
The Role of Regulatory T Cells in Cancer Treatment Resistance
by
 , , and
, , and
Anna Dąbrowska
1,†,
Magdalena Grubba
1,†,
Amar Balihodzic
2,3,
Olga Szot
1,
Bartosz Kamil Sobocki
1,* and
Adrian Perdyan
4,5,* 1
Student Scientific Circle of Oncology and Radiotherapy, Medical University of Gdansk, 80-210 Gdansk, Poland
2
Division of Oncology, Department of Internal Medicine, Comprehensive Cancer Center Graz, Medical University of Graz, 8036 Graz, Austria
3
BioTechMed-Graz, 8010 Graz, Austria
4
3P-Medicine Laboratory, Medical University of Gdansk, 80-210 Gdansk, Poland
5
Department of Biology, Stanford University, Stanford, CA 94305, USA
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2023, 24(18), 14114; https://doi.org/10.3390/ijms241814114
Submission received: 16 August 2023
/
Revised: 6 September 2023
/
Accepted: 7 September 2023
/
Published: 14 September 2023
(This article belongs to the Special Issue Current Research on Cancer Drug Resistance)
Abstract
:Despite tremendous progress in cancer treatment in recent years, treatment resistance is still a major challenge for a great number of patients. One of the main causes is regulatory T lymphocytes (Tregs), which suppress excessive inflammatory responses via the secretion of immunosuppressive cytokines and upregulate the immune checkpoints. Their abundance causes an immunosuppressive reprogramming of the tumor environment, which is ideal for tumor growth and drug inefficiency. Hence, regiments that can regain tumor immunogenicity are a promising strategy to overcome Tregs-mediated drug resistance. However, to develop effective therapeutic regimens, it is essential to understand the molecular mechanisms of Treg-mediated resistance. In this article, we gathered a comprehensive summary of the current knowledge on molecular mechanisms and the role of Tregs in cancer treatment resistance, including cancer immunotherapy, targeted therapy, chemotherapy, and radiotherapy.
1. Introduction
Regulatory T cells (Tregs) are a phenotypically and functionally heterogeneous group of T lymphocytes [1]. Tregs come in many forms, including both CD4+ and CD8+ T cells, and are essentially differentiated mostly by the expression of specific markers. For instance, CD4+ CD25+ Tregs actively contribute to the maintenance of immune homeostasis and immunological self-tolerance [1]. A more detailed classification of CD4+ Tregs distinguishes subsets such as CD4+CD25+FoxP3−, CD4+CD25−FoxP3low, CD4+CD25hiCD125low, CD4+CD25hiCD45ROhi, and CD4+CD25+CD62LlowCD44hi [2]. Forkhead box protein P3 (FoxP3) plays a crucial role in the development and suppressive function of Tregs. It is a significant regulator that distinguishes Tregs from activated CD4+CD25− T cells [3]. CD8+ Tregs also represent a heterogeneous group, which includes the following subsets: CD8+FoxP3+, CD8+CD122+, CD8+CD28−, CD8αα+, CD8+Qa-1-restricted, and CD8+CD45RClow. Nevertheless, the number of studies investigating the immunomodulatory properties of the CD8+ Tregs subset remains insufficient [4]. Tregs are responsible for restoring immune homeostasis after an excessive acute inflammatory response, thus preventing the development of chronic inflammation [5]. Mechanistically, their suppressive properties extend to antigen-presenting cells (APCs), natural killer (NK) cells, and effector T lymphocytes (Teffs) (Figure 1) [1]. Moreover, Tregs exploit other mechanisms to control the excessive inflammatory response and evade tumors (Figure 2). Such mechanisms include modulation of the metabolism of IL-2 and ATP, calcium ions disruption, the secretion of inhibitory cytokines such as granzyme B, IL-10, IL-35, and TGF-β, the inhibition of Teffs and APC through kynurenine production and immune checkpoint (ICs) protein expression, and the secretion of extracellular vesicles (Evs) [6].
Tregs are found in peripheral blood, various tissues, and inflammatory regions, including heterogeneous tumor microenvironment (TME) with other immunosuppressive and immunostimulant cells (Figure 3A). Like other T lymphocytes, Tregs express numerous co-signaling receptors that play critical roles in regulating the immune response (Figure 3B). Some of these receptors, including CTLA-4, GITR, OX40, PD-1, ICOS, TIGIT, LAG-3, TIM-3, and 4-1BB, are expressed on Tregs. The high expression of certain receptors by Tregs and cancer tissues contributes to tumor immune evasion [7,8].
2. Results and Discussion
2.1. Involvement of Tregs in Immunotherapy Resistance
ICs are receptors expressed by immune cells that are significant for T cell functionality. Tumor cells exploit certain interactions with these receptors to maintain immune tolerance [9]. Immune checkpoint inhibitors (ICIs) are cancer immunotherapies that block the receptors on the surface of T-lymphocytes and tumor cells that control immune cell activity. Contemporary, there are three main groups of ICIs: PD-1 inhibitors, PD-L1 inhibitors, and CTLA-4 inhibitors [7].
The introduction of ICIs in cancer treatment was one of the greatest breakthroughs in recent years. However, it quickly turned out that only ~20% of cancer patients respond to the treatment [10,11]. Additionally, a great number of patients will become resistant with time. Multiple resistance mechanisms limit the potential of ICIs, one of which could be the presence of Tregs [12,13]. Specifically, the heterogeneity of TME may play a major role in immunotherapy resistance [14]. Recently, it has been shown that apoptotic Tregs eliminated the effectiveness of PD-L1 blockade in CRC-bearing mice [15]. Moreover, the presence of Tregs results in the immunosuppressive microenvironment, which is a potential resistance mechanism to the ICIs (i.e., an anti-PD-L1 monoclonal antibody (mAb)—atezolizumab used in urothelial carcinoma) [16].
2.1.1. Primary Resistance
Primary resistance to the ICIs is defined as a lack of initial response or a low overall response rate (RR). To enhance the efficacy of ICIs, researchers are exploring the benefits of combination with other ICIs or systemic therapies (e.g., chemotherapy and radiotherapy) and identifying predictive biomarkers, which are critical for predicting positive responses to ICIs [17]. Current predictive biomarkers to ICIs responsiveness, such as total tumor mutational load (TML), density and distribution of CD8+ T lymphocytes, PD-L1 expression, and T cell clonality, have several limitations. For instance, some patients with a high level of TML exhibit ineffective responses, while other patients with a low level of TML may exhibit favorable responses to ICIs [18]. A correlation between the accumulation of FoxP3+ Tregs and poor prognoses in patients with IL12AloTGFβ1lo expression (type A CRC; p = 0.038) was found. In patients with IL12AhiTGFβ1hi (type B CRC) expression, high levels of FoxP3 were associated with a better prognosis, but not significantly (p = 0.34) [19]. Particularly, a higher Tregs/Teffs ratio within tumor tissue is associated with worse outcomes [20]. However, it can be used to predict primary RR to immunotherapy. Moreover, mAb targeting CTLA-4 and PD-1 may elevate the intratumoral Teffs/Tregs ratio, thereby positively affecting the prognosis [21].
2.1.2. Acquired Resistance
Acquired resistance to immunotherapy develops in patients who ultimately experience illness recurrence despite the initial clinical response. Several mechanisms have been identified, including the upregulation of alternative ICs, defective antigen presentation, a lack of IFN-γ response, T cell exclusion [9], and tumor-mediated immunosuppression or exclusion [17]. However, the identification of a specific mechanism is often difficult due to several challenges: the lack of consistent terminology to define and classify acquired resistance; difficulty in collecting optimal tumor samples for analyses; and the limited efficacy of identifying immune resistance mechanisms in the tumor, host, and TME [17].
Nevertheless, several mechanisms have been proposed recently. APLNR, a G-protein-coupled receptor, regulates T cell responses by modulating JAK1 and IFN-γ signaling, which plays a role in acquired resistance to immunotherapy. It has also been shown that activating mutations in Ptpn2 affect resistance to ICIs through resistance to IFN-γ [17]. Ultimately, an increasing number of Tregs contribute to the development of acquired resistance to immunotherapy [22]. Table 1 summarizes other mechanisms of treatment resistance.
Due to Tregs’ involvement in the development of acquired resistance, there are several therapeutic approaches to target Tregs to overcome the resistance. For example, they can be targeted by CD25 antibodies. Radiotherapy combined with the blockade of CD25, PD-L1, and TIM-3 has been shown to be more effective than radiotherapy with anti-PD-L1/TIM-3 alone in HNSCC treatment. However, the caveat is that non-Treg cells also express CD25. Another potential treatment target is FoxP3. It is closely related to the efficiency of Tregs as the expression of FoxP3, and IL-10 is reduced after anti-PD-1 treatment. Furthermore, Mdb2 protein’s binding to TSDR has been linked to TET2-mediated demethylation, leading to increased FoxP3 expression. Importantly, TSDR methylation is more specific than CD25 blockade, which may be a more effective approach for Tregs depletion [22].
2.2. PD-1 and CTLA-4 Expression Ratio between Teffs and Tregs
Tregs are characterized by a low expression of PD-1 in blood and non-cancerous tissues, but a high expression in tumors. Interestingly, around 10% of gastric cancer patients treated with an anti-PD-1 mAb experienced increased tumor infiltration of Tregs and disease progression. Hence, to increase the effectiveness of the therapy, it seems reasonable to deplete the Tregs before starting treatment with the anti-PD-1 mAb [1].
The effectiveness of anti-PD-1/PD-L1 and anti-CTLA-4 mAbs in cancer immunotherapy relies on their interactions with FcƔ receptors (FcƔRs). Due to the high surface expression of CTLA-4 on Tregs and the presence of effector myeloid-expressing, activating FcƔRs in the TME, Tregs are preferentially depleted over Teffs cells. CTLA-4 and PD-1 are expressed on Tregs and Teffs differently, distinguishing the mechanisms of these ICs blocking mAbs. The expression of CTLA-4 is higher on the regulatory tumor-infiltrating lymphocytes (TILs), whereas PD-1 is overexpressed on the effector TILs. Therefore, anti-CTLA-4 or PD-1 depletion of regulatory T cells leads to higher or lower Teffs/Tregs ratios [21].
PD-1/PD-L1 plays a crucial role in tumor evasion of the immune response. Thus, the PD-1/PD-L1 axis is a good target for mAbs as its blockade can return Teffs function and increase the Teffs/Tregs ratio. Anti-PD-1 antibodies, such as pembrolizumab, downregulate FoxP3, which leads to tTregs and pTregs suppression in melanoma patients (Figure 4). In addition, combined anti-CTLA-4 and anti-PD-1 therapy has shown higher effectiveness compared to individual treatments, significantly increasing Teffs infiltration and the Teffs/Tregs ratio within the tumor. The proliferation and immunosuppressive properties of Tregs may be enhanced by PD-1 deficiency. Therefore, a careful balance must be struck to maximize the benefits of anti-PD-1 treatments while minimizing the unintended consequences [16]. The Tregs/Teffs ratio has the potential to become a predictor of OS and chemotherapeutic response. A higher ratio of Tregs/Teffs in a tumor is associated with a worse prognosis for various cancers, including ovarian cancer, lung cancer, and melanoma [22].
2.3. Upregulation of ICs
The primary goal of immunotherapy is to turn on or off the immune checkpoints associated with immune surveillance [29]. Numerous reports have demonstrated that if a single immune checkpoint is blocked, other ICs in the TME may be overexpressed. This phenomenon has been observed in lung cancer between PD-1 and TIM-3 checkpoints [7]. Cancer cells are protected from immune-cell-mediated death by upregulated ICs in TME [30]. In mice models, it has been shown that the expression of ICs can affect the differentiation and other properties of Tregs by increasing the secretion of suppressive cytokines. Specifically, IL-10 and IL-35 produced by Tregs lead to the upregulation of ICs, including PD-1, TIM-3, LAG-3, TIGIT, and the depletion of tumor-infiltrating CD4+ and CD8+ T cells. Additionally, the upregulation of ICs may indirectly inhibit the activation of Teffs by negatively affecting APC function [22].
PD-1 is highly expressed on activated lymphocytes and is upregulated by TILs. T cells with high PD-1 expression decrease the production of pro-inflammatory cytokines (e.g., IFN-γ and IL-2) and increase the secretion of IL-10 via the upregulation of various inhibitory receptors, including CTLA-4 or TIM-3 [25]. Other factors that upregulate PD-1 expression are TGF-β and dexamethasone (Figure 5A) [25,30]. Therefore, the purpose of the PD-1 blockade is an inversion of T cell depletion [21].
Cancer cells acquire immunosuppressive properties by forming suppressive TME, reducing their immunogenicity and attempting to evade immune surveillance. One of the key pathways they exploit is the PD-1 pathway by upregulating PD-L1 expression [21]. It should be noted that the presence of PD-L1 is frequently associated with a poor prognosis. Studies have shown that PD-L1 can weaken the anticancer response by itself. In contrast, PD-L1 overexpression is associated with a better response to the inhibition of the PD-1/PD-L1 axis [29]. PD-L1 expression is regulated by two main mechanisms—innate and adaptive immune response. The first one causes PD-L1 upregulation through the oncogenic NPM/ALK kinase pathway via STAT3. The second, acquired immune response, regulates the expression through pro-inflammatory cytokines (e.g., IFN-γ) [31]. Moreover, cytotoxic T cells play a significant role in PD-L1 expression. They release IFN-γ into the TME, which stimulates signal transducers and activators of signaling pathways, resulting in an increase in PD-L1 expression [14]. Abnormal oncogenic signaling pathways are another factor that promotes cancer progression and increases PD-L1 expression. MYC, HIF-1α, and HIF-2α overexpression are involved in PD-L1 upregulation in melanoma, NSCLC, and HNSCC [32]. Hypoxia, radiotherapy, and chemotherapy are further factors that increase PD-L1 expression. Hsieh et al. showed that in irradiated CRC cells, activated ATR signaling upregulates PD-L1 and CD47. Moreover, irradiated tumor cells use the DNA repair signaling pathway to increase PD-L1 and CD47 expression. This connection has been observed in other solid tumors [33]. Chemotherapy enhances TGF-β expression, also leading to PD-L1 upregulation (Figure 5B) [34].
In many malignancies, the interaction of PD-L1 on cancer cells and PD-1 on TIL promotes tumor immune evasion. Blocking this interaction with mAbs against PD-1 or PD-L1 can reactivate Teffs proliferation and function. This includes cytokine production, such as IFN-γ and IL-2, which decrease the Treg number by increasing the Teffs/Tregs ratio [16]. The PD-1/PD-L1 axis is involved in the induction of Treg expansion through modulating the Notch pathway and asparaginyl endopeptidase (AEP) inactivation. The Notch signaling pathway, which is important for cell–cell communication, regulates the differentiation and function of Tregs, while AEP is a lysosomal cysteine protease responsible for FoxP3 destabilization and antigen processing in dendritic cells (DCs). Therefore, Notch activation and AEP inactivation through PD-1/PD-L1 axis upregulation enhance the immunosuppressive properties of Tregs. Some in vitro studies showed that PD-L1 coated beads have the potential to convert naive CD4+ T cells into Tregs through the downregulation of Akt, mTOR, and ERK2 and the upregulation of PTEN (Figure 6) [35].
FoxP3+ CD4+ Tregs express CTLA-4, and CD4+ and CD8+ Teffs upregulate it. It improves suppressive Tregs activity, while its absence causes unregulated T cell proliferation [7,36]. Therefore, a moderate upregulation of CTLA-4 expression potentially indicates the activation of Tregs [37]. Ipilimumab, an anti-CTLA-4 mAb, can effectively kill CTLA-4-expressing Tregs in TME through antibody-dependent cellular cytotoxicity in melanoma- and CRC-bearing mice [1]. Combining anti-CTLA-4 blockade with radiation-induced Tregs depression may further enhance the removal of suppressor T cells within tumor tissues [37].
2.4. Involvement of Tregs in Chemotherapy and Radiotherapy Resistance
Contemporary, approximately 80 cytotoxic drugs are approved for cancer treatment. They can be divided by the mechanism of action into alkylating agents (e.g., cyclophosphamide (CPA); oxaliplatin (FOLFOX-6)), antimetabolites (e.g., fluorouracil (5-FU)), topoisomerase inhibitors (tubulin/microtubule inhibitors (e.g., paclitaxel and docetaxel)), and DNA binders or cleavers (e.g., bleomycin) [29]. Long-lasting reduction in Tregs was observed after docetaxel administration in NSCLC and after folinic acid/5-FU/FOLFOX-6 in gastric cancer [38,39]. A high dose of CPA depletes the Tregs population, which is consistent with its strong lymphopenic capabilities. A study on mice with fibrosarcoma has shown that Tregs surviving CPA treatment had rapid proliferation after chemotherapy and therefore inhibited the development of antitumor immunity after lymphodepletion [40]. In small amounts over a long period, CPA is capable of selectively reducing the proliferation of Tregs, including those in the TME. This suggests that low-dose CPA administration can enhance antitumor immune responses without strong lymphopenic effects [41]. This finding on CPA extended to other drugs would be a useful tool for chemotherapy antitumor augmentation as well as for lymphodepletion strategies to potentiate adoptive T cell therapy. The majority of studies have reported that patients with a higher percentage of Tregs within the CD4+ T cell population before chemotherapy had worse long-term outcomes. However, Treg abundance has been proposed as a potential predictive biomarker in several cancers, including TNBC and ovarian cancer [22]. Tumor PD-L1 expression increases after platinum-based neoadjuvant chemotherapy, predicting poor clinical outcomes in NSCLC [42]. Nonetheless, high PD-L1 expression can improve RR with the administration of anti-PD1 treatment. Meta-analysis of 22 randomized controlled trials with 4289 patients showed that the combination of ICI and chemotherapy significantly improved ORR and PFS relative to ICI alone in NSCLC [43].
Radiotherapy is an integral part of cancer treatment, but its effectiveness can be compromised by upregulated markers like PD-L1, CTLA-4, TIM-3, and STAT3, which can induce Tregs proliferation, leading to treatment resistance [44,45]. A study on gamma irradiation showed that CTLA-4 was upregulated by a low dose of γ-ray (1.8 Gy), whereas a high dose (30 Gy) decreased CTLA-4 expression and therefore abolished the suppressive capacity of Tregs [45]. This corresponds with a more recent study where high doses (10 Gy) of radiation increased the expression of LAG-3 and decreased the expression of CD25 and CTLA-4 (Figure 7) [46].
TIM-3 is another coinhibitory molecule that was found to be upregulated in response to radiotherapy. A unique feature of TIM-3 is the lack of known inhibitory signaling motifs in its cytoplasmic tail in comparison to classic ICs such as PD-1 and CTLA-4. Sixty percent of all CD4+FOXP3+ TILs co-express TIM-3, according to a study on patients with lung cancer. Tregs do not constitutively express TIM-3. The exception is intratumoral Tregs, suggesting their immune regulatory roles within the TME [16]. During the combined radiotherapy and anti-PD-1 treatment (pembrolizumab) for HNSCC, it was found that TIM-3 was upregulated on CD8+ T cells and Tregs. The expression of IL-10 in TIM-3-positive Tregs is higher than that in TIM-3-negative Tregs, and they have a higher capacity to inhibit the release of IFN-γ and TNF-α by Teffs (Figure 8) [47].
About 60% of all cancer patients will be treated with radiotherapy during their disease. Radiotherapy can increase the immunogenicity of TME through immunogenic cell death, the release of reactive oxygen species (ROS), and damage-associated molecular pattern (DAMPs), therefore transforming “cold” non-immunogenic to a “hot” immune-reactive TME [48]. Despite enhancing immunogenicity, radiotherapy has also been reported to promote immunosuppressive TME by increasing the transcription of HIF-1α, which induces Tregs proliferation, activating latent TGF-β in the TME that polarizes tumor-associated macrophages (TAMs) into immunosuppressive phenotype and converting CD4+ T cells into Tregs. TAMs are one of the main mediators of immunosuppression, next to Tregs [49]. They suppress Teff’s function and promote tumor growth, creating an immunosuppressive environment [49,50]. These properties are typical for alternatively activated M2 macrophages. It has been shown that the presence of Tregs and M2 macrophages is correlated with each other and the stage of nasopharyngeal carcinoma [51]. Tregs and M2 macrophages can mutually increase each other by secreting immunosuppressive cytokines [50]. The presence of M2 macrophages was associated with increased tumor invasion and therefore with a poorer prognosis in various cancers, including nasopharyngeal carcinoma and CRC [50,51]. Using the small animal radiation research platform, it was demonstrated that radiotherapy resulted in an increase in tumor-infiltrating Tregs that, among other receptors, exhibited a higher expression of CTLA-4 compared with Tregs in non-irradiated tumors, including melanoma, CRC, and renal cell carcinoma [52]. A study on rodents revealed that Tregs are more resistant to radioactivity, less susceptible to radiation-induced cell death, and have a higher frequency of repopulation than CD4+Foxp3 cells. However, Tregs that have been irradiated exhibit functional impairment and a diminished suppressive capacity [37]. Similar results were obtained in human T cell samples, which indicated that both nTregs and iTregs are more resistant to cell death by radiation than CD4+ Tconvs. However, pTregs showed a more robust decrease in Foxp3 expression than tTregs, suggesting that they are more sensitive to the effects of radiation [46]. Tregs in radioresistance have been studied in a small number of cancers, but the available results are consistent. An increased presence of Tregs in TME or peripheral blood correlates with poor response to radiotherapy in NSCLC [53]. Furthermore, Tregs can play an important role in modulating radiation resistance in HNSCC [54]. These results are also supported by the fact that Tregs depletion enhances radiotherapy outcomes in breast cancer and CRC, which is a reason for combined Tregs-targeted drugs and radiotherapy [55,56].
2.5. Molecular Mechanisms of Treg-Mediated Treatment Resistance
2.5.1. Apoptosis
A study on human ovarian cancer ascites showed that oxidative stress plays a significant role by triggering the apoptosis of Tregs via ROS in TME. This is attributed to lower amounts of the transcription factor NRF2 in Tregs, which is responsible for the regulation of the cellular antioxidant system, resulting in increased susceptibility to ROS in TME [15,36].
CD39+CD73+ live Tregs can convert ATP to adenosine. Thus, Teffs can be inhibited via an adenosinergic pathway. Apoptotic Tregs also mediate suppression via the A2A pathway through the expression of CD39 and CD73. Additionally, they release higher levels of ATP through pannexin-1-dependent channels, which effectively intensify the suppression of Teffs [15,35]. Therefore, CD39, CD73, and pannexin-1-dependent channels are potential targets for novel treatment. The administration of two inhibitors partially eliminated the immunosuppressive effect of apoptotic Tregs and enhanced the efficacy of PD-L1 blockade in CRC (Figure 9) [15].
2.5.2. TME Metabolism
Hypoxia upregulates HIF-1α in various cells of TME, which then promotes the expression of CD39/CD73 and adenosine receptors A2AR and A2BR. That leads to a significant extracellular adenosine concentration and intracellular cAMP accumulation, which create a beneficial environment for Tregs recruitment [57]. Despite cellular adaptations to hypoxia, reduced oxygenation in TME has far-reaching consequences, particularly in terms of chemoresistance. Firstly, intravenous drug delivery limits the efficacy of chemotherapy. Secondly, hypoxia influences cellular uptake, also impairing the effectiveness of anticancer drugs through associated acidity and drug efflux pump expression (e.g., Pgp). Moreover, the lack of oxygen in the hypoxic TME hampers the induction of cytotoxicity by chemotherapeutics [58]. The necrosis of tumor cells during chemotherapy results in an increased extracellular potassium concentration. This subsequently suppresses the Akt/mTOR pathway, preventing the transformation of resting CD4+ T cells into Teffs and promoting the development of Tregs in melanoma-bearing mice (Figure 10) [59]. Table 2 provides a broader overview of the mechanisms of the TME metabolism involved in Treg-mediated treatment resistance.
2.5.3. TGF-β-Dependent Upregulation of Tregs
TGF-β is a multifunctional cytokine produced by various cells, such as fibroblasts, macrophages, platelets, Tregs, and tumor cells (Figure 11—left side). Tregs are the significant source of latent TGF-β isoform (i.e., TGF-β1) and can activate it through the expression of cell surface docking receptor GARP and αv integrins [65]. On the other side, TGF-β, along with IL-2, is essential to induce CD4+ T cells to express FoxP3, which is crucial for Tregs development [66]. High levels of TGF-β expressed by tumor cells contribute to the establishment of the local immunosuppressive environment. It is achieved by blocking naive T cell differentiation into a Th1 effector phenotype, promoting their conversion into the Tregs subset, and abolishing antigen-presenting functions of DCs [30]. Both Tregs and TGF-β expression or activation increases in irradiated tissues, but available data are inconsistent about the underlying mechanism [52]. For instance, in melanoma, kidney cancer, and CRC models, stereotactic radiotherapy elevates the levels of intratumoral Tregs in a non-TGF-β-dependent manner, while in a murine prostate cancer model, TGF-β1 mediates Tregs elevation in response to radiation [52,67]. The regrowth of irradiated tumors was significantly correlated with TGF- β1 levels and Treg accumulation when mouse models of prostate cancer were exposed to sub-lethal doses of radiation. Moreover, the inhibition of TGF-1 led to a reduction in Treg accumulation and tumor regrowth after treatment [67]. It should be underlined that radiation-induced TGF-β production depends on dose, time, and tissue [68]. Further investigation is needed to fully understand the precise role of TGF-β in the accumulation of Tregs in various tumor tissues (e.g., lung, prostate, HNSCC) [68,69]. Chemotherapy effects are correlated with epithelial–mesenchymal transition (EMT), the process that contributes to stem cell generation, anticancer drug resistance, genomic instability, and localized immunosuppression [34]. The mechanisms in which cytotoxic agents (e.g., cisplatin) increase TGF-β expression are led through the activation of intracellular transcriptional effectors SMAD (Figure 11) [70]. Table 3 summarizes other mechanisms of the TGF-β-dependent upregulation of Tregs.
3. Materials and Methods
This narrative review was conducted according to the SANRA guidelines (https://researchintegrityjournal.biomedcentral.com/articles/10.1186/s41073-019-0064-8; accessed on 5 October 2022). In October 2022, we performed a search using PubMed, Scopus, and Google Scholar. We used the following search queries: “regulatory T cell”, “treatment resistance”, “immunotherapy”, “chemotherapy”, “radiotherapy”. Additionally, more articles were found in the references section of the included articles. Studies in languages other than English were excluded from this narrative review.
4. Conclusions
Tregs play an important role in suppressing the antitumor response due to their intrinsic immunosuppressive properties. A high level of tumor-infiltrating Tregs in TME is a negative prognostic factor for various types of cancer. Understanding the role of this subset of T cells and their interactions with other cells and molecules is crucial for the development of effective therapeutic regimens. In this review, we have collected and presented available information on the essential roles of Tregs in cancer treatment resistance. In addition, we have summarized and explained how available therapeutics and their combinations may enhance Tregs targeting that could overcome Tregs-mediated resistance and eventually improve patient outcomes. For example, combining different ICIs (e.g., anti-CTLA-4 and anti-PD-L1) leads to an increased Teffs to Tregs ratio and enhanced immunogenic cell death. Various therapeutic methods that increase the immunogenicity of tumors (e.g., chemo- or radiotherapy) with ICIs can also increase their overall effectiveness. Moreover, we recognized the crucial molecular mechanisms of Treg-mediated resistance that are potential targets for novel therapeutics. Undoubtedly, numerous combination possibilities will exist in the future. However, Tregs-targeted therapies require further extensive research to develop the most optimal, safe, and effective therapeutic strategies to reduce side effects and overcome cancer treatment resistance.
Author Contributions
Conceptualization, B.K.S. and A.P.; methodology, A.D., M.G., B.K.S. and A.P.; validation, A.D., M.G., A.B., O.S., B.K.S. and A.P.; formal analysis, B.K.S. and A.P.; investigation, A.D., M.G., A.B., O.S., B.K.S. and A.P.; resources, A.D., M.G., A.B., O.S., B.K.S. and A.P.; data curation, A.D., M.G., A.B., B.K.S. and A.P.; writing—original draft preparation, A.D., M.G., A.B., O.S., B.K.S. and A.P.; writing—review and editing, A.D., M.G., A.B., O.S., B.K.S. and A.P.; visualization, A.D., M.G. and O.S.; supervision, A.B., B.K.S. and A.P.; project administration, B.K.S. and A.P.; funding acquisition, B.K.S. and A.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Foundation for Polish Science under the International Research Agendas Program financed from the Smart Growth Operational Program 2014–2020 (Grant Agreement No. MAB/2018/6). At the time of manuscript preparation A.P. was supported with Walczak’s Scholarship funded by NAWA-Polish National Agency for Academic Exchange (Scholarship Agreement No. BPN/WAL/2022/1/00024/U/00001).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 4-1BB | tumor necrosis factor receptor superfamily 9 |
| 5-FU | fluorouracil |
| A2AR | adenosine A2A receptor |
| A2BR | adenosine A2B receptor |
| AEP | asparaginyl endopeptidase |
| Akt | cellular homolog of murine thymoma virus Akt8 oncogene |
| ALK | anaplastic lymphoma kinase |
| APC | antigen-presenting cell |
| ATP | adenosine triphosphate |
| ATR | ataxia telangiectasia and RAD3-related protein |
| B2M | β2-microglobulin |
| cAMP | cyclic adenosine monophosphate |
| CD25 | interleukin-2 receptor alpha chain |
| CD39 | ectonucleoside triphosphate diphosphohydrolase-1 |
| CD47 | cluster of differentiation 47 |
| CD73 | ecto-5′-nucleotidase |
| CD80 | cluster of differentiation 80 |
| CD86 | cluster of differentiation 86 |
| CPA | cyclophosphamide |
| CRC | colorectal cancer |
| CTLA-4 | cytotoxic-T-lymphocyte-associated antigen 4 |
| DAMP | damage-associated molecular pattern |
| DC | dendritic cell |
| EMT | epithelial–mesenchymal transition |
| ERK2 | extracellular-signal-regulated kinase 2 |
| Evs | extracellular vesicles |
| FcƔR | Fc-gamma receptor |
| FoxP3 | forkhead box P3 |
| Gal-1 | galectin-1 |
| GARP | glycoprotein A repetitions predominant |
| GITR | glucocorticoid-induced tumor necrosis factor receptor family-related gene |
| GM1 | monosialotetrahexosylganglioside |
| GTP | guanosine triphosphate |
| HIF-1α | hypoxia-inducible factor 1α |
| HIF-2α | hypoxia-inducible factor 2α |
| HNSCC | head and neck squamous cell carcinoma |
| IC | immune checkpoint |
| ICI | immune checkpoint inhibitor |
| ICOS | inducible T cell co-stimulator |
| IDO | indoleamine 2,3-dioxygenase |
| IFN-γ | interferon gamma |
| IL-2 | interleukin 2 |
| IL-6 | interleukin 6 |
| IL-10 | interleukin 10 |
| IL-35 | interleukin 35 |
| iTreg | induced Treg |
| JAK | Janus kinase |
| LAG-3 | lymphocyte activation gene-3 |
| mAb | monoclonal antibody |
| MCT4 | monocarboxylate transporter 4 |
| MHC | histocompatibility complex |
| miRNA | microRNA |
| mTOR | mammalian target of rapamycin |
| NK | natural killer |
| NPM | nucleophosmin |
| NRF2 | nuclear factor erythroid 2-related factor |
| NSCLC | non-small lung cancer |
| nTreg | natural Treg |
| ORR | objective response rate |
| OS | overall survival |
| OX40 | tumor necrosis factor receptor superfamily, member 4 |
| OXPHOS | oxidative phosphorylation |
| PD-1 | programmed cell death 1 |
| PD-L1 | programmed cell death ligand-1 |
| PFS | progression-free survival |
| Pgp | permeability glycoprotein |
| PI3K | phosphoinositide 3-kinase |
| PTEN | phosphatase and tensin homolog deleted on chromosome 10 |
| Ptpn2 | protein tyrosine phosphatase non-receptor type 2 |
| pTreg | peripherally derived Treg |
| ROS | reactive oxygen species |
| RR | response rate |
| SHP2 | Src homology phosphotyrosyl phosphatase 2 |
| SMAD | suppressor od mothers against decapentaplegic |
| STAT | signal transducer and activator of transcription protein |
| TAM | tumor-associated macrophage |
| Tconv | conventional T cell |
| TCR | T cell receptor |
| Teff | effector T cell |
| TET2 | tet metylcytosine dioxygenase 2 |
| TGF-β | transforming growth factor β |
| TIGIT | T cell immunoreceptor with Ig and ITIM domains |
| TIL | Tumor-infiltrating lymphocyte |
| TIM-3 | T cell immunoglobulin and mucin-domain containing-3 protein |
| TME | tumor microenvironment |
| TML | tumor mutational load |
| TNBC | triple-negative breast cancer |
| TNF-α | tumor necrosis factor α |
| Treg | regulatory T cell |
| TRPC5 | transient receptor potential channel 5 |
| tTreg | thymus-derived Treg |
| TSDR | Treg-specific demethylated region |
| VISTA | V-domain immunoglobulin suppressor of T cell activation |
| Wnt | Wingless/Integrated |
References
- Sakaguchi, S.; Mikami, N.; Wing, J.B.; Tanaka, A.; Ichiyama, K.; Ohkura, N. Regulatory T Cells and Human Disease. Annu. Rev. Immunol. 2020, 38, 541–566. [Google Scholar] [CrossRef] [PubMed]
- “Regulatory T Cells Overview|Thermo Fisher Scientific—PL”. Available online: https://www.thermofisher.com/pl/en/home/life-science/cell-analysis/cell-analysis-learning-center/immunology-at-work/regulatory-t-cell-overview.html (accessed on 11 July 2023).
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. J. Immunol. 2017, 198, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Niederlova, V.; Tsyklauri, O.; Chadimova, T.; Stepanek, O. CD8+ Tregs revisited: A heterogeneous population with different phenotypes and properties. Eur. J. Immunol. 2021, 51, 512–530. [Google Scholar] [CrossRef] [PubMed]
- Gershon, R.K.; Kondo, K. Cell Interactions in the Induction of Tolerance: The Role of Thymic Lymphocytes. Immunology 1970, 18, 723–737. [Google Scholar]
- Shevyrev, D.; Tereshchenko, V. Treg Heterogeneity, Function, and Homeostasis. Front. Immunol. 2020, 10, 3100. [Google Scholar] [CrossRef]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef]
- Kumar, P.; Bhattacharya, P.; Prabhakar, B.S. A comprehensive review on the role of co-signaling receptors and Treg homeostasis in autoimmunity and tumor immunity. J. Autoimmun. 2018, 95, 77–99. [Google Scholar] [CrossRef]
- Johnson, D.B.; Nebhan, C.A.; Moslehi, J.J.; Balko, J.M. Immune-checkpoint inhibitors: Long-term implications of toxicity. Nat. Rev. Clin. Oncol. 2022, 19, 254–267. [Google Scholar] [CrossRef]
- Tan, S.; Li, D.; Zhu, X. Cancer immunotherapy: Pros, cons and beyond. Biomed. Pharmacother. 2020, 124, 109821. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Long, G.V.; Scolyer, R.A.; Teng, M.W.; Smyth, M.J. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat. Rev. 2017, 52, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Huang, X.; Zhang, G.; Hong, Z.; Bai, X.; Liang, T. Advantages of targeting the tumor immune microenvironment over blocking immune checkpoint in cancer immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 72. [Google Scholar] [CrossRef] [PubMed]
- Maj, T.; Wang, W.; Crespo, J.; Zhang, H.; Wang, W.; Wei, S.; Zhao, L.; Vatan, L.; Shao, I.; Szeliga, W.; et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 2017, 18, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors in cancer therapy: A focus on T-regulatory cells. Immunol. Cell Biol. 2018, 96, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, A.J.; Hellmann, M.D. Acquired Resistance to Immune Checkpoint Inhibitors. Cancer Cell 2020, 37, 443–455. [Google Scholar] [CrossRef]
- Wu, C.C.; Wang, Y.A.; Livingston, J.A.; Zhang, J.; Futreal, P.A. Prediction of biomarkers and therapeutic combinations for anti-PD-1 immunotherapy using the global gene network association. Nat. Commun. 2022, 13, 42. [Google Scholar] [CrossRef]
- Saito, T.; Nishikawa, H.; Wada, H.; Nagano, Y.; Sugiyama, D.; Atarashi, K.; Maeda, Y.; Hamaguchi, M.; Ohkura, N.; Sato, E.; et al. Two FOXP3 + CD4 + T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat. Med. 2016, 22, 679–684. [Google Scholar] [CrossRef]
- Chaudhary, B.; Elkord, E. Regulatory T cells in the tumor microenvironment and cancer progression: Role and therapeutic targeting. Vaccines 2016, 4, 28. [Google Scholar] [CrossRef]
- Dahan, R.; Sega, E.; Engelhardt, J.; Selby, M.; Korman, A.J.; Ravetch, J.V. FcγRs Modulate the Anti-tumor Activity of Antibodies Targeting the PD-1/PD-L1 Axis. Cancer Cell 2015, 28, 285–295. [Google Scholar] [CrossRef]
- Saleh, R.; Elkord, E. FoxP3+ T regulatory cells in cancer: Prognostic biomarkers and therapeutic targets. Cancer Lett. 2020, 490, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, Z.-H.; Gabius, H.-J.; Rohowsky-Kochan, C.; Ledeen, R.W.; Wu, G. Cross-Linking of GM1 Ganglioside by Galectin-1 Mediates Regulatory T Cell Activity Involving TRPC5 Channel Activation: Possible Role in Suppressing Experimental Autoimmune Encephalomyelitis. J. Immunol. 2009, 182, 4036–4045. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Chen, X.; Zhu, W.; Yu, J.; Li, W.; Li, Y.; Li, Z.; Olsen, N.; Liang, D.; Zheng, S.G. The cAMP–Adenosine Feedback Loop Maintains the Suppressive Function of Regulatory T Cells. J. Immunol. 2019, 203, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Li, H.X.; Zheng, C.; Han, J.; Zhu, J.; Liu, S.; Jin, T. PD-1/PD-L1 Axis as a Potential Therapeutic Target for Multiple Sclerosis: A T Cell Perspective. Front. Cell. Neurosci. 2021, 15, 716747. [Google Scholar] [CrossRef]
- Wardell, C.M.; MacDonald, K.N.; Levings, M.K.; Cook, L. Cross talk between human regulatory T cells and antigen-presenting cells: Lessons for clinical applications. Eur. J. Immunol. 2021, 51, 27–38. [Google Scholar] [CrossRef]
- Routy, J.P.; Routy, B.; Graziani, G.M.; Mehraj, V. The kynurenine pathway is a double-edged sword in immune-privileged sites and in cancer: Implications for immunotherapy. Int. J. Tryptophan Res. 2016, 9, 67–77. [Google Scholar] [CrossRef]
- Tung, S.L.; Boardman, D.A.; Sen, M.; Letizia, M.; Peng, Q.; Cianci, N.; Dioni, L.; Carlin, L.; Lechler, R.; Bollati, V.; et al. Regulatory T cell-derived extracellular vesicles modify dendritic cell function. Sci. Rep. 2018, 8, 6065. [Google Scholar] [CrossRef]
- Bailly, C.; Thuru, X.; Quesnel, B. Survey and summary: Combined cytotoxic chemotherapy and immunotherapy of cancer: Modern times. NAR Cancer 2020, 2, zcaa002. [Google Scholar] [CrossRef]
- Stuelten, C.H.; Zhang, Y.E. Transforming Growth Factor-β: An Agent of Change in the Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9, 764727. [Google Scholar] [CrossRef]
- Li, H.; van der Merwe, P.A.; Sivakumar, S. Biomarkers of response to PD-1 pathway blockade. Br. J. Cancer 2022, 126, 1663–1675. [Google Scholar] [CrossRef]
- Yadollahi, P.; Jeon, Y.K.; Ng, W.L.; Choi, I. Current understanding of cancer-intrinsic PD-L1: Regulation of expression and its protumoral activity. BMB Rep. 2021, 54, 12–20. [Google Scholar] [CrossRef]
- Hsieh, R.C.E.; Krishnan, S.; Wu, R.C.; Boda, A.R.; Liu, A.; Winkler, M.; Hsu, W.-H.; Lin, S.H.; Hung, M.-C.; Chan, L.-C.; et al. ATR-mediated CD47 and PD-L1 up-regulation restricts radiotherapy-induced immune priming and abscopal responses in colorectal cancer. Sci. Immunol. 2022, 7, eabl9330. [Google Scholar] [CrossRef]
- Funaki, S.; Shintani, Y.; Ura, T.K.; Kanzaki, R.; Minami, M.; Okumura, M. Chemotherapy enhances programmed cell death 1/ligand 1 expression via TGF-β induced epithelial mesenchymal transition in non-small cell lung cancer. Oncol. Rep. 2017, 38, 2277–2284. [Google Scholar] [CrossRef]
- Cai, J.; Wang, D.; Zhang, G.; Guo, X. The role of PD-1/PD-L1 axis in treg development and function: Implications for cancer immunotherapy. Onco Targets Ther. 2019, 12, 8437–8445. [Google Scholar] [CrossRef] [PubMed]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Balogh, A.; Persa, E.; Bogdándi, E.N.; Benedek, A.; Hegyesi, H.; Sáfrány, G.; Lumniczky, K. The effect of ionizing radiation on the homeostasis and functional integrity of murine splenic regulatory T cells. Inflamm. Res. 2013, 62, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Y.; Duan, X.-F.; Wang, L.-P.; Xu, Y.-J.; Huang, L.; Zhang, T.-F.; Liu, J.-Y.; Li, F.; Zhang, Z.; Yue, D.-L.; et al. Selective depletion of regulatory t cell subsets by docetaxel treatment in patients with nonsmall cell lung cancer. J. Immunol. Res. 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, D.; Ren, H.; Chen, Y. Effects of modified FOLFOX-6 chemotherapy on cellular immune function in patients with gastric cancer. Oncol. Lett. 2018, 15, 8635–8640. [Google Scholar] [CrossRef]
- Saida, Y.; Watanabe, S.; Tanaka, T.; Baba, J.; Sato, K.; Shoji, S.; Igarashi, N.; Kondo, R.; Okajima, M.; Koshio, J.; et al. Critical Roles of Chemoresistant Effector and Regulatory T Cells in Antitumor Immunity after Lymphodepleting Chemotherapy. J. Immunol. 2015, 195, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017, 27, 109–118. [Google Scholar] [CrossRef]
- Shin, J.; Chung, J.H.; Kim, S.H.; Lee, K.S.; Suh, K.J.; Lee, J.Y.; Lee, J.-O.; Kim, J.-W.; Kim, Y.-J.; Lee, K.-W.; et al. Effect of platinum-based chemotherapy on PD-L1 expression on tumor cells in non-small cell lung cancer. Cancer Res. Treat. 2019, 51, 1086–1097. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Han, H.; Zhang, F.; Lv, T.; Zhan, P.; Ye, M.; Song, Y.; Liu, H. Immune checkpoint inhibitors alone vs. immune checkpoint inhibitors—Combined chemotherapy for NSCLC patients with high PD-L1 expression: A network meta-analysis. Br. J. Cancer 2022, 127, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Knitz, M.W.; Bickett, T.E.; Darragh, L.B.; Oweida, A.J.; Bhatia, S.; Van Court, B.; Bhuvane, S.; Piper, M.; Gadwa, J.; Mueller, A.C.; et al. Targeting resistance to radiation-immunotherapy in cold HNSCCs by modulating the Treg-dendritic cell axis. J. Immunother. Cancer 2021, 9, e001955. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Cabrera, R.; Xu, Y.; Liu, C.; Nelson, D. Gamma irradiation alters the phenotype and function of CD4+CD25+ regulatory T cells. Cell Biol. Int. 2019, 33, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Beauford, S.S.; Kumari, A.; Garnett-Benson, C. Ionizing radiation modulates the phenotype and function of human CD4+ induced regulatory T cells. BMC Immunol. 2020, 21, 18. [Google Scholar] [CrossRef] [PubMed]
- Oweida, A.; Hararah, M.K.; Phan, A.; Binder, D.; Bhatia, S.; Lennon, S.; Bukkapatnam, S.; Van Court, B.; Uyanga, N.; Darragh, L.; et al. Resistance to radiotherapy and PD-L1 blockade is mediated by TIM-3 upregulation and regulatory T-cell infiltration. Clin. Cancer Res. 2018, 24, 5368–5380. [Google Scholar] [CrossRef]
- Zhu, L.; Yu, X.; Wang, L.; Liu, J.; Qu, Z.; Zhang, H.; Li, L.; Chen, J.; Zhou, Q. Angiogenesis and immune checkpoint dual blockade in combination with radiotherapy for treatment of solid cancers: Opportunities and challenges. Oncogenesis 2021, 10, 47. [Google Scholar] [CrossRef]
- Joshi, S.; Durden, D.L. Combinatorial Approach to Improve Cancer Immunotherapy: Rational Drug Design Strategy to Simultaneously Hit Multiple Targets to Kill Tumor Cells and to Activate the Immune System. J. Oncol. 2019, 2019, 1–18. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, Y.; Ma, X.; Wang, Y.; Liu, J.; Yang, C.; Wang, Y.; Bao, C.; Song, X.; Feng, Y.; et al. A study on the correlation between M2 macrophages and regulatory T cells in the progression of colorectal cancer. Int. J. Biol. Markers 2022, 37, 412–420. [Google Scholar] [CrossRef]
- Aliyah, S.H.; Ardiyan, Y.N.; Mardhiyah, I.; Herdini, C.; Dwianingsih, E.K.; Aning, S.; Handayani, N.S.N.; Asmara, W.; Fachiroh, J.; Paramita, D.K. The Distribution of M2 Macrophage and Treg in Nasopharyngeal Carcinoma Tumor Tissue and the Correlation with TNM Status and Clinical Stage. Asian Pac. J. Cancer Prev. 2021, 22, 3447–3453. [Google Scholar] [CrossRef]
- Muroyama, Y.; Nirschl, T.R.; Kochel, C.M.; Lopez-Bujanda, Z.; Theodros, D.; Mao, W.; Carrera-Haro, M.A.; Ghasemzadeh, A.; Marciscano, A.E.; Velarde, E.; et al. Stereotactic radiotherapy increases functionally suppressive regulatory T cells in the tumor microenvironment. Cancer Immunol. Res. 2017, 5, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wu, S.; Meng, X.; Liu, G.; Chen, D.; Cong, Y.; Shen, G.; Sun, B.; Wang, W.; Wang, Q.; et al. Predictive value of peripheral regulatory T cells in non-small cell lung cancer patients undergoing radiotherapy. Oncotarget 2017, 8, 43427–43438. [Google Scholar] [CrossRef] [PubMed]
- Oweida, A.; Darragh, L.; Bhatia, S.; Raben, D.; Heasley, L.; Nemenoff, R.; Clambey, E.; Karam, S. Regulatory T cells mediate resistance to radiotherapy in head and neck squamous cell carcinoma. J. Clin. Oncol. 2019, 37, 70. [Google Scholar] [CrossRef]
- Bos, P.D.; Plitas, G.; Rudra, D.; Lee, S.Y.; Rudensky, A.Y. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J. Exp. Med. 2013, 210, 2435–2466. [Google Scholar] [CrossRef]
- Ji, D.; Song, C.; Li, Y.; Xia, J.; Wu, Y.; Jia, J.; Cui, X.; Yu, S.; Gu, J. Combination of radiotherapy and suppression of Tregs enhances abscopal antitumor effect and inhibits metastasis in rectal cancer. J. Immunother. Cancer 2020, 8, e000826. [Google Scholar] [CrossRef]
- Feng, L.L.; Cai, Y.Q.; Zhu, M.C.; Xing, L.-J.; Wang, X. The yin and yang functions of extracellular ATP and adenosine in tumor immunity. Cancer Cell Int. 2020, 20, 110. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef]
- Conforti, L. Potassium channels of T lymphocytes take center stage in the fight against cancer. J. Immunother. 2017, 5, 2. [Google Scholar] [CrossRef]
- Sebastian, C.; Ferrer, C.; Serra, M.; Choi, J.-E.; Ducano, N.; Mira, A.; Shah, M.S.; Stopka, S.A.; Perciaccante, A.J.; Isella, C.; et al. A non-dividing cell population with high pyruvate dehydrogenase kinase activity regulates metabolic heterogeneity and tumorigenesis in the intestine. Nat. Commun. 2022, 13, 1503. [Google Scholar] [CrossRef]
- van Gisbergen, M.W.; Zwilling, E.; Dubois, L.J. Metabolic Rewiring in Radiation Oncology Toward Improving the Therapeutic Ratio. Front. Oncol. 2021, 11, 653621. [Google Scholar] [CrossRef]
- Raychaudhuri, D.; Bhattacharya, R.; Sinha, B.P.; Liu, C.S.C.; Ghosh, A.R.; Rahaman, O.; Bandopadhyay, P.; Sarif, J.; D’Rozario, R.; Paul, S.; et al. Lactate induces pro-tumor reprogramming in intratumoral plasmacytoid dendritic cells. Front. Immunol. 2019, 10, 1878. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Vuckovic, I.; Zhang, S.; Xiong, Y.; Carlson, B.L.; Jacobs, J.; Olson, I.; Petterson, X.-M.; Macura, S.I.; Sarkaria, J.; et al. Radiation Induced Metabolic Alterations Associate with Tumor Aggressiveness and Poor Outcome in Glioblastoma. Front. Oncol. 2020, 10, 535. [Google Scholar] [CrossRef] [PubMed]
- Ring, S.; Enk, A.H.; Mahnke, K. ATP Activates Regulatory T Cells In Vivo during Contact Hypersensitivity Reactions. J. Immunol. 2010, 184, 3408–3416. [Google Scholar] [CrossRef]
- Moreau, J.M.; Velegraki, M.; Bolyard, C.; Rosenblum, M.D.; Li, Z. Transforming growth factor–β1 in regulatory T cell biology. Sci. Immunol. 2022, 7, eabi4613. [Google Scholar] [CrossRef]
- Zheng, S.G.; Wang, J.; Wang, P.; Gray, J.D.; Horwitz, D.A. IL-2 Is Essential for TGF-β to Convert Naive CD4+CD25− Cells to CD25+Foxp3+ Regulatory T Cells and for Expansion of These Cells. J. Immunol. 2007, 178, 2018–2027. [Google Scholar] [CrossRef]
- Wu, C.T.; Hsieh, C.C.; Yen, T.C.; Chen, W.-C.; Chen, M.-F. TGF-β1 mediates the radiation response of prostate cancer. J. Mol. Med. 2014, 93, 73–82. [Google Scholar] [CrossRef]
- Guo, T.; Zou, L.; Ni, J.; Zhou, Y.; Ye, L.; Yang, X.; Zhu, Z. Regulatory T cells: An emerging player in radiation-induced lung injury. Front. Immunol. 2020, 11, 1769. [Google Scholar] [CrossRef]
- Liu, S.; Sun, X.; Luo, J.; Zhu, H.; Yang, X.; Guo, Q.; Song, Y.; Sun, X. Effects of radiation on T regulatory cells in normal states and cancer: Mechanisms and clinical implications. Am. J. Cancer Res. 2015, 5, 3276–3285. [Google Scholar]
- Zheng, H.; Kang, Y. Multilayer control of the EMT master regulators. Oncogene 2014, 33, 1755–1763. [Google Scholar] [CrossRef]
- Oshimori, N.; Oristian, D.; Fuchs, E. TGF-β Promotes Heterogeneity and Drug Resistance in Squamous Cell Carcinoma. Cell 2015, 160, 963–976. [Google Scholar] [CrossRef]
- Neel, J.C.; Humbert, L.; Lebrun, J.J. The Dual Role of TGFβ in Human Cancer: From Tumor Suppression to Cancer Metastasis. ISRN Mol. Biol. 2012, 2012, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Lainé, A.; Labiad, O.; Hernandez-Vargas, H.; This, S.; Sanlaville, A.; Léon, S.; Dalle, S.; Sheppard, D.; Travis, M.A.; Paidassi, H.; et al. Regulatory T cells promote cancer immune-escape through integrin αvβ8-mediated TGF-β activation. Nat. Commun. 2021, 12, 6228. [Google Scholar] [CrossRef] [PubMed]
- Quan, Q.; Zhong, F.; Wang, X.; Chen, K.; Guo, L. PAR2 inhibition enhanced the sensitivity of colorectal cancer cells to 5-FU and reduced EMT signaling. Oncol. Res. 2019, 27, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, Z.; Wang, Z.; Du, G.; Lun, L. TGF-beta signaling in cancer radiotherapy. Cytokine 2021, 148, 155709. [Google Scholar] [CrossRef]
- Liu, R.M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef]
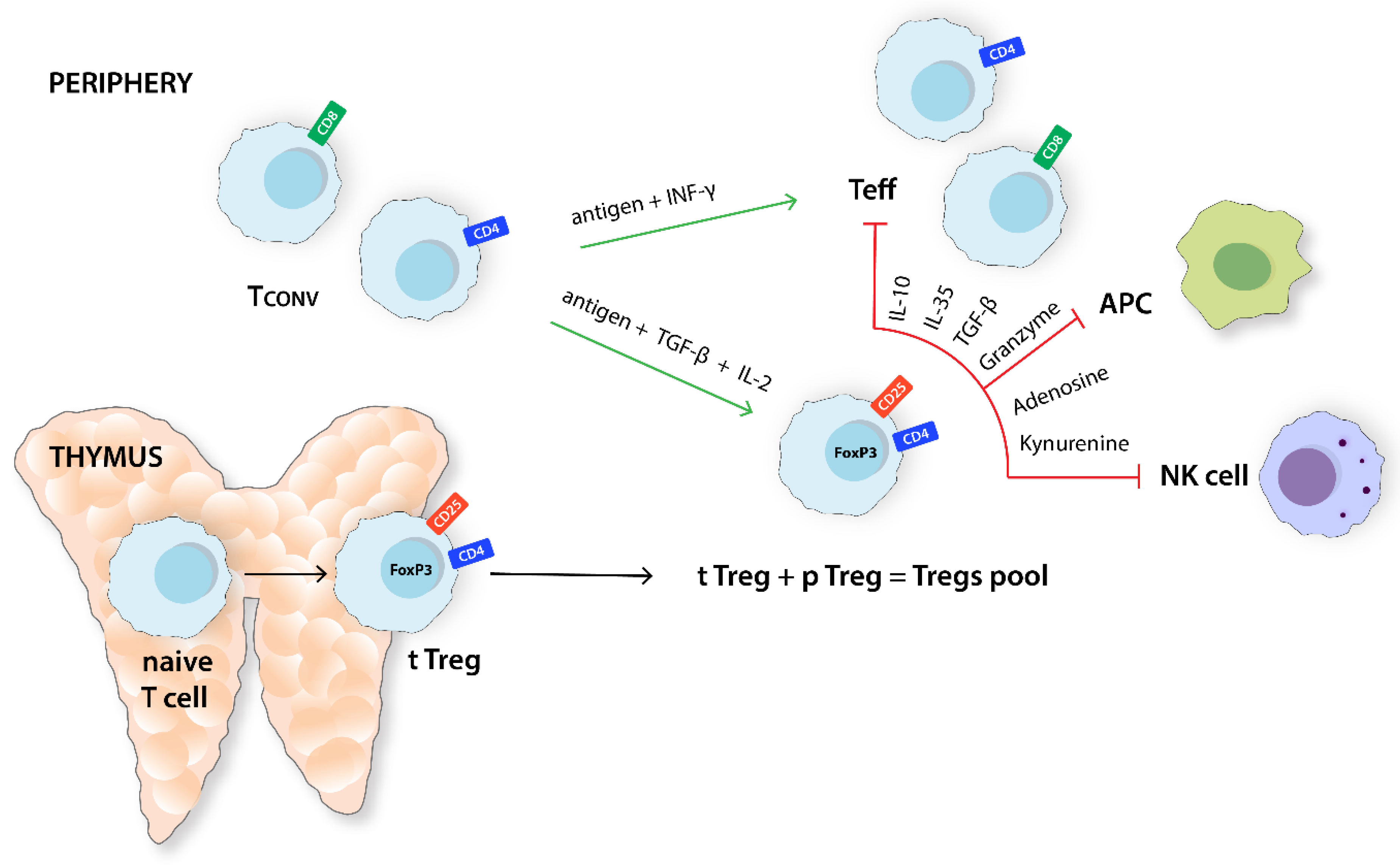
Figure 1.
Differentiation and the main function of Tregs. CD4+CD25+FoxP3+Tregs population consists of fractions derived from the thymus and those arising from peripheral Tconv in the presence of environmental antigen stimulation, TGF-β and IL-2. Tregs decrease excessive inflammation, inhibiting the activity of Teffs, APC, and NK cells. Tconv, T conventional cell; IFN-γ, interferon γ; Teff, effector T cell; TGF-β, transforming growth factor β; APC, antigen-presenting cell; NK cell, natural killer cell; FoxP3, forkhead box P3; tTreg, thymus-derived Treg; pTreg, peripherally derived Treg.
Figure 1.
Differentiation and the main function of Tregs. CD4+CD25+FoxP3+Tregs population consists of fractions derived from the thymus and those arising from peripheral Tconv in the presence of environmental antigen stimulation, TGF-β and IL-2. Tregs decrease excessive inflammation, inhibiting the activity of Teffs, APC, and NK cells. Tconv, T conventional cell; IFN-γ, interferon γ; Teff, effector T cell; TGF-β, transforming growth factor β; APC, antigen-presenting cell; NK cell, natural killer cell; FoxP3, forkhead box P3; tTreg, thymus-derived Treg; pTreg, peripherally derived Treg.

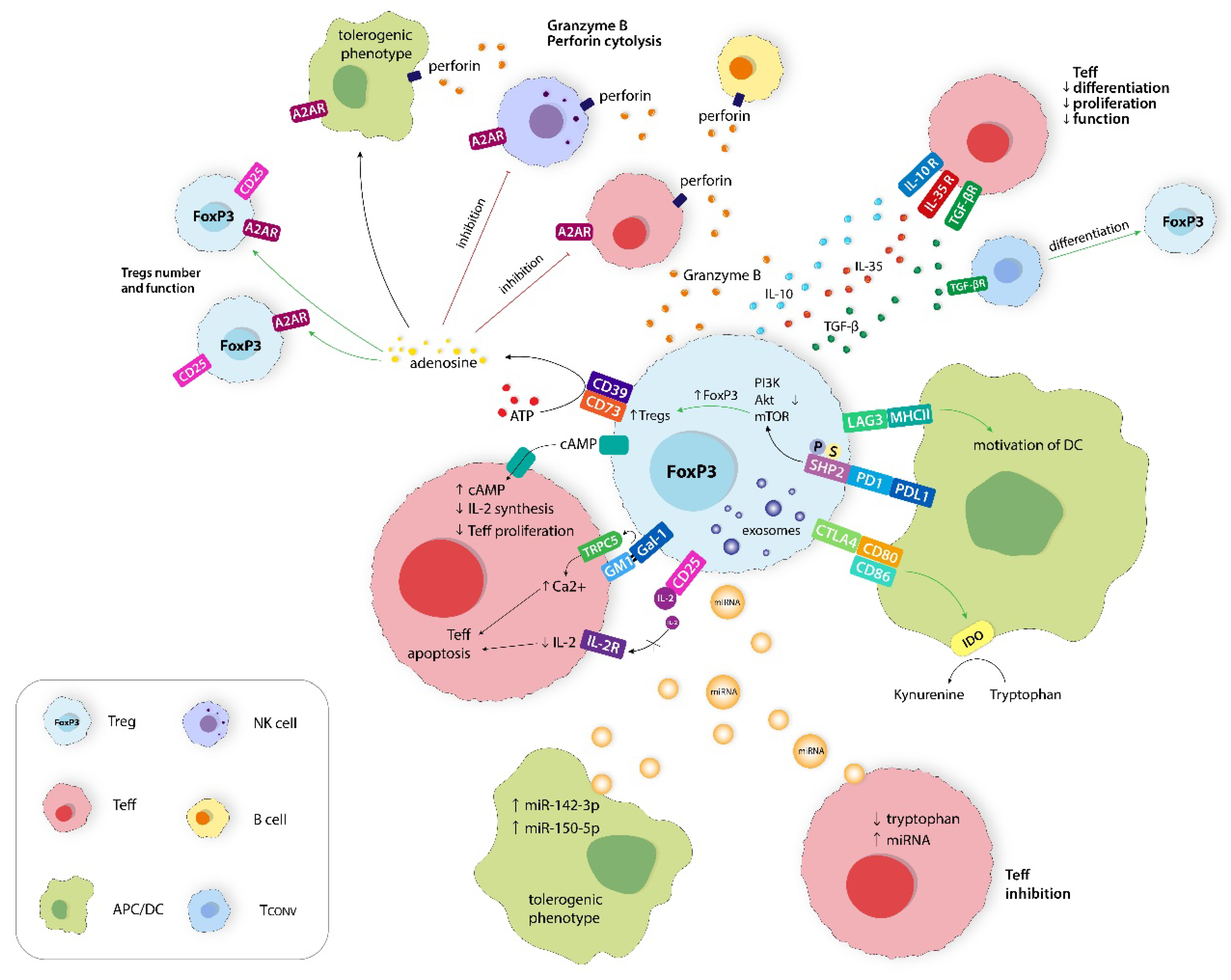
Figure 2.
Immunosuppression mechanisms of Tregs are directed to inhibit Teffs, NK cells, B cells, and APC functions. This includes metabolic disruption on Teffs, expression of inhibitory receptors, production of immunosuppressive molecules, and secretion of extracellular vesicles (Evs). A2AR, adenosine 2A receptor; PI3K, phosphoinositide 3-kinase; Akt, cellular homolog of murine thymoma virus Akt8 oncogene; mTOR, mammalian (or mechanistic) target of rapamycin; SHP2, Src homology phosphotyrosyl phosphatase 2; IDO, indolamine 2,3-dioksygenase 1; miRNA, microRNA; Gal-1, Galectin-1; TRPC5, transient receptor potential channel 5; Ca2+, calcium ion; GM1, monosialotetrahexosylganglioside; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; APC/DC, antigen-presenting cell/dendritic cell; Tconv, T conventional lymphocyte.
Figure 2.
Immunosuppression mechanisms of Tregs are directed to inhibit Teffs, NK cells, B cells, and APC functions. This includes metabolic disruption on Teffs, expression of inhibitory receptors, production of immunosuppressive molecules, and secretion of extracellular vesicles (Evs). A2AR, adenosine 2A receptor; PI3K, phosphoinositide 3-kinase; Akt, cellular homolog of murine thymoma virus Akt8 oncogene; mTOR, mammalian (or mechanistic) target of rapamycin; SHP2, Src homology phosphotyrosyl phosphatase 2; IDO, indolamine 2,3-dioksygenase 1; miRNA, microRNA; Gal-1, Galectin-1; TRPC5, transient receptor potential channel 5; Ca2+, calcium ion; GM1, monosialotetrahexosylganglioside; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; APC/DC, antigen-presenting cell/dendritic cell; Tconv, T conventional lymphocyte.

Figure 3.
(A) Examples of immunosuppressive and immunostimulatory cells in TME. (B) Tregs are an immunosuppressive cell subset, which can express multiple co-signaling receptors, both inhibitory and stimulatory, all important for Treg homeostasis. CTLA-4, OX-40, GITR, and BTLA are constitutively expressed by Tregs, while PD-1, TIGIT, TIM-3, LAG-3, and 4-1BB are preferentially overexpressed on tumor-infiltrating Tregs. Activation of coinhibitory receptors through ligation with corresponding ligands leads to enhanced Treg function, resulting in tumor progression. Activation of costimulatory receptors on Tregs can both promote and diminish their suppressive function [9]. MDSC, myeloid-derived suppressor cell; MC, mast cell; ILC2/3, innate lymphoid cell type 2 and 3; M2-like tumor-associated macrophages; TIL, tumor-infiltrating CD4+ and CD8+ lymphocytes; NK, natural killer cell; DC, dendritic cell; BTLA, B, and T lymphocyte attenuator; HVEM, herpesvirus entry mediator; LAG-3, lymphocyte-activation gene 3; MHCII, major histocompatibility complex II; TIM-3, T cell immunoglobulin and mucin-domain containing-3; Gal-9, Galectin 9; TIGIT, T cell immunoreceptor with Ig and ITIM domains; PD-1, programmed cell death; PD-L1, programmed cell death ligand; CTLA-4, cytotoxic T cell antigen 4; 4-1BB/4-1BBL, tumor necrosis factor receptor superfamily 9 and ligand; GITR/GITRL, glucocorticoid-induced TNFR-related protein and ligand; OX40/OX40L, tumor necrosis factor receptor superfamily, member 4/ligand; ICOS/ICOSL, inducible T cell co-stimulator/ligand; APC, antigen-presenting cell.
Figure 3.
(A) Examples of immunosuppressive and immunostimulatory cells in TME. (B) Tregs are an immunosuppressive cell subset, which can express multiple co-signaling receptors, both inhibitory and stimulatory, all important for Treg homeostasis. CTLA-4, OX-40, GITR, and BTLA are constitutively expressed by Tregs, while PD-1, TIGIT, TIM-3, LAG-3, and 4-1BB are preferentially overexpressed on tumor-infiltrating Tregs. Activation of coinhibitory receptors through ligation with corresponding ligands leads to enhanced Treg function, resulting in tumor progression. Activation of costimulatory receptors on Tregs can both promote and diminish their suppressive function [9]. MDSC, myeloid-derived suppressor cell; MC, mast cell; ILC2/3, innate lymphoid cell type 2 and 3; M2-like tumor-associated macrophages; TIL, tumor-infiltrating CD4+ and CD8+ lymphocytes; NK, natural killer cell; DC, dendritic cell; BTLA, B, and T lymphocyte attenuator; HVEM, herpesvirus entry mediator; LAG-3, lymphocyte-activation gene 3; MHCII, major histocompatibility complex II; TIM-3, T cell immunoglobulin and mucin-domain containing-3; Gal-9, Galectin 9; TIGIT, T cell immunoreceptor with Ig and ITIM domains; PD-1, programmed cell death; PD-L1, programmed cell death ligand; CTLA-4, cytotoxic T cell antigen 4; 4-1BB/4-1BBL, tumor necrosis factor receptor superfamily 9 and ligand; GITR/GITRL, glucocorticoid-induced TNFR-related protein and ligand; OX40/OX40L, tumor necrosis factor receptor superfamily, member 4/ligand; ICOS/ICOSL, inducible T cell co-stimulator/ligand; APC, antigen-presenting cell.

Figure 4.
PD-1/PD-L1 axis blockade leads to restoration of Teffs and Tregs suppression, resulting in increased Teffs/Tregs ratio. Anti-PD-1 Ab, anti-PD-1 antibody; anti-PD-L1 Ab, anti-PD-L1 antibody; FoxP3, forkhead box P3; PD-1, programmed cell death; PD-L1, programmed cell death ligand; Teffs, effector T cells; TME, tumor microenvironment; Tregs, regulatory T cells.
Figure 4.
PD-1/PD-L1 axis blockade leads to restoration of Teffs and Tregs suppression, resulting in increased Teffs/Tregs ratio. Anti-PD-1 Ab, anti-PD-1 antibody; anti-PD-L1 Ab, anti-PD-L1 antibody; FoxP3, forkhead box P3; PD-1, programmed cell death; PD-L1, programmed cell death ligand; Teffs, effector T cells; TME, tumor microenvironment; Tregs, regulatory T cells.

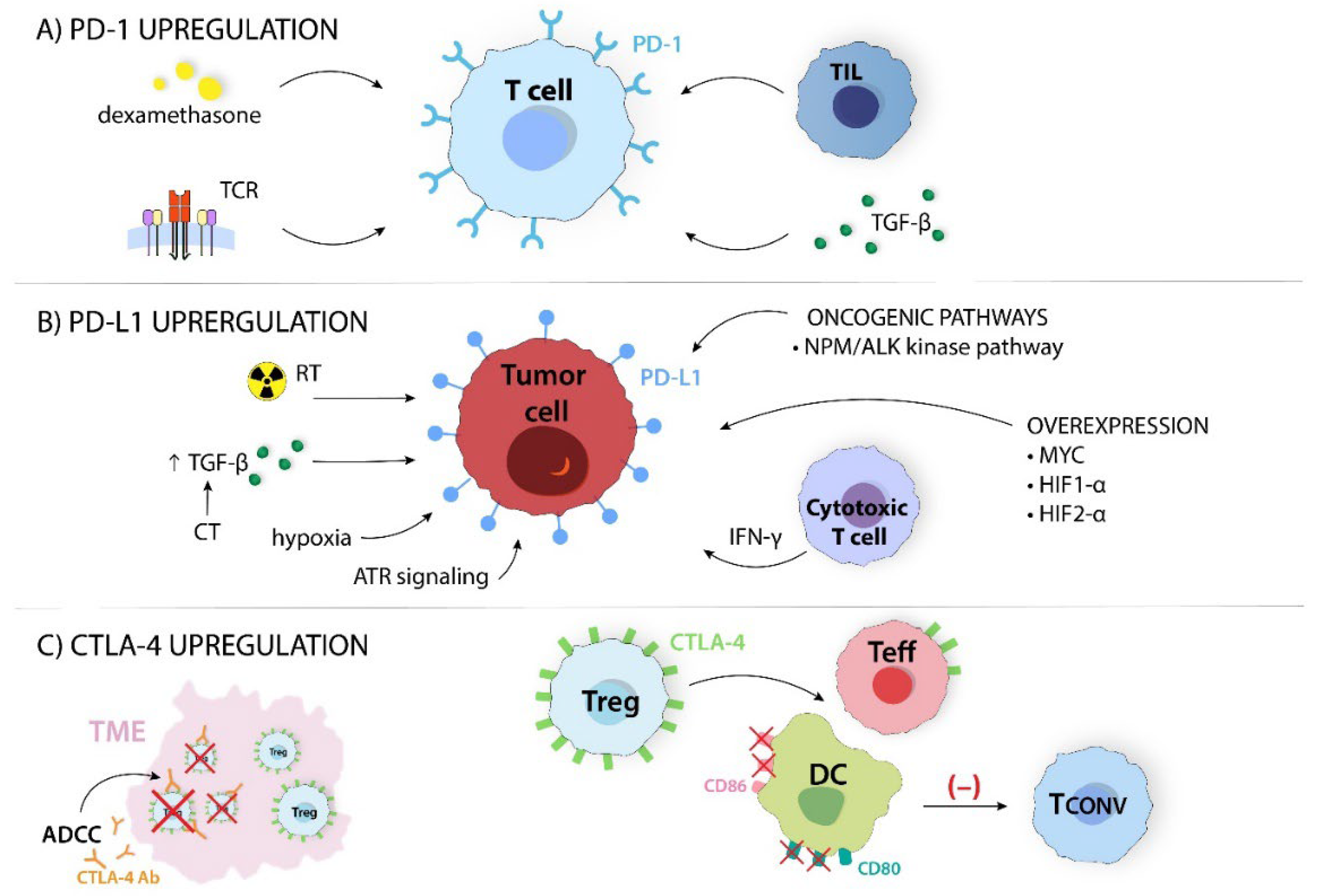
Figure 5.
Many ICs expression mechanisms can affect the properties of immune cells. (A) PD-1, programmed cell death; TCR, T cell receptor; TGF-β, transforming growth factor β; TIL, tumor-infiltrating lymphocyte; (B) ATR, ataxia telangiectasia and RAD3-related protein; CT, chemotherapy; HIF-1α, hypoxia-inducible factor 1α; HIF-2α, hypoxia-inducible factor 2α; IFNγ, interferon γ; NPM/ALK, nucleophosmin/anaplastic lymphoma kinase; PD-L1, programmed cell death ligand; RT, radiotherapy; TGF-β, transforming growth factor β; (C) ADCC, antibody-dependent cellular cytotoxicity; CD80, cluster of differentiation 80; CD86, cluster of differentiation 86; CTLA-4, cytotoxic-T-lymphocyte-associated antigen 4; CTLA-4 Ab, cytotoxic-T-lymphocyte-associated antigen 4 antibody; DC, dendritic cell; Tconv, conventional T cell; Teff, effector T cell; TME, tumor microenvironment; Treg, regulatory T cell.
Figure 5.
Many ICs expression mechanisms can affect the properties of immune cells. (A) PD-1, programmed cell death; TCR, T cell receptor; TGF-β, transforming growth factor β; TIL, tumor-infiltrating lymphocyte; (B) ATR, ataxia telangiectasia and RAD3-related protein; CT, chemotherapy; HIF-1α, hypoxia-inducible factor 1α; HIF-2α, hypoxia-inducible factor 2α; IFNγ, interferon γ; NPM/ALK, nucleophosmin/anaplastic lymphoma kinase; PD-L1, programmed cell death ligand; RT, radiotherapy; TGF-β, transforming growth factor β; (C) ADCC, antibody-dependent cellular cytotoxicity; CD80, cluster of differentiation 80; CD86, cluster of differentiation 86; CTLA-4, cytotoxic-T-lymphocyte-associated antigen 4; CTLA-4 Ab, cytotoxic-T-lymphocyte-associated antigen 4 antibody; DC, dendritic cell; Tconv, conventional T cell; Teff, effector T cell; TME, tumor microenvironment; Treg, regulatory T cell.

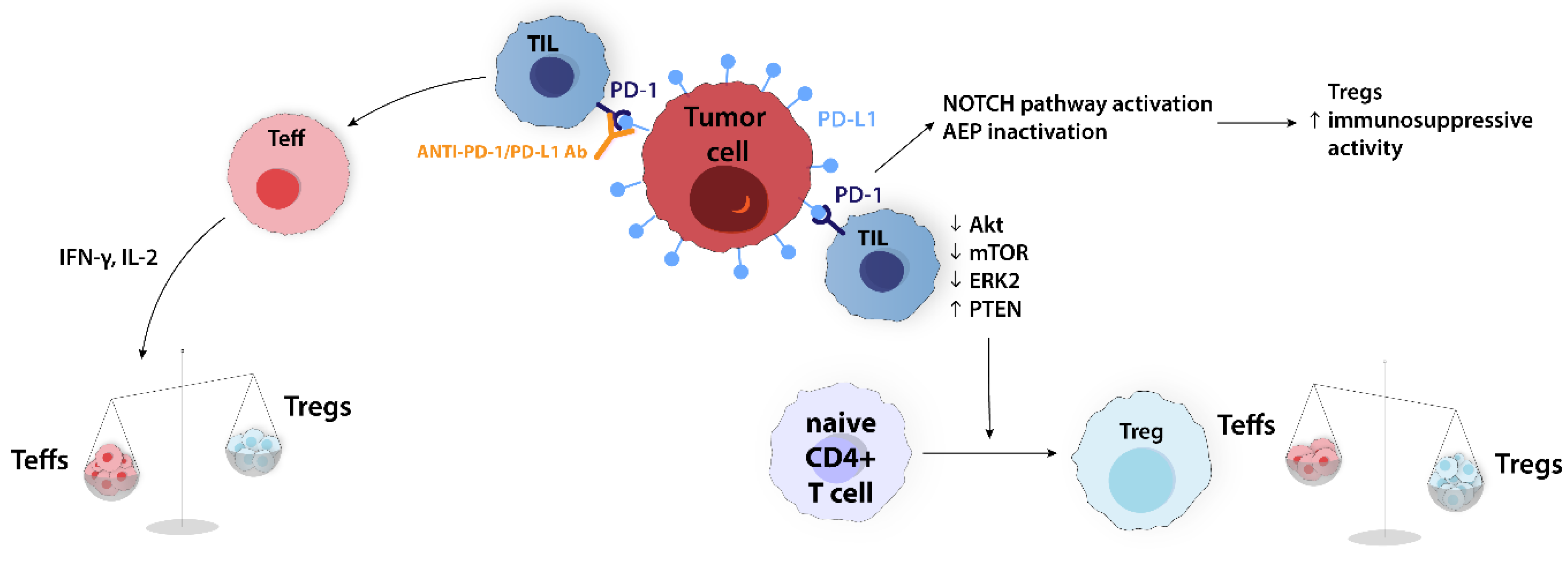
Figure 6.
PD-1/PD-L1 axis affects Tregs and Teffs through different mechanisms. NOTCH pathway activation and AEP inactivation induce Tregs immunosuppressive activity. Downregulation of AKT, mTOR, ERK2, and upregulation of PTEN mediates the conversion of the naïve CD4+ T cells into Tregs. Moreover, blockade of PD-1/PD-L1 interaction can increase Teffs proliferation and restore their secretory function. AEP, asparaginyl endopeptidase; Akt, cellular homolog of murine thymoma virus Akt8 oncogene; anti-PD-1/PD-L1 Ab, anti-PD-1/PD-L1 antibody; ERK2, extracellular-signal-regulated kinase 2; mTOR, mammalian target of rapamycin; PD-1, programmed cell death; PD-L1, programmed cell death ligand; PTEN, phosphatase and tensin homolog deleted on chromosome 10; Teff, effector T cell; TIL, tumor-infiltrated T cell; Treg, regulatory T cell.
Figure 6.
PD-1/PD-L1 axis affects Tregs and Teffs through different mechanisms. NOTCH pathway activation and AEP inactivation induce Tregs immunosuppressive activity. Downregulation of AKT, mTOR, ERK2, and upregulation of PTEN mediates the conversion of the naïve CD4+ T cells into Tregs. Moreover, blockade of PD-1/PD-L1 interaction can increase Teffs proliferation and restore their secretory function. AEP, asparaginyl endopeptidase; Akt, cellular homolog of murine thymoma virus Akt8 oncogene; anti-PD-1/PD-L1 Ab, anti-PD-1/PD-L1 antibody; ERK2, extracellular-signal-regulated kinase 2; mTOR, mammalian target of rapamycin; PD-1, programmed cell death; PD-L1, programmed cell death ligand; PTEN, phosphatase and tensin homolog deleted on chromosome 10; Teff, effector T cell; TIL, tumor-infiltrated T cell; Treg, regulatory T cell.

Figure 7.
Radiation affects the expression of ICs differently depending on the dose. Low-dose radiation upregulates CTLA-4 and CD25 expression. High-dose radiation upregulates LAG-3 and downregulates CTLA-4 and CD25 expression. Radiation, regardless of the dose, upregulates TIM-3 expression. CD25, interleukin 2 receptor alpha chain; CTLA-4, cytotoxic-T-lymphocyte-associated antigen 4; LAG-3, lymphocyte activation gene-3; TIM-3, T cell immunoglobulin and mucin-domain containing-3 protein.
Figure 7.
Radiation affects the expression of ICs differently depending on the dose. Low-dose radiation upregulates CTLA-4 and CD25 expression. High-dose radiation upregulates LAG-3 and downregulates CTLA-4 and CD25 expression. Radiation, regardless of the dose, upregulates TIM-3 expression. CD25, interleukin 2 receptor alpha chain; CTLA-4, cytotoxic-T-lymphocyte-associated antigen 4; LAG-3, lymphocyte activation gene-3; TIM-3, T cell immunoglobulin and mucin-domain containing-3 protein.

Figure 8.
Tregs do not express TIM-3 constitutively, except the intratumoral Tregs. TIM-3-positive Tregs secrete higher amounts of IL-10 compared to TIM-3-negative Tregs. A higher level of IL-10 effectively inhibits IFN-γ and TNF-α secretion by Teffs. IFN-γ, interferon γ; IL-10, interleukin 10; Teff, effector T cell; TIM-3, T cell immunoglobulin and mucin-domain containing-3 protein; TNF-α, tumor necrosis factor α; Treg, regulatory T cell.
Figure 8.
Tregs do not express TIM-3 constitutively, except the intratumoral Tregs. TIM-3-positive Tregs secrete higher amounts of IL-10 compared to TIM-3-negative Tregs. A higher level of IL-10 effectively inhibits IFN-γ and TNF-α secretion by Teffs. IFN-γ, interferon γ; IL-10, interleukin 10; Teff, effector T cell; TIM-3, T cell immunoglobulin and mucin-domain containing-3 protein; TNF-α, tumor necrosis factor α; Treg, regulatory T cell.

Figure 9.
Tregs convert ATP to adenosine, suppressing Teffs. Apoptotic Tregs release more ATP through pannexin-1-dependent channels, which causes more effective suppression of Teffs. ATP, adenosine triphosphate; CD39, ectonucleoside triphosphate diphosphohydrolase-1; CD73, ecto-5′-nucleotidase; Teff, effector T cell; Treg, regulatory T cell.
Figure 9.
Tregs convert ATP to adenosine, suppressing Teffs. Apoptotic Tregs release more ATP through pannexin-1-dependent channels, which causes more effective suppression of Teffs. ATP, adenosine triphosphate; CD39, ectonucleoside triphosphate diphosphohydrolase-1; CD73, ecto-5′-nucleotidase; Teff, effector T cell; Treg, regulatory T cell.

Figure 10.
Numerous processes occur in TME that induce the immunosuppressive properties of Tregs. Hypoxia increases HIF-1α and glycolysis, and decreases OXPHOS, which enhances the amount of lactic acid (LA) in TME. LA stimulates the conversion of tryptophan into kynurenine, which stimulates Tregs. Ionizing radiation causes cancer cell degradation, which increases the amount of potassium in TME, stimulating the conversion of CD4+ T cells into Tregs. ATP, also released from tumor cells, is converted to cAMP, which leads to Tregs recruitment. A2AR, adenosine A2A receptor; A2BR, adenosine A2B receptor; Akt, cellular homolog of murine thymoma virus Akt8 oncogene; ASC, ascorbate; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; CD39, ectonucleoside triphosphate diphosphohydrolase-1; CD73, ecto-5′-nucleotidase; DC, dendritic cell; GSH, glutathione; GTP, guanosine triphosphate; HIF-1α, hypoxia-inducible factor 1α; IDO, indoleamine 2,3-dioxygenase; IR, ionizing radiation; LA, lactic acid; MCT-4, monocarboxylate transporter 4; mTOR, mammalian target of rapamycin; TME, tumor microenvironment; Treg, regulatory T cell.
Figure 10.
Numerous processes occur in TME that induce the immunosuppressive properties of Tregs. Hypoxia increases HIF-1α and glycolysis, and decreases OXPHOS, which enhances the amount of lactic acid (LA) in TME. LA stimulates the conversion of tryptophan into kynurenine, which stimulates Tregs. Ionizing radiation causes cancer cell degradation, which increases the amount of potassium in TME, stimulating the conversion of CD4+ T cells into Tregs. ATP, also released from tumor cells, is converted to cAMP, which leads to Tregs recruitment. A2AR, adenosine A2A receptor; A2BR, adenosine A2B receptor; Akt, cellular homolog of murine thymoma virus Akt8 oncogene; ASC, ascorbate; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; CD39, ectonucleoside triphosphate diphosphohydrolase-1; CD73, ecto-5′-nucleotidase; DC, dendritic cell; GSH, glutathione; GTP, guanosine triphosphate; HIF-1α, hypoxia-inducible factor 1α; IDO, indoleamine 2,3-dioxygenase; IR, ionizing radiation; LA, lactic acid; MCT-4, monocarboxylate transporter 4; mTOR, mammalian target of rapamycin; TME, tumor microenvironment; Treg, regulatory T cell.

Figure 11.
Examples of TGF-β-producing cells (left side). The role of TGF-β in TME is multimodal. TGF-β can stimulate tumor progression in advanced stages. It also promotes the conversion of naive T cells into Tregs and inhibits conversion into Teffs. TGF-β upregulates PD-L1 expression and mediates SMAD activation and abolishes antigen-presenting functions of DCs. CAF, cancer-associated fibroblast; CHT, chemotherapy; DC, dendritic cell; EMT, epithelial–mesenchymal transition; GARP, glycoprotein A repetitions predominant; IL-2, interleukin 2; PD-L1, programmed cell death ligand; RT, radiotherapy; SMAD, suppressor od mothers against decapentaplegic; TCR, T cell receptor; Teff, effector T cell; TGF-β, transforming growth factor β; Treg, regulatory T cell.
Figure 11.
Examples of TGF-β-producing cells (left side). The role of TGF-β in TME is multimodal. TGF-β can stimulate tumor progression in advanced stages. It also promotes the conversion of naive T cells into Tregs and inhibits conversion into Teffs. TGF-β upregulates PD-L1 expression and mediates SMAD activation and abolishes antigen-presenting functions of DCs. CAF, cancer-associated fibroblast; CHT, chemotherapy; DC, dendritic cell; EMT, epithelial–mesenchymal transition; GARP, glycoprotein A repetitions predominant; IL-2, interleukin 2; PD-L1, programmed cell death ligand; RT, radiotherapy; SMAD, suppressor od mothers against decapentaplegic; TCR, T cell receptor; Teff, effector T cell; TGF-β, transforming growth factor β; Treg, regulatory T cell.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Mechanisms of treatment resistance.
| Reference | Type of Resistance | Factor | Mechanism |
|---|---|---|---|
| Wang et al. [23] | Primary | Reduction in IL-2 available for Teffs | IL-2 preferentially binds to CD25 on Tregs |
| Wang et al. [23] | Primary | Cell-bound Gal-1 ligation to GM1 on Teffs | Induces Ca2+ influx via TRPC5 channels, leading to growth arrest |
| Su et al. [24] | Primary | Inhibition of Teffs proliferation and IL-2 synthesis | By directly transferring inhibitory cAMP through the gap junctions of Teffs |
| Li et al. [25] | Primary | Induction of cytolysis of B cells, NK cells, and CD8+ T cells | In a granzyme B- and perforin-dependent manner |
| Li et al. [25] | Primary | Immunosuppressive cytokines such as TGF-β, IL-10, and IL-35 derived from Tregs | Inhibit the differentiation, proliferation, and functions of Teffs |
| Li et al. [25] | Primary | TGF-β | Promotes the conversion of activated Tconv into cells with an immunosuppressive phenotype |
| Li et al. [25] | Primary | LAG-3 expression on Tregs | Binds to MHC class II molecules on immature DC, blocking their maturation and limiting T-cell-mediated immune responses |
| Li et al. [25] | Primary | Recruitment of Src homology phosphotyrosyl phosphatase 2 (SHP2) by PD-1/PD-L1 interaction | Decreases the activity of the PI3K/Akt/mTOR signaling pathway and upregulates the expression of FoxP3, thereby inducing Tregs differentiation |
| Wardell et al. [26], Routy et al. [27] | Primary | CTLA-4 binding to CD80/CD86 on APCs | Limits T cell activation, inhibits T cell responses, induces a CTLA-4-mediated increase in IDO, and lowers the concentration of tryptophan, which is necessary for Teffs to proliferate |
| Routy et al. [27] | Primary | Kynurenine | Protects tissue from inflammation-mediated damage and participates in cancer immune escape |
| Tung et al. [28] | Primary | Releasing Evs, such as exosomes, stocked with miRNA | Inhibit target cells such as Teffs |
| Tung et al. [28] | Primary | Transferring Evs to DC with the induction of a tolerogenic phenotype | Increased immunosuppressive IL-10 and decreased IL-6 production |
| Schoenfeld et al. [17] | Acquired | T-cell-mediated immune activation | Antigen recognition in MHCs presented by APCs |
| Schoenfeld et al. [17] | Acquired | Loss of MHC class I, resulting in the loss of β2-microglobulin (B2M) and MHC class I expression and eventually an acquired defect in antigen presentation | B2M gene mutation |
| Schoenfeld et al. [17] | Acquired | Disorders in inducing tumor cell death | A defect of IFN-γ response triggers a signaling cascade in tumor cells through the activation of the JAK/STAT pathway that mediates MHC class I and PD-L1 expression |
| Schoenfeld et al. [17] | Acquired | Tumor progression despite receiving ICIs | Inactivating mutations in JAK/STAT components |
| Schoenfeld et al. [17] | Acquired | Increased expression of immunosuppressive cytokines and decreased IFN-γ, leading to inhibition of T-cell-mediated infiltration and immunity | Loss of the tumor suppressor PTEN |
| Schoenfeld et al. [17] | Acquired | The effect of WNT/β-catenin | Increased levels of immunosuppressive cytokines, promotion of Tregs, and lack of significant T cell infiltration |
| Schoenfeld et al. [17] | Acquired | Additional inhibitory checkpoints | Upregulated expression of various T cell checkpoints such as TIM-3, LAG-3, and VISTA |
Table 2.
Examples of mechanisms of TME metabolism involved in Treg-mediated treatment resistance.
| Reference | Factor | Role in TME | Mechanism |
|---|---|---|---|
| Sebastian et al. [60], van Gisbergen et al. [61] | Aerobic glycolysis | Gain energy that supports biomass production and rapid proliferation | Altered vascularization and OXPHOS-dependent increased oxygen consumption rate cause hypoxia areas in TME with following HIF-1α upregulation |
| van Gisbergen et al. [61] | Activated HIF-1α | Supports the depletion of ROS and promotes angiogenesis | Promotes aerobic glycolysis and downregulates OXPHOS |
| Raychaudhuri et al. [62] | Lactate(glycolysis end product) | Contributes to the induction of FoxP3+ CD4+ Tregs; increases acidification of the TME | Enhances tryptophan metabolism and kynurenine production by DCs; enhances expression of MCT4 |
| Gupta et al. [63] | Irradiation | It is associated with tumor aggressiveness and poor outcome | Induces tumor-permissive changes, such as reduced levels of antioxidants, glutathione, and ascorbate, and elevated levels of energy carriers, such as extracellular ATP and GTP |
| Ring et al. [64] | ATP | Strong DAMP | Activates Tregs and stimulates their suppressive function by producing immunosuppressive adenosine via the ectonucleotides CD39 and CD73 |
| Feng et al. [57] | Hypoxia | Generates high extracellular adenosine concentration and accumulation of intracellular cAMP, which leads to the Tregs recruitment | Upregulates HIF-1α in TME cells, which then promotes CD39/CD73 and adenosine receptors A2AR and A2BR expression |
| Conforti [59] | Necrosis of tumor cells | Inhibits the polarization of resting CD4+ T cells into effector cells and promotes the Tregs development | It results in an increased extracellular potassium concentration, which subsequently suppresses the Akt/mTOR pathway |
Table 3.
Mechanisms of TGF-β-dependent upregulation of Tregs.
| Reference | Factor | Role | Mechanism |
|---|---|---|---|
| Oshimori et al. [71] | TGF-β during tumorigenesis | TGF-β-responding tumor cells are responsible for drug resistance and tumor recurrence | Induces apoptosis and inhibits the proliferation of cancer cells; in advanced stages, it can stimulate tumor progression |
| Neel et al. [72,73], Lainé et al. [72,73] | Tregs and TGF-β mutual relation | Promotes tumor immune escape | (1) Tregs development requires at least TCR stimulation and the TGF-β and IL-2 supply; (2) Tregs secrete latent form of TGF-β; (3) Treg cell-integrin expression is essential to activate TGF-β produced by cancer cells. |
| Funaki et al. [34] | Chemotherapy | CD8+ T lymphocytes repression | Increases TGF-β levels with following enhanced PD-L1 expression on cancer cells; elevates EMT markers |
| Funaki et al. [34], Quan et al. [74] | EMT | Contributes to stem cell generation, anticancer drug resistance, genomic instability, and localized immunosuppression | Elevated levels of E-cadherin and vimentin |
| Wang et al. [75] | TGF-β and the JAK/STAT axis | Promotes radioresistance | Induces the EMT, cancer stem cells, and cancer-associated fibroblasts |
| Liu et al. [76] | ROS | Metastasis | Mediate TGF-induced EMT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dąbrowska, A.; Grubba, M.; Balihodzic, A.; Szot, O.; Sobocki, B.K.; Perdyan, A. The Role of Regulatory T Cells in Cancer Treatment Resistance. Int. J. Mol. Sci. 2023, 24, 14114. https://doi.org/10.3390/ijms241814114
AMA Style
Dąbrowska A, Grubba M, Balihodzic A, Szot O, Sobocki BK, Perdyan A. The Role of Regulatory T Cells in Cancer Treatment Resistance. International Journal of Molecular Sciences. 2023; 24(18):14114. https://doi.org/10.3390/ijms241814114
Chicago/Turabian StyleDąbrowska, Anna, Magdalena Grubba, Amar Balihodzic, Olga Szot, Bartosz Kamil Sobocki, and Adrian Perdyan. 2023. "The Role of Regulatory T Cells in Cancer Treatment Resistance" International Journal of Molecular Sciences 24, no. 18: 14114. https://doi.org/10.3390/ijms241814114
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.