
Genetic Control of GCF Exudation: Innate Immunity Genes and Periodontitis Susceptibility




Abstract
:1. Introduction
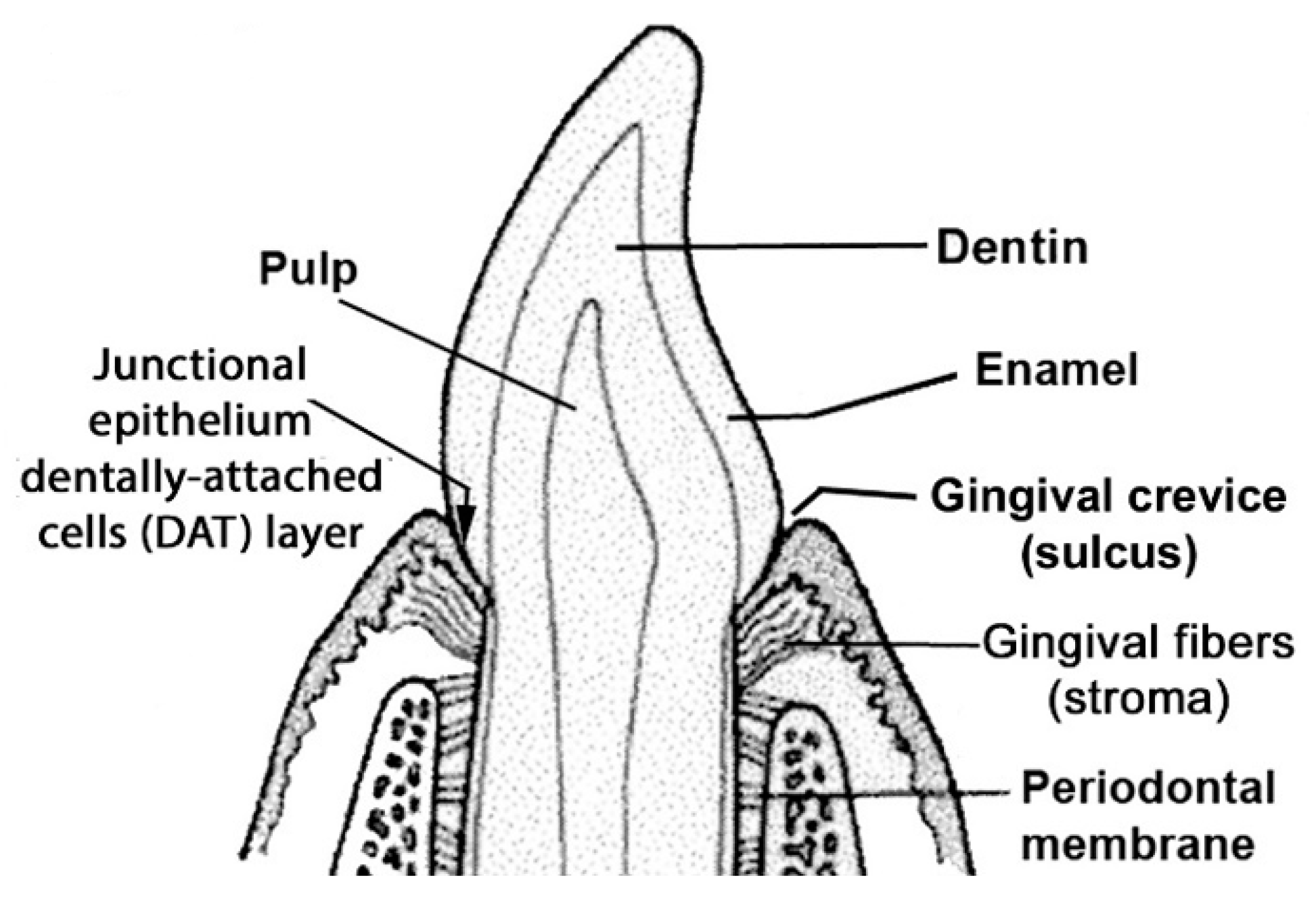
1.1. Periodontal Disease
1.2. Evidence for Strong and Weak Inflammatory Responses to Oral Bacteria in EG

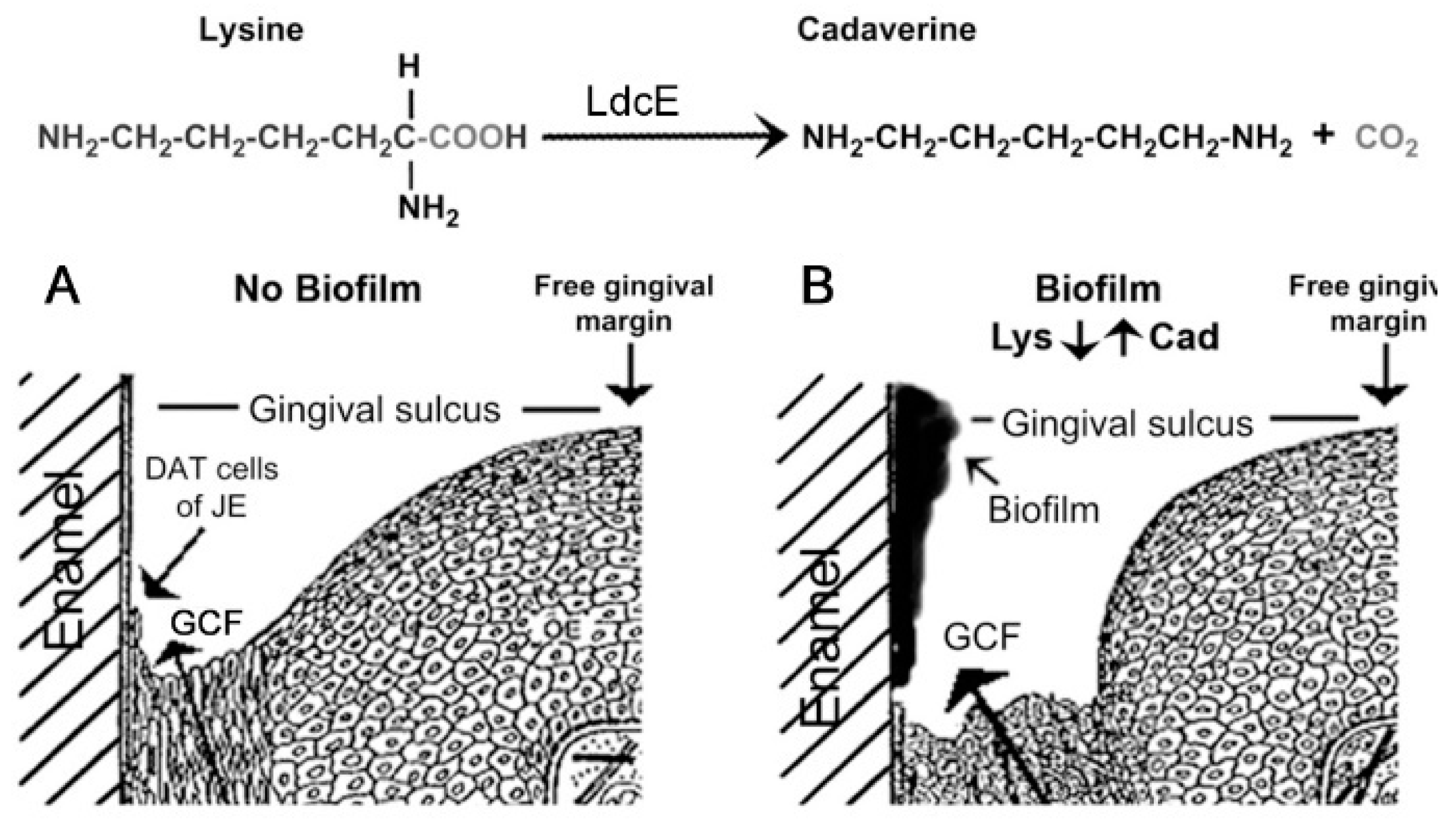
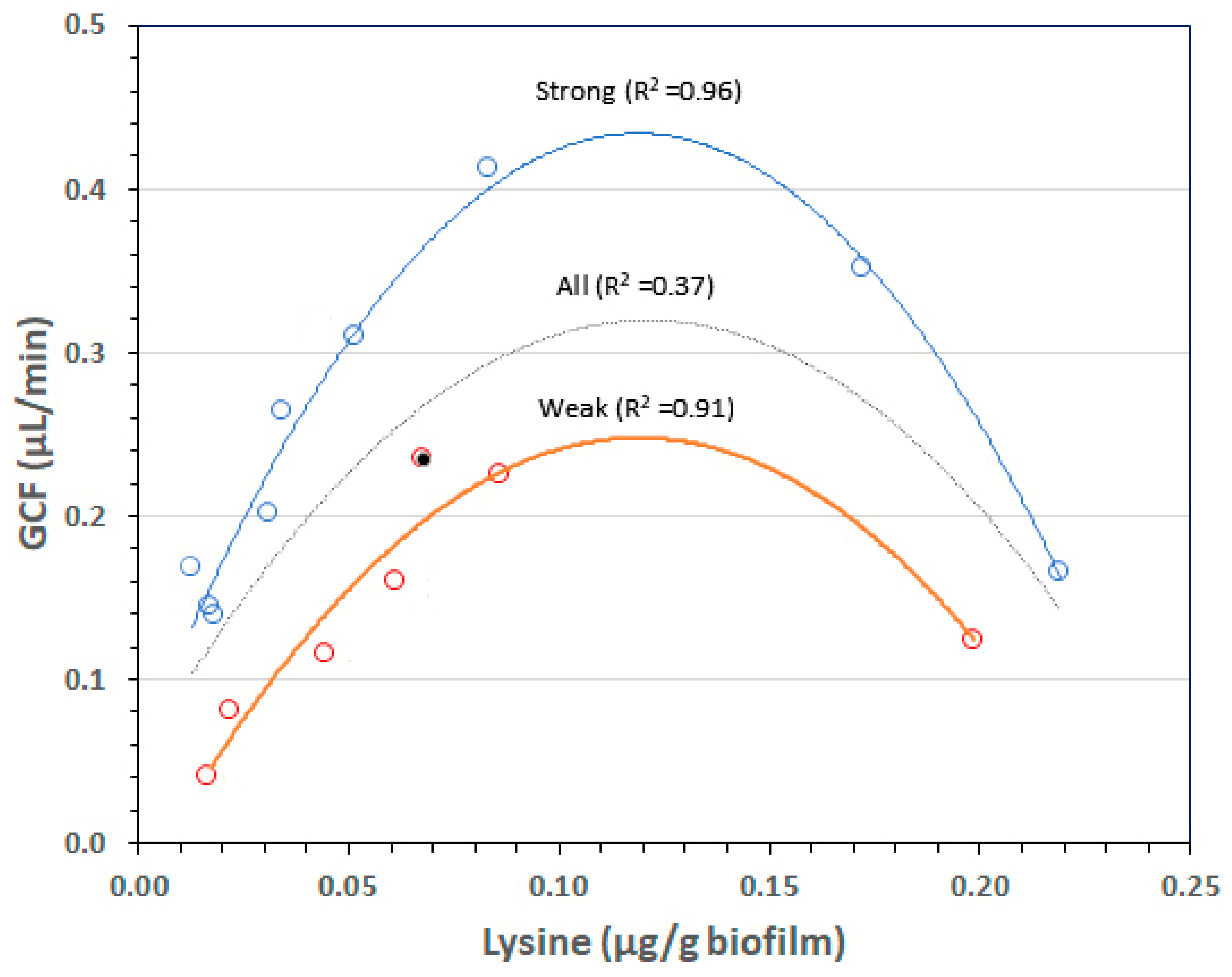
1.3. Biofilm Lysine Concentrations Determine GCF Responses after a Week of EG
1.4. Rationale and Aim
2. Results
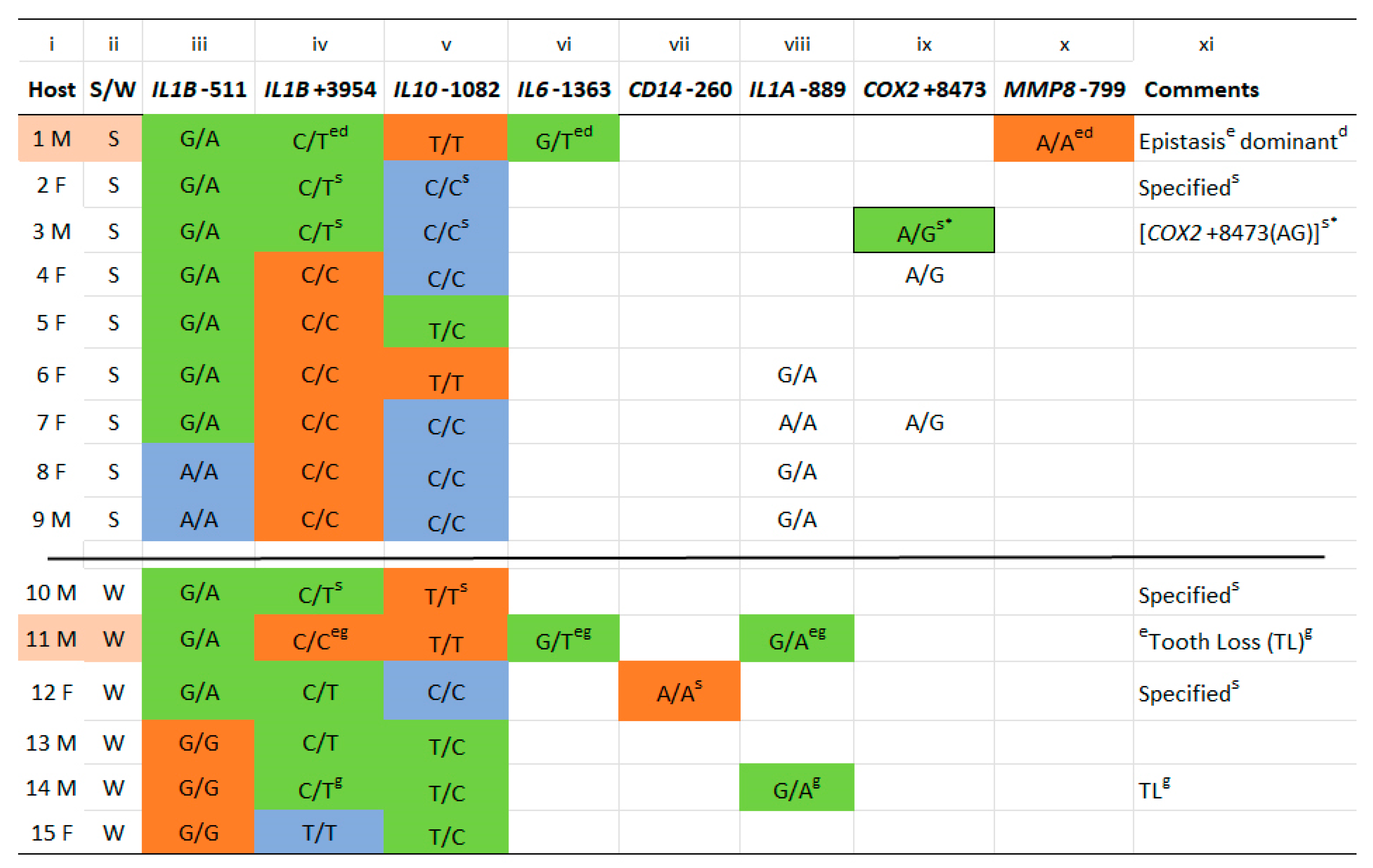
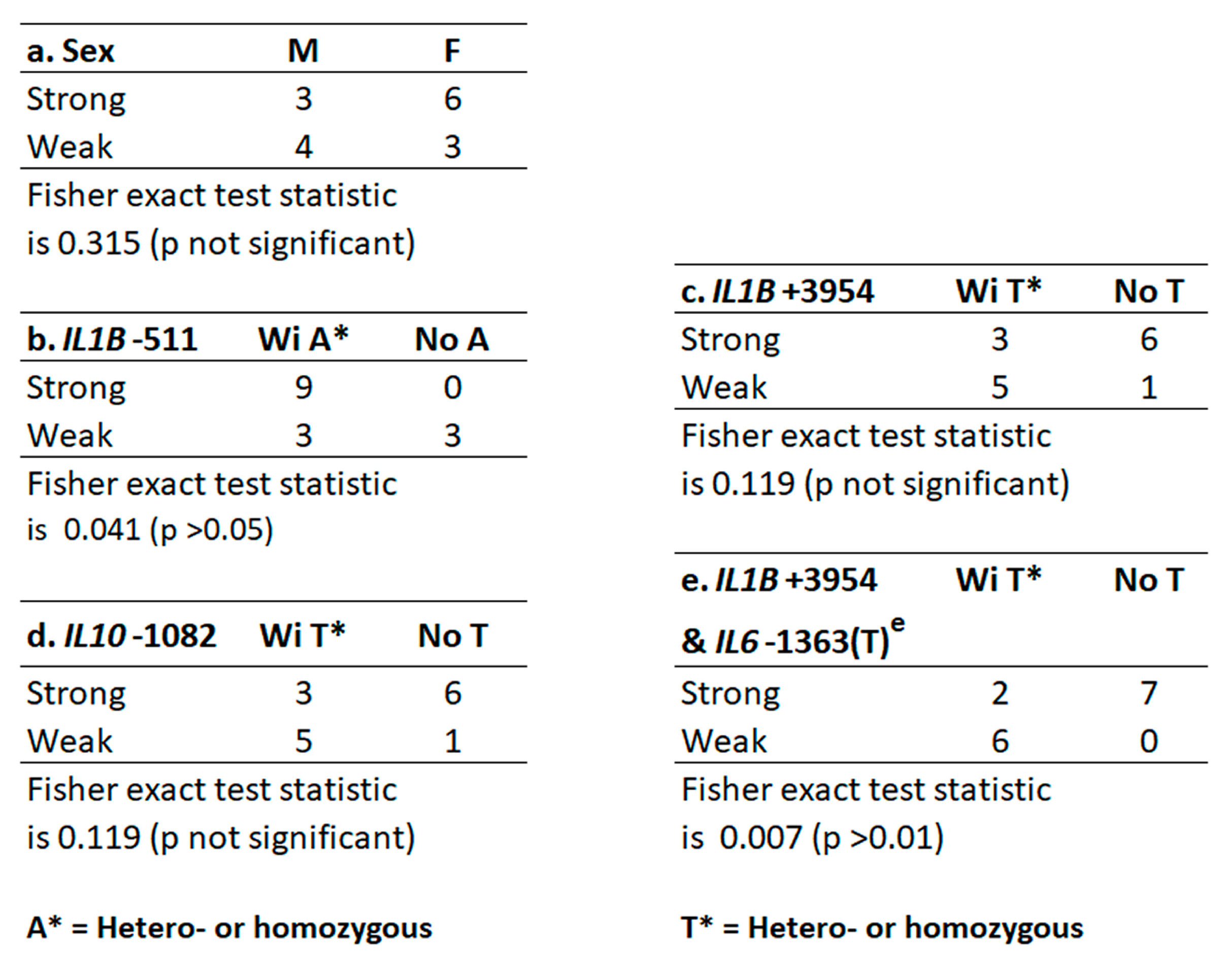
2.1. Host Gene Identification, Allele Distribution, and Relationship to Strength of GCF Exudation
2.2. IL6-1363(T) Acts on Alleles at IL1B+3954 to Reverse the Expected GCF Response (Epistasis)
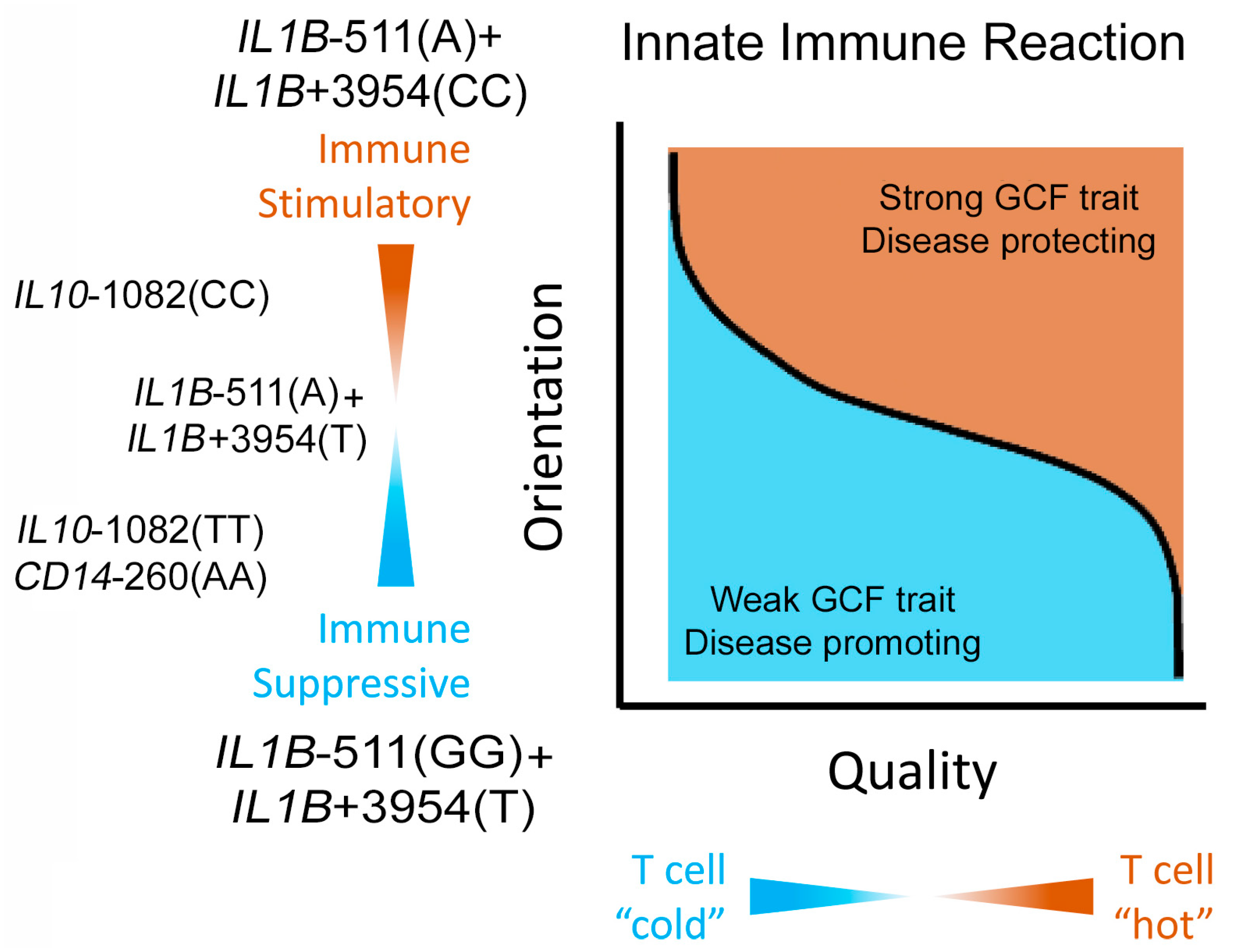
2.3. IL1B Alleles Alone or with Alleles of IL6, IL10 and CD14 Determine Strong or Weak GCF Response Traits to Biofilm Lysine Content in EG
3. Discussion
3.1. Strong and Weak GCF Responses to Biofilm Lysine Are Genetically Determined Phenotypic Traits
3.2. Greater Expression of the Selected Genes Promotes the Weak GCF Trait
3.3. How Weak GCF Exudation Promotes P. gingivalis Infection and Periodontitis
3.4. Weak GCF Trait, Periodontitis, CVD and Dementia
3.5. Using GCF Phenotypic Traits to Develop New Methods to Prevent or Control Periodontitis
4. Materials and Methods
4.1. Subject Selection and Follow-up
4.2. Measurement of Alleles of Genes Associated with Periodontitis
4.3. Statistical Analyses of GCF Responses to Biofilm Lysine after a Week of EG
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Genco, R.J.; Borgnakke, W.S. Risk factors for periodontal disease. Periodontology 2000 2013, 62, 59–94. [Google Scholar] [CrossRef] [PubMed]
- Liccardo, D.; Cannavo, A.; Spagnuolo, G.; Ferrara, N.; Cittadini, A.; Rengo, C.; Rengo, G. Periodontal disease: A risk factor for diabetes and cardiovascular disease. Int. J. Mol. Sci. 2019, 20, 1414. [Google Scholar] [CrossRef] [PubMed]
- Sandal, I.; Karydis, A.; Luo, J.; Prislovsky, A.; Whittington, K.B.; Rosloniec, E.F.; Dong, C.; Novack, D.V.; Mydel, P.; Zheng, S.G.; et al. Bone loss and aggravated autoimmune arthritis in HLA-DRbeta1-bearing humanized mice following oral challenge with Porphyromonas gingivalis. Arthritis Res. Ther. 2016, 18, 249. [Google Scholar] [CrossRef]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef]
- Guo, H.; Chang, S.; Pi, X.; Hua, F.; Jiang, H.; Liu, C.; Du, M. The effect of periodontitis on dementia and cognitive impairment: A meta-analysis. Int. J. Environ. Res. Public. Health 2021, 18, 6823. [Google Scholar] [CrossRef]
