
Applications and Research Advances in the Delivery of CRISPR/Cas9 Systems for the Treatment of Inherited Diseases

Abstract
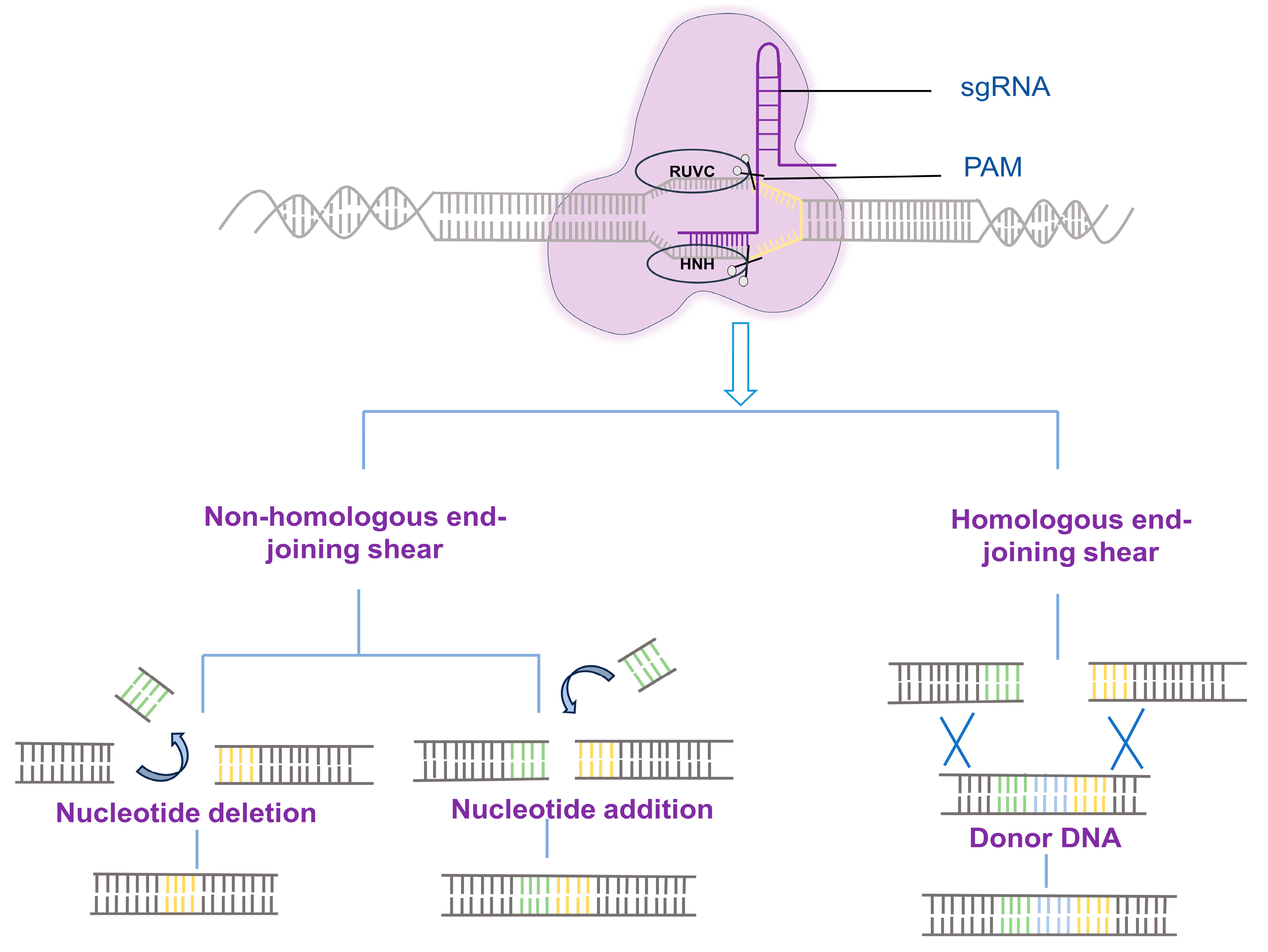
:1. Introduction
2. CRISPR-Associated Transposase
3. Disease Modeling and Gene Therapy
3.1. Duchenne Muscular Dystrophy
3.2. Hemophilia
3.3. Cystic Fibrosis
3.4. Thalassemia
3.5. Familial Hypercholesterolemia
3.6. Diabetic Retinopathy
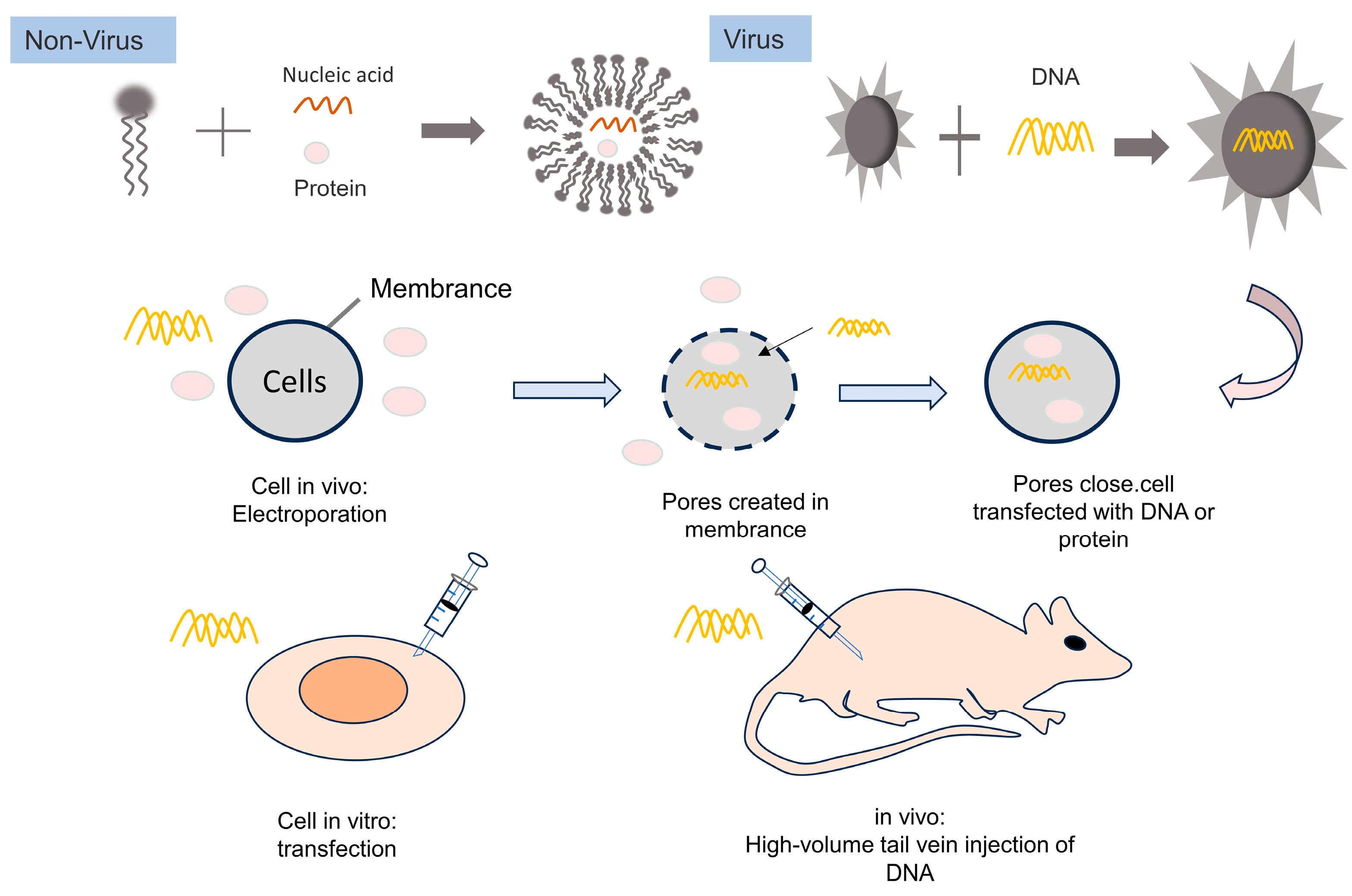
4. Delivery of Biomacromolecules
4.1. Duchenne Muscular Dystrophy
4.1.1. Adeno-Associated Virus (AAV)
4.1.2. Lentiviral Vector (LV Vector)
4.2. Non-Viral Delivery of Genome-Editing Systems
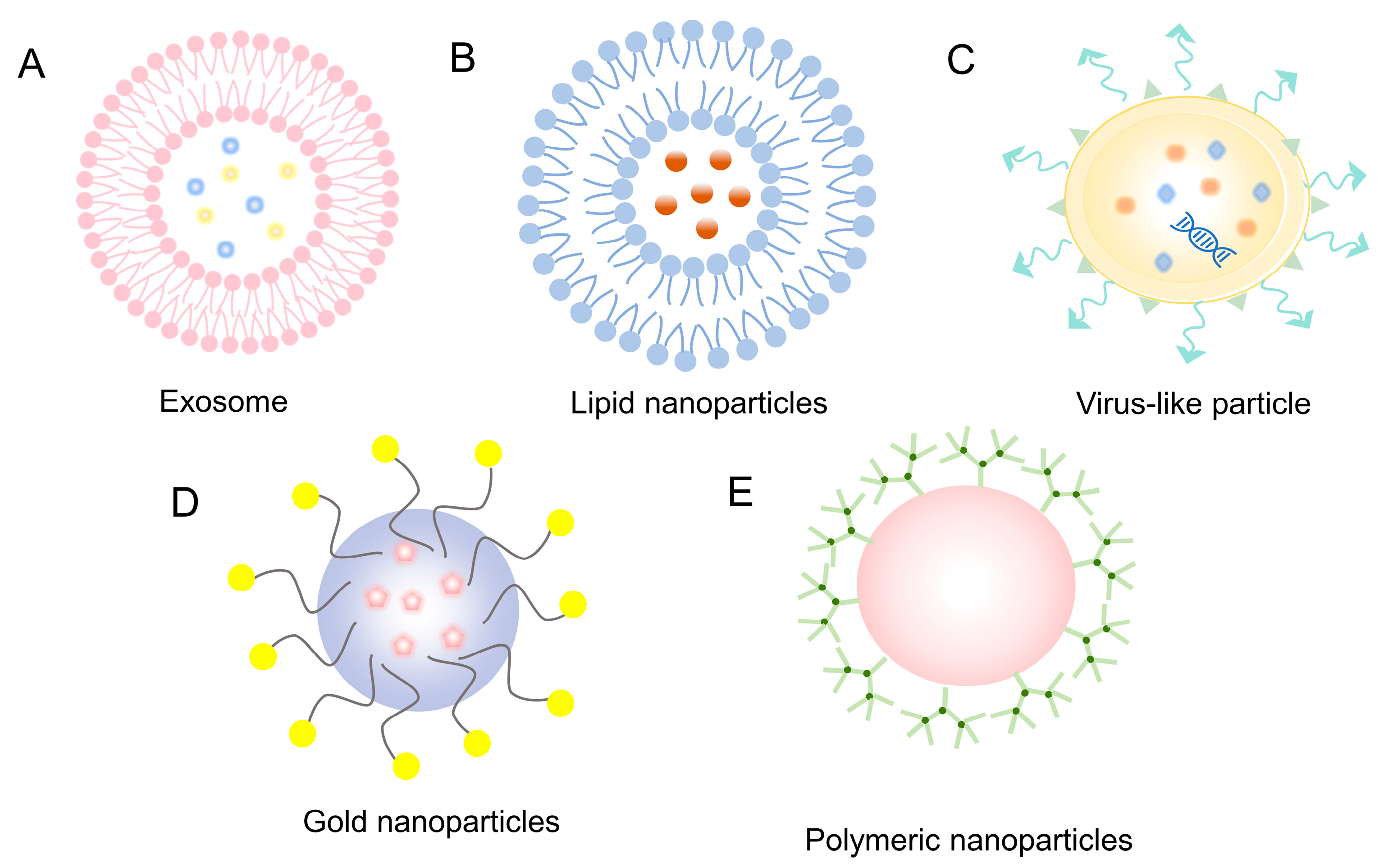
4.2.1. Exosomes
4.2.2. Lipid Nanoparticles
4.2.3. Virus-like Particle
4.2.4. Gold Nanoparticles
4.2.5. Polymeric Nanoparticles
5. Current Status of Genome-Editing Clinical Trials
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wada, N.; Osakabe, K.; Osakabe, Y. Expanding the plant genome editing toolbox with recently developed CRISPR-Cas systems. Plant Physiol. 2022, 188, 1825–1837. [Google Scholar] [CrossRef] [PubMed]
- Kostyusheva, A.; Brezgin, S.; Babin, Y.; Vasilyeva, I.; Glebe, D.; Kostyushev, D.; Chulanov, V. CRISPR-Cas systems for diagnosing infectious diseases. Methods 2022, 203, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.G.; Doudna, J.A. CRISPR-Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wen, Y.; Guo, X. CRISPR/Cas9 for genome editing: Progress, implications and challenges. Hum. Mol. Genet. 2014, 23, R40–R46. [Google Scholar] [CrossRef]
- Yin, K.; Gao, C.; Qiu, J.L. Progress and prospects in plant genome editing. Nat. Plants 2017, 3, 17107. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Zhang, F.; Ding, Y. CRISPR/Cas9 Delivery System Engineering for Genome Editing in Therapeutic Applications. Pharmaceutics 2021, 13, 1649. [Google Scholar] [CrossRef]
- Sharma, G.; Sharma, A.R.; Bhattacharya, M.; Lee, S.S.; Chakraborty, C. CRISPR-Cas9: A Preclinical and Clinical Perspective for the Treatment of Human Diseases. Mol. Ther. 2021, 29, 571–586. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, R.; Dai, Z. Key considerations in designing CRISPR/Cas9-carrying nanoparticles for therapeutic genome editing. Nanoscale 2020, 12, 21001–21014. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Deng, L.; Li, S. Application of nanoparticles in CRISPR/Cas9-based gene therapy. Sheng Wu Gong Cheng Xue Bao/Chin. J. Biotechnol. 2022, 38, 2087–2104. [Google Scholar] [CrossRef]
- Yip, B.H. Recent Advances in CRISPR/Cas9 Delivery Strategies. Biomolecules 2020, 10, 839. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Kantor, B. Lentiviral Vectors for Delivery of Gene-Editing Systems Based on CRISPR/Cas: Current State and Perspectives. Viruses 2021, 13, 1288. [Google Scholar] [CrossRef]
- Asmamaw, M.; Zawdie, B. Mechanism and Applications of CRISPR/Cas-9-Mediated Genome Editing. Biologics 2021, 15, 353–361. [Google Scholar] [CrossRef]
- Le Rhun, A.; Escalera-Maurer, A.; Bratovič, M.; Charpentier, E. CRISPR-Cas in Streptococcus pyogenes. RNA Biol. 2019, 16, 380–389. [Google Scholar] [CrossRef]
- Ronzitti, G.; Gross, D.A.; Mingozzi, F. Human Immune Responses to Adeno-Associated Virus (AAV) Vectors. Front. Immunol. 2020, 11, 670. [Google Scholar] [CrossRef]
- Ismail, A.M.; Cui, T.; Dommaraju, K.; Singh, G.; Dehghan, S.; Seto, J.; Shrivastava, S.; Fedorova, N.B.; Gupta, N.; Stockwell, T.B.; et al. Genomic analysis of a large set of currently-and historically-important human adenovirus pathogens. Emerg. Microbes Infect. 2018, 7, 10. [Google Scholar] [CrossRef]
- Stephens, C.J.; Kashentseva, E.; Everett, W.; Kaliberova, L.; Curiel, D.T. Targeted in vivo knock-in of human alpha-1-antitrypsin cDNA using adenoviral delivery of CRISPR/Cas9. Gene Ther. 2018, 25, 139–156. [Google Scholar] [CrossRef]
- Zhao, Q.; Allen, M.J.; Wang, Y.; Wang, B.; Wang, N.; Shi, L.; Sitrin, R.D. Disassembly and reassembly improves morphology and thermal stability of human papillomavirus type 16 virus-like particles. Nanomedicine 2012, 8, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.M.; Foley, J.; Tison, T.; Silva, R.; Ogembo, J.G. Novel Epstein-Barr virus-like particles incorporating gH/gL-EBNA1 or gB-LMP2 induce high neutralizing antibody titers and EBV-specific T-cell responses in immunized mice. Oncotarget 2017, 8, 19255–19273. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Kaur, J.; Wuest, M.; Wuest, F. In situ click chemistry generation of cyclooxygenase-2 inhibitors. Nat. Commun. 2017, 8, 1. [Google Scholar] [CrossRef]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef]
- Chen, W.; Jiang, J.; Gong, L.; Shu, Z.; Xiang, D.; Zhang, X.; Bi, K.; Diao, H. Hepatitis B virus P protein initiates glycolytic bypass in HBV-related hepatocellular carcinoma via a FOXO3/miRNA-30b-5p/MINPP1 axis. J. Exp. Clin. Cancer Res. 2021, 40, 1. [Google Scholar] [CrossRef]
- Majeau, N.; Fortin-Archambault, A.; Gérard, C.; Rousseau, J.; Yaméogo, P.; Tremblay, J.P. Serum extracellular vesicles for delivery of CRISPR-CAS9 ribonucleoproteins to modify the dystrophin gene. Mol. Ther. 2022, 30, 2429–2442. [Google Scholar] [CrossRef]
- George, A.; Shah, P.A.; Shrivastav, P.S. Natural biodegradable polymers based nano-formulations for drug delivery: A review. Int. J. Pharm. 2019, 561, 244–264. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.M.; Zhao, Y.; Xiao, R.L.; Zhang, S.Q.; Tian, B.H.; Li, X.X.; Zhang, R.; Ma, R.S.; Liang, H.X. Nuclear-targeted carbon quantum dot mediated CRISPR/Cas9 delivery for fluorescence visualization and efficient editing. Nanoscale 2022, 14, 14645–14660. [Google Scholar] [CrossRef]
- Luther, D.C.; Huang, R.; Jeon, T.; Zhang, X.; Lee, Y.W.; Nagaraj, H.; Rotello, V.M. Delivery of drugs, proteins, and nucleic acids using inorganic nanoparticles. Adv. Drug Deliv. Rev. 2020, 156, 188–213. [Google Scholar] [CrossRef] [PubMed]
- Paschon, D.E.; Lussier, S.; Wangzor, T.; Xia, D.F.; Li, P.W.; Hinkley, S.J.; Scarlott, N.A.; Lam, S.C.; Waite, A.J.; Truong, L.N.; et al. Diversifying the structure of zinc finger nucleases for high-precision genome editing. Nat. Commun. 2019, 10, 1133. [Google Scholar] [CrossRef] [PubMed]
- Carroll, D. Progress and prospects: Zinc-finger nucleases as gene therapy agents. Gene Ther. 2008, 15, 1463–1468. [Google Scholar] [CrossRef]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet 2010, 11, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Cermak, T.; Doyle, E.L.; Christian, M.; Wang, L.; Zhang, Y.; Schmidt, C.; Baller, J.A.; Somia, N.V.; Bogdanove, A.J.; Voytas, D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011, 39, e82. [Google Scholar] [CrossRef] [PubMed]
- Schmid-Burgk, J.L.; Schmidt, T.; Kaiser, V.; Höning, K.; Hornung, V. A ligation-independent cloning technique for high-throughput assembly of transcription activator–like effector genes. Nat. Biotechnol. 2013, 31, 76–81. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, L.; Liu, H.; Cheng, K. Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications. J. Control. Release 2017, 266, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.; Huang, J.; Xiao, Q.; He, B.; Dong, X.; Liu, Y.; Yang, X.; Han, D.; Wang, Z.; Wang, X.; et al. High-fidelity Cas13 variants for targeted RNA degradation with minimal collateral effects. Nat. Biotechnol. 2023, 41, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Xu, Z.; Xue, Y.; Xu, C.; Han, L.; Liu, Y.; Wang, F.; Zhang, R.; Han, S.; Wang, X.; et al. Rescue of autosomal dominant hearing loss by in vivo delivery of mini dCas13X-derived RNA base editor. Sci. Transl. Med. 2022, 14, eabn0449. [Google Scholar] [CrossRef]
- Thuronyi, B.W.; Koblan, L.W.; Levy, J.M.; Yeh, W.H.; Zheng, C.; Newby, G.A.; Wilson, C.; Bhaumik, M.; Shubina-Oleinik, O.; Holt, J.R.; et al. Continuous evolution of base editors with expanded target compatibility and improved activity. Nat. Biotechnol. 2019, 37, 1070–1079. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Gao, X.D.; Podracky, C.J.; Nelson, A.T.; Koblan, L.W.; Raguram, A.; Levy, J.M.; Mercer, J.A.M.; Liu, D.R. Programmable deletion, replacement, integration and inversion of large DNA sequences with twin prime editing. Nat. Biotechnol. 2022, 40, 731–740. [Google Scholar] [CrossRef]
- Gainetdinov, I.; Vega-Badillo, J.; Cecchini, K.; Bagci, A.; Colpan, C.; De, D.; Bailey, S.; Arif, A.; Wu, P.H.; MacRae, I.J.; et al. Relaxed targeting rules help PIWI proteins silence transposons. Nature 2023, 619, 394–402. [Google Scholar] [CrossRef]
- Singh, H.; Huls, H.; Kebriaei, P.; Cooper, L.J. A new approach to gene therapy using Sleeping Beauty to genetically modify clinical-grade T cells to target CD19. Immunol. Rev. 2014, 257, 181–190. [Google Scholar] [CrossRef]
- Kovač, A.; Miskey, C.; Menzel, M.; Grueso, E.; Gogol-Döring, A.; Ivics, Z. RNA-guided retargeting of Sleeping Beauty transposition in human cells. Elife 2020, 9, e53868. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Chalmers, R. Targeted DNA transposition in vitro using a dCas9-transposase fusion protein. Nucleic Acids Res. 2019, 47, 8126–8135. [Google Scholar] [CrossRef] [PubMed]
- Strecker, J.; Ladha, A.; Gardner, Z.; Schmid-Burgk, J.L.; Makarova, K.S.; Koonin, E.V.; Zhang, F. RNA-guided DNA insertion with CRISPR-associated transposases. Science 2019, 365, 48–53. [Google Scholar] [CrossRef]
- Muto, V.; Benigni, F.; Magliocca, V.; Borghi, R.; Flex, E.; Pallottini, V.; Rosa, A.; Compagnucci, C.; Tartaglia, M. CRISPR/Cas9 and piggyBac Transposon-Based Conversion of a Pathogenic Biallelic TBCD Variant in a Patient-Derived iPSC Line Allows Correction of PEBAT-Related Endophenotypes. Int. J. Mol. Sci. 2023, 24, 7988. [Google Scholar] [CrossRef]
- Zhao, S.; Jiang, E.; Chen, S.; Gu, Y.; Shangguan, A.J.; Lv, T.; Luo, L.; Yu, Z. PiggyBac transposon vectors: The tools of the human gene encoding. Transl. Lung Cancer Res. 2016, 5, 120–125. [Google Scholar] [CrossRef]
- Jin, Y.; Chen, Y.; Zhao, S.; Guan, K.L.; Zhuang, Y.; Zhou, W.; Wu, X.; Xu, T. DNA-PK facilitates piggyBac transposition by promoting paired-end complex formation. Proc. Natl. Acad. Sci. USA 2017, 114, 7408–7413. [Google Scholar] [CrossRef]
- Pallarès-Masmitjà, M.; Ivančić, D.; Mir-Pedrol, J.; Jaraba-Wallace, J.; Tagliani, T.; Oliva, B.; Rahmeh, A.; Sánchez-Mejías, A.; Güell, M. Find and cut-and-transfer (FiCAT) mammalian genome engineering. Nat. Commun. 2021, 12, 7071. [Google Scholar] [CrossRef] [PubMed]
- Ousterout, D.G.; Kabadi, A.M.; Thakore, P.I.; Majoros, W.H.; Reddy, T.E.; Gersbach, C.A. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 2015, 6, 6244. [Google Scholar] [CrossRef]
- Zhao, X.; Qu, K.; Curci, B.; Yang, H.; Bolund, L.; Lin, L.; Luo, Y. Comparison of In-Frame Deletion, Homology-Directed Repair, and Prime Editing-Based Correction of Duchenne Muscular Dystrophy Mutations. Biomolecules 2023, 13, 870. [Google Scholar] [CrossRef]
- Choi, E.; Koo, T. CRISPR technologies for the treatment of Duchenne muscular dystrophy. Mol. Ther. 2021, 29, 3179–3191. [Google Scholar] [CrossRef]
- Erkut, E.; Yokota, T. CRISPR Therapeutics for Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2022, 23, 1832. [Google Scholar] [CrossRef]
- Aslesh, T.; Erkut, E.; Yokota, T. Restoration of dystrophin expression and correction of Duchenne muscular dystrophy by genome editing. Expert Opin. Biol. Ther. 2021, 21, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wei, T.; Yang, H.; Li, G.; Li, H. CRISPR-Based Therapeutic Gene Editing for Duchenne Muscular Dystrophy: Advances, Challenges and Perspectives. Cells 2022, 11, 2964. [Google Scholar] [CrossRef] [PubMed]
- Tabebordbar, M.; Zhu, K.; Cheng, J.K.W.; Chew, W.L.; Widrick, J.J.; Yan, W.X.; Maesner, C.; Wu, E.Y.; Xiao, R.; Ran, F.A.; et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016, 351, 407–411. [Google Scholar] [CrossRef]
- Birch, S.M.; Lawlor, M.W.; Conlon, T.J.; Guo, L.J.; Crudele, J.M.; Hawkins, E.C.; Nghiem, P.P.; Ahn, M.; Meng, H.; Beatka, M.J.; et al. Assessment of systemic AAV-microdystrophin gene therapy in the GRMD model of Duchenne muscular dystrophy. Sci. Transl. Med. 2023, 15, eabo1815. [Google Scholar] [CrossRef]
- Bengtsson, N.E.; Crudele, J.M.; Klaiman, J.M.; Halbert, C.L.; Hauschka, S.D.; Chamberlain, J.S. Comparison of dystrophin expression following gene editing and gene replacement in an aged preclinical DMD animal model. Mol. Ther. 2022, 30, 2176–2185. [Google Scholar] [CrossRef]
- Egorova, T.V.; Polikarpova, A.V.; Vassilieva, S.G.; Dzhenkova, M.A.; Savchenko, I.M.; Velyaev, O.A.; Shmidt, A.A.; Soldatov, V.O.; Pokrovskii, M.V.; Deykin, A.V.; et al. CRISPR-Cas9 correction in the DMD mouse model is accompanied by upregulation of Dp71f protein. Mol. Therapy. Methods Clin. Dev. 2023, 30, 161–180. [Google Scholar] [CrossRef]
- Duan, D.S.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Fujimoto, N.; Sasakawa, N.; Ohinata, Y.; Shima, M.; Yamanaka, S.; Sugimoto, M.; Hotta, A. Delivery of full-length factor VIII using a piggyBac transposon vector to correct a mouse model of hemophilia A. PLoS ONE 2014, 9, e104957. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Tonnu, N.; Tachikawa, K.; Limphong, P.; Vega, J.B.; Karmali, P.P.; Chivukula, P.; Verma, I.M. Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc. Natl. Acad. Sci. USA 2017, 114, E1941–E1950. [Google Scholar] [CrossRef]
- Paunovska, K.; Loughrey, D.; Dahlman, J.E. Drug delivery systems for RNA therapeutics. Nat. Rev. Genet. 2022, 23, 265–280. [Google Scholar] [CrossRef]
- Lee, J.H.; Han, J.P.; Song, D.W.; Lee, G.S.; Choi, B.S.; Kim, M.; Lee, Y.; Kim, S.; Lee, H.; Yeom, S.C. Invivo genome editing for hemophilia B therapy by the combination of rebalancing and therapeutic gene knockin using a viral and non-viral vector. Mol. Ther. Nucleic Acids 2023, 32, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.Y.; Hu, Z.Q.; Zhao, J.Y.; Zhou, T.; Tang, S.Q.; Wang, P.Y.; Xiao, R.; Chen, Y.; Wu, L.Q.; Zhou, M.J.; et al. CRISPR-Mediated In Situ Introduction or Integration of F9-Padua in Human iPSCs for Gene Therapy of Hemophilia B. Int. J. Mol. Sci. 2023, 24, 9013. [Google Scholar] [CrossRef]
- Yan, Z.; McCray, P.B., Jr.; Engelhardt, J.F. Advances in gene therapy for cystic fibrosis lung disease. Hum. Mol. Genet. 2019, 28, R88–R94. [Google Scholar] [CrossRef]
- Suzuki, S.; Crane, A.M.; Anirudhan, V.; Barillà, C.; Matthias, N.; Randell, S.H.; Rab, A.; Sorscher, E.J.; Kerschner, J.L.; Yin, S.; et al. Highly Efficient Gene Editing of Cystic Fibrosis Patient-Derived Airway Basal Cells Results in Functional CFTR Correction. Mol. Ther. 2020, 28, 1684–1695. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A. CFTR: More than just a chloride channel. Pediatr. Pulmonol. 2005, 39, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Amistadi, S.; Maule, G.; Ciciani, M.; Ensinck, M.M.; De Keersmaecker, L.; Ramalho, A.S.; Guidone, D.; Buccirossi, M.; Galietta, L.J.V.; Carlon, M.S.; et al. Functional restoration of a CFTR splicing mutation through RNA delivery of CRISPR adenine base editor. Mol. Ther. 2023, 31, 1647–1660. [Google Scholar] [CrossRef]
- Wang, G.S. Genome Editing for Cystic Fibrosis. Cells 2023, 12, 1555. [Google Scholar] [CrossRef]
- Krishnamurthy, S.; Wohlford-Lenane, C.; Kandimalla, S.; Sartre, G.; Meyerholz, D.K.; Théberge, V.; Hallée, S.; Duperré, A.M.; Del’Guidice, T.; Lepetit-Stoffaes, J.P.; et al. Engineered amphiphilic peptides enable delivery of proteins and CRISPR-associated nucleases to airway epithelia. Nat. Commun. 2019, 10, 4906. [Google Scholar] [CrossRef]
- McCarron, A.; Cmielewski, P.; Reyne, N.; McIntyre, C.; Finnie, J.; Craig, F.; Rout-Pitt, N.; Delhove, J.; Schjenken, J.E.; Chan, H.Y.; et al. Phenotypic Characterization and Comparison of Cystic Fibrosis Rat Models Generated Using CRISPR/Cas9 Gene Editing. Am. J. Pathol. 2020, 190, 977–993. [Google Scholar] [CrossRef]
- Yan, Z.; Vorhies, K.; Feng, Z.; Park, S.Y.; Choi, S.H.; Zhang, Y.; Winter, M.; Sun, X.; Engelhardt, J.F. Recombinant Adeno-Associated Virus-Mediated Editing of the G551D Cystic Fibrosis Transmembrane Conductance Regulator Mutation in Ferret Airway Basal Cells. Hum. Gene Ther. 2022, 33, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Meisel, R. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2021, 384, e91. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, M.A.; Abbasalipour, M.; Concordet, J.P.; Berg, J.V.; Zeinali, S.; Arashkia, A.; Azadmanesh, K.; Buch, T.; Karimipoor, M. Targeted deletion of BCL11A gene by CRISPR-Cas9 system for fetal hemoglobin reactivation: A promising approach for gene therapy of beta thalassemia disease. Eur. J. Pharmacol. 2019, 854, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Quagliano, A.; Acevedo, D.; Hardigan, P.; Prasad, S. Using Clustered Regularly Interspaced Short Palindromic Repeats gene editing to induce permanent expression of fetal hemoglobin in β-thalassemia and sickle cell disease: A comparative meta-analysis. Front. Med. 2022, 9, 943631. [Google Scholar] [CrossRef]
- Pavani, G.; Fabiano, A.; Laurent, M.; Amor, F.; Cantelli, E.; Chalumeau, A.; Maule, G.; Tachtsidi, A.; Concordet, J.P.; Cereseto, A.; et al. Correction of β-thalassemia by CRISPR/Cas9 editing of the α-globin locus in human hematopoietic stem cells. Blood Adv. 2021, 5, 1137–1153. [Google Scholar] [CrossRef]
- Mettananda, S.; Fisher, C.A.; Hay, D.; Badat, M.; Quek, L.; Clark, K.; Hublitz, P.; Downes, D.; Kerry, J.; Gosden, M.; et al. Editing an α-globin enhancer in primary human hematopoietic stem cells as a treatment for β-thalassemia. Nat. Commun. 2017, 8, 424. [Google Scholar] [CrossRef]
- Bhatia, M.; Walters, M.C. Hematopoietic cell transplantation for thalassemia and sickle cell disease: Past, present and future. Bone Marrow Transpl. 2008, 41, 109–117. [Google Scholar] [CrossRef]
- Rui, Y.; Wilson, D.R.; Green, J.J. Non-Viral Delivery To Enable Genome Editing. Trends Biotechnol. 2019, 37, 281–293. [Google Scholar] [CrossRef]
- Ban, Q.; Lee, J.S.; Shi, Z.N.; Lu, D.Q.; Qiao, L.; Yang, P.; Li, X.; Cheng, H.Y.; Zhang, M.; Hou, J.B.; et al. Intraosseous injection of SMNP vectors enables CRISPR/Cas9-mediated knock-in of HBB gene into hematopoietic stem and progenitor cells. Nano Today 2022, 47, 101659. [Google Scholar] [CrossRef]
- Xu, S.; Luk, K.; Yao, Q.; Shen, A.H.; Zeng, J.; Wu, Y.; Luo, H.Y.; Brendel, C.; Pinello, L.; Chui, D.H.K.; et al. Editing aberrant splice sites efficiently restores β-globin expression in β-thalassemia. Blood 2019, 133, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Paschoudi, K.; Yannaki, E.; Psatha, N. Precision Editing as a Therapeutic Approach for β-Hemoglobinopathies. Int. J. Mol. Sci. 2023, 24, 9527. [Google Scholar] [CrossRef] [PubMed]
- Ali, G.; Tariq, M.A.; Shahid, K.; Ahmad, F.J.; Akram, J. Advances in genome editing: The technology of choice for precise and efficient β-thalassemia treatment. Gene Ther. 2021, 28, 6–15. [Google Scholar] [CrossRef]
- Yang, Z.; Ji, P.; Li, Z.; Zhang, R.; Wei, M.; Yang, Y.; Yuan, L.; Han, Y.; Yang, G. Improved extracellular vesicle-based mRNA delivery for familial hypercholesterolemia treatment. Theranostics 2023, 13, 3467–3479. [Google Scholar] [CrossRef]
- Langsted, A.; Kamstrup, P.R.; Benn, M.; Tybjærg-Hansen, A.; Nordestgaard, B.G. High lipoprotein(a) as a possible cause of clinical familial hypercholesterolaemia: A prospective cohort study. Lancet Diabetes Endocrinol. 2016, 4, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Dharmayat, K.I.; Stevens, C.A.T.; Sharabiani, M.T.A.; Jones, R.S.; Watts, G.F.; Genest, J.; Ray, K.K.; Vallejo-Vaz, A.J. Prevalence of Familial Hypercholesterolemia Among the General Population and Patients With Atherosclerotic Cardiovascular Disease: A Systematic Review and Meta-Analysis. Circulation 2020, 141, 1742–1759. [Google Scholar] [CrossRef]
- Vallejo-Vaz, A.J.; De Marco, M.; Stevens, C.A.T.; Akram, A.; Freiberger, T.; Hovingh, G.K.; Kastelein, J.J.P.; Mata, P.; Raal, F.J.; Santos, R.D.; et al. Overview of the current status of familial hypercholesterolaemia care in over 60 countries—The EAS Familial Hypercholesterolaemia Studies Collaboration (FHSC). Atherosclerosis 2018, 277, 234–255. [Google Scholar] [CrossRef]
- Abifadel, M.; Varret, M.; Rabès, J.P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef]
- Li, Z.F.; Wu, N.Q. The Progression of Treatment for Refractory Hypercholesterolemia: Focus on the Prospect of Gene Therapy. Front. Genet. 2022, 13, 911429. [Google Scholar] [CrossRef]
- Ebenezer, O.; Comoglio, P.; Wong, G.K.; Tuszynski, J.A. Development of Novel siRNA Therapeutics: A Review with a Focus on Inclisiran for the Treatment of Hypercholesterolemia. Int. J. Mol. Sci. 2023, 24, 4019. [Google Scholar] [CrossRef]
- Kassim, S.H.; Li, H.; Vandenberghe, L.H.; Hinderer, C.; Bell, P.; Marchadier, D.; Wilson, A.; Cromley, D.; Redon, V.; Yu, H.; et al. Gene therapy in a humanized mouse model of familial hypercholesterolemia leads to marked regression of atherosclerosis. PLoS ONE 2010, 5, e13424. [Google Scholar] [CrossRef] [PubMed]
- Si-Tayeb, K.; Noto, F.K.; Nagaoka, M.; Li, J.; Battle, M.A.; Duris, C.; North, P.E.; Dalton, S.; Duncan, S.A. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 2010, 51, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Kawashiri, M.A.; Nohara, A.; Noguchi, T.; Tada, H.; Nakanishi, C.; Mori, M.; Konno, T.; Hayashi, K.; Fujino, N.; Inazu, A.; et al. Efficacy and safety of coadministration of rosuvastatin, ezetimibe, and colestimide in heterozygous familial hypercholesterolemia. Am. J. Cardiol. 2012, 109, 364–369. [Google Scholar] [CrossRef]
- Okada, H.; Nakanishi, C.; Yoshida, S.; Shimojima, M.; Yokawa, J.; Mori, M.; Tada, H.; Yoshimuta, T.; Hayashi, K.; Yamano, T.; et al. Function and Immunogenicity of Gene-corrected iPSC-derived Hepatocyte-Like Cells in Restoring Low Density Lipoprotein Uptake in Homozygous Familial Hypercholesterolemia. Sci. Rep. 2019, 9, 4695. [Google Scholar] [CrossRef] [PubMed]
- Kankkonen, H.M.; Vähäkangas, E.; Marr, R.A.; Pakkanen, T.; Laurema, A.; Leppänen, P.; Jalkanen, J.; Verma, I.M.; Ylä-Herttuala, S. Long-term lowering of plasma cholesterol levels in LDL-receptor-deficient WHHL rabbits by gene therapy. Mol. Ther. 2004, 9, 548–556. [Google Scholar] [CrossRef]
- Parsamanesh, N.; Kooshkaki, O.; Siami, H.; Santos, R.D.; Jamialahmadi, T.; Sahebkar, A. Gene and cell therapy approaches for familial hypercholesterolemia: An update. Drug Discov. Today 2023, 28, 103470. [Google Scholar] [CrossRef]
- Sosnowska, B.; Adach, W.; Surma, S.; Rosenson, R.S.; Banach, M. Evinacumab, an ANGPTL3 Inhibitor, in the Treatment of Dyslipidemia. J. Clin. Med. 2022, 12, 168. [Google Scholar] [CrossRef]
- Vanhoye, X.; Janin, A.; Caillaud, A.; Rimbert, A.; Venet, F.; Gossez, M.; