D and 3D In Vitro Models for Anticancer Drug Screening. In Lung Cancer: Strategies for Diagnosis and Treatment; Fabiola Costa Torres, A., Ed.; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef]
- Han, H.; Xu, X. MiR-205 Promotes the Viability, Migration, and Tube Formation of Cervical Cancer Cells In Vitro by Targeting GATA3. Cancer Biother. Radiopharm. 2022, 37, 779–791. [Google Scholar] [CrossRef]
- Li, L.; Li, S. miR-205-5p inhibits cell migration and invasion in prostatic carcinoma by targeting ZEB1. Oncol. Lett. 2018, 16, 1715–1721. [Google Scholar] [CrossRef]
- Fan, Y.; Wang, K. miR-205 suppresses cell migration, invasion and EMT of colon cancer by targeting mouse double minute 4. Mol. Med. Rep. 2020, 22, 633–642. [Google Scholar] [CrossRef]
- Chen, W.; Kong, K.-K.; Xu, X.-K.; Chen, C.; Li, H.; Wang, F.-Y.; Peng, X.-F.; Zhang, Z.; Li, P.; Li, J.-L.; et al. Downregulation of miR-205 is associated with glioblastoma cell migration, invasion, and the epithelial-mesenchymal transition, by targeting ZEB1 via the Akt/mTOR signaling pathway. Int. J. Oncol. 2017, 52, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Malek, R.; Wang, H.; Taparra, K.; Tran, P.T. Therapeutic Targeting of Epithelial Plasticity Programs: Focus on the Epithelial-Mesenchymal Transition. Cells Tissues Organs 2017, 203, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Wang, P.; Toh, A.; Thompson, E.W. New Insights Into the Role of Phenotypic Plasticity and EMT in Driving Cancer Progression. Front. Mol. Biosci. 2020, 7, 71. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.-J.; Wong, M.K.; Tan, T.Z.; Kuay, K.T.; Ng, A.H.C.; Chung, V.Y.; Chu, Y.-S.; Matsumura, N.; Lai, H.-C.; Lee, Y.F.; et al. An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e-cadherin restoration by a src-kinase inhibitor, saracatinib (AZD0530). Cell Death Dis. 2013, 4, e915. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef]
- Lee, A.-F.; Chen, M.-C.; Chen, C.-J.; Yang, C.-J.; Huang, M.-S.; Liu, Y.-P. Reverse epithelial-mesenchymal transition contributes to the regain of drug sensitivity in tyrosine kinase inhibitor-resistant non-small cell lung cancer cells. PLoS ONE 2017, 12, e0180383. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Kotchetkov, R.; Cinatl, J.; Blaheta, R.; Vogel, J.-U.; Karaskova, J.; Squire, J.; Driever, P.H.; Klingebiel, T.; Cinatl, J. Development of resistance to vincristine and doxorubicin in neuroblastoma alters malignant properties and induces additional karyotype changes: A preclinical model. Int. J. Cancer 2002, 104, 36–43. [Google Scholar] [CrossRef]
- Balcells, I.; Cirera, S.; Busk, P.K. Specific and sensitive quantitative RT-PCR of miRNAs with DNA primers. BMC Biotechnol. 2011, 11, 70. [Google Scholar] [CrossRef]
- Andersen, C.L.; Jensen, J.L.; Ørntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper--Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Thery, C.; Clayton, A.; Amigorena, S.; Raposo, G. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 3, 3–22. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Fold Change | FDR p-Value | p-Value | Up-/Down- Regulation |
|---|---|---|---|---|
| hsa-miR-205-5p | −151.9 | 1.37 × 106 | 5.34 × 108 | Down |
| hsa-miR-200b-3p | −87.6 | 8.51 × 1013 | 4.07 × 1015 | Down |
| hsa-miR-203a-3p | −85.7 | 8.51 × 1013 | 3.10 × 1015 | Down |
| hsa-miR-200a-5p | −81.0 | 5.27 × 1013 | 8.40 × 1016 | Down |
| hsa-miR-655-3p | −72.4 | 7.23 × 105 | 5.53 × 106 | Down |
| hsa-miR-375-3p | −64.9 | 9.99 × 1012 | 9.56 × 1014 | Down |
| hsa-miR-432-5p | −33.1 | 5.63 × 1010 | 7.19 × 1012 | Down |
| hsa-miR-431-3p | −31.6 | 7.96 × 1012 | 6.35 × 1014 | Down |
| hsa-miR-429 | −30.6 | 3.35 × 108 | 9.61 × 1010 | Down |
| hsa-miR-2276-3p | −28.1 | 6.74 × 105 | 4.84 × 106 | Down |
| hsa-miR-99a-5p | 133.2 | 1.99 × 108 | 4.76 × 1010 | Up |
| hsa-miR-1246 | 111.0 | 3.44 × 107 | 1.15 × 108 | Up |
| hsa-miR-767-5p | 93.8 | 1.13 × 103 | 1.63 × 104 | Up |
| hsa-miR-1290 | 89.5 | 6.71 × 106 | 3.32 × 107 | Up |
| hsa-miR-4508 | 70.6 | 1.41 × 109 | 2.48 × 1011 | Up |
| hsa-miR-9901 | 63.2 | 2.00 × 109 | 3.83 × 1011 | Up |
| hsa-miR-4497 | 27.8 | 9.06 × 104 | 1.24 × 104 | Up |
| hsa-miR-4455 | 25.6 | 7.32 × 104 | 9.58 × 105 | Up |
| hsa-miR-99a-3p | 25.1 | 6.96 × 105 | 5.22 × 106 | Up |
| hsa-miR-3182 | 25.0 | 1.45 × 1010 | 1.62 × 1012 | Up |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gohlke, L.; Alahdab, A.; Oberhofer, A.; Worf, K.; Holdenrieder, S.; Michaelis, M.; Cinatl, J., Jr.; Ritter, C.A. Loss of Key EMT-Regulating miRNAs Highlight the Role of ZEB1 in EGFR Tyrosine Kinase Inhibitor-Resistant NSCLC. Int. J. Mol. Sci. 2023, 24, 14742. https://doi.org/10.3390/ijms241914742
Gohlke L, Alahdab A, Oberhofer A, Worf K, Holdenrieder S, Michaelis M, Cinatl J Jr., Ritter CA. Loss of Key EMT-Regulating miRNAs Highlight the Role of ZEB1 in EGFR Tyrosine Kinase Inhibitor-Resistant NSCLC. International Journal of Molecular Sciences. 2023; 24(19):14742. https://doi.org/10.3390/ijms241914742
Chicago/Turabian StyleGohlke, Linus, Ahmad Alahdab, Angela Oberhofer, Karolina Worf, Stefan Holdenrieder, Martin Michaelis, Jindrich Cinatl, Jr., and Christoph A Ritter. 2023. "Loss of Key EMT-Regulating miRNAs Highlight the Role of ZEB1 in EGFR Tyrosine Kinase Inhibitor-Resistant NSCLC" International Journal of Molecular Sciences 24, no. 19: 14742. https://doi.org/10.3390/ijms241914742