- Nara, P.L.; Sindelar, D.; Penn, M.S.; Potempa, J.; Griffin, W.S.T. Porphyromonas gingivalis outer membrane vesicles as the major driver of and explanation for neuropathogenesis, the cholinergic hypothesis, iron dyshomeostasis, and salivary lactoferrin in Alzheimer’s disease. J. Alzheimers Dis. 2021, 82, 1417–1450. [Google Scholar] [CrossRef] [PubMed]
- Michalowicz, B.S.; Diehl, S.R.; Gunsolley, J.C.; Sparks, B.S.; Brooks, C.N.; Koertge, T.E.; Califano, J.V.; Burmeister, J.A.; Schenkein, H.A. Evidence of a substantial genetic basis for risk of adult periodontitis. J. Periodontol. 2000, 71, 1699–1707. [Google Scholar] [CrossRef] [PubMed]
- Giannobile, W.V.; Braun, T.M.; Caplis, A.K.; Doucette-Stamm, L.; Duff, G.W.; Kornman, K.S. Patient stratification for preventive care in dentistry. J. Dent. Res. 2013, 92, 694–701. [Google Scholar] [CrossRef]
- Trombelli, L. Susceptibility to gingivitis: A way to predict periodontal disease? Oral. Health Prev. Dent. 2004, 2 (Suppl. 1), 265–269. [Google Scholar]
- Trombelli, L.; Tatakis, D.N.; Scapoli, C.; Bottega, S.; Orlandini, E.; Tosi, M. Modulation of clinical expression of plaque-induced gingivitis. II. Identification of “high-responder” and “low-responder” subjects. J. Clin. Periodontol. 2004, 31, 239–252. [Google Scholar] [CrossRef]
- Lohinai, Z.; Keremi, B.; Szoko, E.; Tabi, T.; Szabo, C.; Tulassay, Z.; Levine, M. Bacterial lysine decarboxylase influences human dental biofilm lysine content, biofilm accumulation, and subclinical gingival inflammation. J. Periodontol. 2012, 83, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, G.G.; Baelum, V.; Sorsa, T.; Tervahartiala, T.; Skottrup, P.D.; López, R. Salivary levels of MPO, MMP-8 and TIMP-1 are associated with gingival inflammation response patterns during experimental gingivitis. Cytokine 2019, 115, 135–141. [Google Scholar] [CrossRef]
- Nascimento, G.G.; Danielsen, B.; Baelum, V.; Lopez, R. Identification of inflammatory response patterns in experimental gingivitis studies. Eur. J. Oral. Sci. 2019, 127, 33–39. [Google Scholar] [CrossRef]
- Silbereisen, A.; Hallak, A.K.; Nascimento, G.G.; Sorsa, T.; Belibasakis, G.N.; Lopez, R.; Bostanci, N. Regulation of PGLYRP1 and TREM-1 during progression and resolution of gingival inflammation. JDR Clin. Trans. Res. 2019, 4, 2380084419844937. [Google Scholar] [CrossRef] [PubMed]
- Bamashmous, S.; Kotsakis, G.A.; Kerns, K.A.; Leroux, B.G.; Zenobia, C.; Chen, D.; Trivedi, H.M.; McLean, J.S.; Darveau, R.P. Human variation in gingival inflammation. Proc. Natl. Acad. Sci. USA 2021, 118, e2012578118. [Google Scholar] [CrossRef] [PubMed]
- Evavold, C.L.; Kagan, J.C. How inflammasomes inform adaptive immunity. J. Mol. Biol. 2018, 430, 217–237. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and related cytokines in the regulation of inflammation and immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Dutzan, N.; Abusleme, L.; Bridgeman, H.; Greenwell-Wild, T.; Zangerle-Murray, T.; Fife, M.E.; Bouladoux, N.; Linley, H.; Brenchley, L.; Wemyss, K.; et al. On-going mechanical damage from mastication drives homeostatic th17 cell responses at the oral barrier. Immunity 2017, 46, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.; Lohinai, Z.M. Resolving the contradictory functions of lysine decarboxylase and butyrate in periodontal and intestinal diseases. J. Clin. Med. 2021, 10, 2360. [Google Scholar] [CrossRef] [PubMed]
- Bamashmous, S.; Kotsakis, G.A.; Jain, S.; Chang, A.M.; McLean, J.S.; Darveau, R.P. Clinically healthy human gingival tissues show significant inter-individual variability in GCF chemokine expression and subgingival plaque microbial composition. Front. Oral. Health 2021, 2, 689475. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Li, R.; Zeng, X.; He, T.; Zhao, H.; Chang, A.; Bo, C.; Chen, J.; Yang, F.; Knight, R.; et al. Predictive modeling of gingivitis severity and susceptibility via oral microbiota. ISME J. 2014, 8, 1768–1780. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, H.E.; Listgarten, M.A. Fine Structure of the Developing Epithelial Attachment of Human Teeth, Revised ed.; Monographs in Developmental Biology; Wolsky, A., Ed.; S. Karger: Tarrytown, NY, USA, 1977; Volume 2. [Google Scholar]
- Kolenbrander, P.E.; Palmer, R.J., Jr.; Periasamy, S.; Jakubovics, N.S. Oral multispecies biofilm development and the key role of cell-cell distance. Nat. Rev. Microbiol. 2010, 8, 471–480. [Google Scholar] [CrossRef]
- Levine, M.; Collins, L.M.; Lohinai, Z. Zinc chloride inhibits lysine decarboxylase production from Eikenella corrodens in vitro and its therapeutic implications. J. Dent. 2021, 104, 103533. [Google Scholar] [CrossRef] [PubMed]
- Lohinai, Z.; Keremi, B.; Szoko, E.; Tabi, T.; Szabo, C.; Tulassay, Z.; DiCesare, J.C.; Davis, C.A.; Collins, L.M.; Levine, M. Biofilm lysine decarboxylase, a new therapeutic target for periodontal inflammation. J. Periodontol. 2015, 86, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Tabi, T.; Lohinai, Z.; Palfi, M.; Levine, M.; Szoko, E. CE-LIF determination of salivary cadaverine and lysine concentration ratio as an indicator of lysine decarboxylase enzyme activity. Anal. Bioanal. Chem. 2008, 391, 647–651. [Google Scholar] [CrossRef]
- Van Wuyckhuyse, B.C.; Perinpanayagam, H.E.; Bevacqua, D.; Raubertas, R.F.; Billings, R.J.; Bowen, W.H.; Tabak, L.A. Association of free arginine and lysine concentrations in human parotid saliva with caries experience. J. Dent. Res. 1995, 74, 686–690. [Google Scholar] [CrossRef]
- Tan, I.K.; Gajra, B. Plasma and urine amino acid profiles in a healthy adult population of Singapore. Ann. Acad. Med. Singapore 2006, 35, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, R.; Vercelli, D.; Palmer, L.J.; Klimecki, W.J.; Silverman, E.K.; Richter, B.; Riva, A.; Ramoni, M.; Martinez, F.D.; Weiss, S.T.; et al. Single nucleotide polymorphisms in innate immunity genes: Abundant variation and potential role in complex human disease. Immunol. Rev. 2002, 190, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Socransky, S.S.; Smith, C.; Haffajee, A.D. Subgingival microbial profiles in refractory periodontal disease. J. Clin. Periodontol. 2002, 29, 260–268. [Google Scholar] [CrossRef]
- Diaz, P.I.; Hoare, A.; Hong, B.Y. Subgingival microbiome shifts and community dynamics in periodontal diseases. J. Calif. Dent. Assoc. 2016, 44, 421–435. [Google Scholar] [CrossRef]
- Ferreira, S.B., Jr.; Trombone, A.P.; Repeke, C.E.; Cardoso, C.R.; Martins, W., Jr.; Santos, C.F.; Trevilatto, P.C.; Avila-Campos, M.J.; Campanelli, A.P.; Silva, J.S.; et al. An interleukin-1beta (IL-1beta) single-nucleotide polymorphism at position 3954 and red complex periodontopathogens independently and additively modulate the levels of IL-1beta in diseased periodontal tissues. Infect. Immun. 2008, 76, 3725–3734. [Google Scholar] [CrossRef] [PubMed]
- Kornman, K.S.; Crane, A.; Wang, H.Y.; di Giovine, F.S.; Newman, M.G.; Pirk, F.W.; Wilson, T.G., Jr.; Higginbottom, F.L.; Duff, G.W. The interleukin-1 genotype as a severity factor in adult periodontal disease. J. Clin. Periodontol. 1997, 24, 72–77. [Google Scholar] [CrossRef] [PubMed]
- McDevitt, M.J.; Wang, H.Y.; Knobelman, C.; Newman, M.G.; di Giovine, F.S.; Timms, J.; Duff, G.W.; Kornman, K.S. Interleukin-1 genetic association with periodontitis in clinical practice. J. Periodontol. 2000, 71, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Karimbux, N.Y.; Saraiya, V.M.; Elangovan, S.; Allareddy, V.; Kinnunen, T.; Kornman, K.S.; Duff, G.W. Interleukin-1 gene polymorphisms and chronic periodontitis in adult whites: A systematic review and meta-analysis. J. Periodontol. 2012, 83, 1407–1419. [Google Scholar] [CrossRef] [PubMed]
- Rogus, J.; Beck, J.D.; Offenbacher, S.; Huttner, K.; Iacoviello, L.; Latella, M.C.; de Gaetano, M.; Wang, H.Y.; Kornman, K.S.; Duff, G.W. IL1B gene promoter haplotype pairs predict clinical levels of interleukin-1beta and C-reactive protein. Hum. Genet. 2008, 123, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.C.; Ooi, Y.; Pulikkotil, S.J.; Naing, C. The role of three interleukin 10 gene polymorphisms (- 1082 A > G, - 819 C > T, - 592 A > C) in the risk of chronic and aggressive periodontitis: A meta-analysis and trial sequential analysis. BMC Oral. Health 2018, 18, 171. [Google Scholar] [CrossRef]
- Nicu, E.A.; Laine, M.L.; Morré, S.A.; Van der Velden, U.; Loos, B.G. Soluble CD14 in periodontitis. Innate Immun. 2009, 15, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Nibali, L.; Griffiths, G.S.; Donos, N.; Parkar, M.; D’Aiuto, F.; Tonetti, M.S.; Brett, P.M. Association between interleukin-6 promoter haplotypes and aggressive periodontitis. J. Clin. Periodontol. 2008, 35, 193–198. [Google Scholar] [CrossRef]
- Prakash, G.; Umar, M.; Ajay, S.; Bali, D.; Upadhyay, R.; Gupta, K.K.; Dixit, J.; Mittal, B. COX-2 gene polymorphisms and risk of chronic periodontitis: A case-control study and meta-analysis. Oral Dis. 2015, 21, 38–45. [Google Scholar] [CrossRef]
- Izakovicova Holla, L.; Hrdlickova, B.; Vokurka, J.; Fassmann, A. Matrix metalloproteinase 8 (MMP8) gene polymorphisms in chronic periodontitis. Arch. Oral. Biol. 2012, 57, 188–196. [Google Scholar] [CrossRef]
- Starr, T.N.; Thornton, J.W. Epistasis in protein evolution. Protein Sci. 2016, 25, 1204–1218. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhu, Y.; Singh, P.; Ajmera, D.H.; Song, J.; Ji, P. Association of common variants in MMPs with periodontitis risk. Dis. Markers 2016, 2016, 1545974. [Google Scholar] [CrossRef] [PubMed]
- Molnar, E.; Lohinai, Z.; Demeter, A.; Mikecs, B.; Toth, Z.; Vag, J. Assessment of heat provocation tests on the human gingiva: The effect of periodontal disease and smoking. Acta Physiol. Hung. 2015, 102, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Naginyte, M.; Do, T.; Meade, J.; Devine, D.A.; Marsh, P.D. Enrichment of periodontal pathogens from the biofilms of healthy adults. Sci. Rep. 2019, 9, 5491. [Google Scholar] [CrossRef] [PubMed]
- Brill, N. Removal of particles and bacteria from gingival pockets by tissue fluid. Acta Odontol. Scand. 1959, 17, 431–440. [Google Scholar] [CrossRef]
- Mackay, T.F. Epistasis and quantitative traits: Using model organisms to study gene-gene interactions. Nat. Rev. Genet. 2014, 15, 22–33. [Google Scholar] [CrossRef]
- Miton, C.M.; Buda, K.; Tokuriki, N. Epistasis and intramolecular networks in protein evolution. Curr. Opin. Struct. Biol. 2021, 69, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Tsalikis, L.; Parapanisiou, E.; Bata-Kyrkou, A.; Polymenides, Z.; Konstantinidis, A. Crevicular fluid levels of interleukin-1alpha and interleukin-1beta during experimental gingivitis in young and old adults. J. Int. Acad. Periodontol. 2002, 4, 5–11. [Google Scholar] [PubMed]
- Pani, P.; Tsilioni, I.; McGlennen, R.; Brown, C.A.; Hawley, C.E.; Theoharides, T.C.; Papathanasiou, E. IL-1B(3954) polymorphism and red complex bacteria increase IL-1β (GCF) levels in periodontitis. J. Periodontal. Res. 2021, 56, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.Y.; Furtado Araujo, M.V.; Strausbaugh, L.D.; Terzi, E.; Ioannidou, E.; Diaz, P.I. Microbiome profiles in periodontitis in relation to host and disease characteristics. PLoS ONE 2015, 10, e0127077, Erratum in PLoS ONE 2016, 11, e0148893. [Google Scholar] [CrossRef]
- Chang, R.B.; Beatty, G.L. The interplay between innate and adaptive immunity in cancer shapes the productivity of cancer immunosurveillance. J. Leukoc. Biol. 2020, 108, 363–376. [Google Scholar] [CrossRef]
- Fine, N.; Barzilay, O.; Sun, C.; Wellappuli, N.; Tanwir, F.; Chadwick, J.W.; Oveisi, M.; Tasevski, N.; Prescott, D.; Gargan, M.; et al. Primed PMNs in healthy mouse and human circulation are first responders during acute inflammation. Blood Adv. 2019, 3, 1622–1637. [Google Scholar] [CrossRef] [PubMed]
- Fine, N.; Chadwick, J.W.; Sun, C.; Parbhakar, K.K.; Khoury, N.; Barbour, A.; Goldberg, M.; Tenenbaum, H.C.; Glogauer, M. Periodontal inflammation primes the systemic innate immune response. J. Dent. Res. 2021, 100, 318–325. [Google Scholar] [CrossRef]
- Hidalgo, A.; Libby, P.; Soehnlein, O.; Aramburu, I.V.; Papayannopoulos, V.; Silvestre-Roig, C. Neutrophil extracellular traps: From physiology to pathology. Cardiovasc. Res. 2022, 118, 2737–2753. [Google Scholar] [CrossRef]
- Genco, R.J.; Van Dyke, T.E. Prevention: Reducing the risk of CVD in patients with periodontitis. Nat. Rev. Cardiol. 2010, 7, 479–480. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Li, Y.; Liu, X.; Cui, Y.; Zhao, Y.; Sun, S.; Jia, Q.; Chai, Q.; Gong, G.; Zhang, H.; et al. Assessing the effectiveness of statin therapy for alleviating cerebral small vessel disease progression in people ≥75 years of age. BMC Geriatr. 2020, 20, 292. [Google Scholar] [CrossRef] [PubMed]
- Pinal-Fernandez, I.; Casal-Dominguez, M.; Mammen, A.L. Statins: Pros and cons. Med. Clin. 2018, 150, 398–402. [Google Scholar] [CrossRef]
- Torrandell-Haro, G.; Branigan, G.L.; Vitali, F.; Geifman, N.; Zissimopoulos, J.M.; Brinton, R.D. Statin therapy and risk of Alzheimer’s and age-related neurodegenerative diseases. Alzheimers Dement. 2020, 6, e12108. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Harms, A.C.; Hankemeier, T.; Kindt, A.; de Lange, E.C.M. Status of metabolomic measurement for insights in Alzheimer’s disease progression—What is missing? Int. J. Mol. Sci. 2023, 24, 4960. [Google Scholar] [CrossRef]
- Huang, C.W.; Hsu, S.W.; Tsai, S.J.; Chen, N.C.; Liu, M.E.; Lee, C.C.; Huang, S.H.; Chang, W.N.; Chang, Y.T.; Tsai, W.C.; et al. Genetic effect of interleukin-1 beta (C-511T) polymorphism on the structural covariance network and white matter integrity in Alzheimer’s disease. J. Neuroinflamm. 2017, 14, 12. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Yamazaki, K.; Baba, Y.; Ito, H.; Loos, B.G.; Takahashi, K. The Stages and Grades of Periodontitis Are Risk Indicators for Peri-Implant Diseases-A Long-Term Retrospective Study. J. Pers. Med. 2022, 12, 1723. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Severity | Prevalence |
|---|---|
| No disease | 52.80% |
| Mild | 8.70% |
| Moderate | 30.00% |
| Severe | 8.50% |
| Total | 100.00% |
| Gene, Site, (SNP) a | rs Number b | MAFα Euro c |
|---|---|---|
| IL1A-889(G/A) | rs1800587 | 0.285 |
| IL1B+3954(C/T) | rs1143634 | 0.237 |
| IL1B-511(G/A) | rs16944 | 0.335 |
| IL6-1363(G/T) | rs2069827 | 0.085 |
| IL10-1082(T/C) | rs1800896 | 0.527 d |
| CD14-260(A/G) | rs2569190 | 0.485 d |
| COX2+8473(G/A) | rs5275 | 0.337 |
| MMP8-799(G/A) | rs11225395 | 0.464 d |
| Host | Sex | Trait | Genotype | 2nd Gene | Comments | |
|---|---|---|---|---|---|---|
| 1 | M | S | *IL1B+3954(CT)e | IL1B-511(GA) | IL6-1363GTe | Epistasise |
| 2 | F | S | *IL1B+3954(CT) | IL1B-511(GA) | IL10-1082(CC)s | Specifieds |
| 3 | M | S | *IL1B+3954(CT) | IL1B-511(GA) | IL10-1082(CC)s | [COX2+8473(AG)]s* |
| 4 | F | S | IL1B+3954(CC) | IL1B-511(GA) | ||
| 5 | F | S | IL1B+3954(CC) | IL1B-511(GA) | ||
| 6 | F | S | IL1B+3954(CC) | IL1B-511(GA) | ||
| 7 | F | S | IL1B+3954(CC) | IL1B-511(GA) | ||
| 8 | F | S | IL1B+3954(CC) | IL1B-511(GA) | ||
| 9 | M | S | IL1B+3954(CC) | IL1B-511(GA) | ||
| 10 | M | W | *IL1B+3954(CT) | IL1B-511(GA) | IL10-1082(TT)s | |
| 11 | M | W | IL1B+3954(CC)eg | IL1B-511(GA) | IL6-1363(GT)eg | e[IL1A-889(GA)]g |
| 12 | F | W | *IL1B+3954(CT)s | IL1B-511(GA) | CD14-260(AA)s | Specifieds |
| 13 | M | W | IL1B+3954(CT) | IL1B-511(GG) | ||
| 14 | M | W | IL1B+3954(CT)g | IL1B-511(GG) | IL1A-889(GA)g | Genotypeg |
| 15 | F | W | IL1B+3954(TT) | IL1B-511(GG) | ||
| # of Hosts | IL1B+3954 | IL1B-511 | Predicted 1 | Periodontitis 2 | |
|---|---|---|---|---|---|
| 7 | 47% | IL1B+3954(CC) | IL1B-511(GA) | 6S+1We | Protected |
| 3 | 20% | IL1B+3954(CT) | IL1B-511(GG) | 3W | Susceptible |
| 5 | 33% | IL1B+3954(CT) | IL1B-511(GA) | 2Ss+1Se+2Ws | 2nd Gene 3 |
| 0 | 0 | IL1B+3954(CC) | IL1B-511(GG) | Not present | Not available |
| Groups | Strong or weak GCF Traits from Allelic Genotypes |
|---|---|
| A. | IL1B+3954(CC)+IL1B-511(GA) = strong (6 hosts) |
| B. | IL1B+3954(CT)+IL1B-511(GG) = weak (3 hosts) |
| B1. | IL1B+3954(T)+IL1B-511(GG)+IL1A-889(A) = weak (tooth loss gene) |
| C. | IL6-1363(T) reverses phenotype, specific epistasis (2 hosts) |
| C1. | IL1B+3954(CC)+IL1B-511(GA) = strong |
| C2. | IL1B+3954(CT)+IL1B-511(GA) = weak |
| C2.1. | IL1B+3954(CC)+IL1B-511(GA)+IL6-1363(T)+IL1A-889(GA) = weak |
| (Epistasis with tooth loss gene) | |
| D. | IL1B+3954(CT) and IL1B-511(GA) conflicted, but with: |
| D1. | Presence of IL10-1082(CC) or COX2+8473(AG) = strong (2 hosts). |
| D2. | Presence of IL10-1082(TT) or CD14-260(AA) = weak (2 hosts). |
| E. | IL1B+3954(CC)+IL1B-511(GG) = GCF trait uninterpretable (no hosts) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lohinai, Z.M.; Ruksakiet, K.; Földes, A.; Dinya, E.; Levine, M. Genetic Control of GCF Exudation: Innate Immunity Genes and Periodontitis Susceptibility. Int. J. Mol. Sci. 2023, 24, 14249. https://doi.org/10.3390/ijms241814249
Lohinai ZM, Ruksakiet K, Földes A, Dinya E, Levine M. Genetic Control of GCF Exudation: Innate Immunity Genes and Periodontitis Susceptibility. International Journal of Molecular Sciences. 2023; 24(18):14249. https://doi.org/10.3390/ijms241814249
Chicago/Turabian StyleLohinai, Zsolt M., Kasidid Ruksakiet, Anna Földes, Elek Dinya, and Martin Levine. 2023. "Genetic Control of GCF Exudation: Innate Immunity Genes and Periodontitis Susceptibility" International Journal of Molecular Sciences 24, no. 18: 14249. https://doi.org/10.3390/ijms241814249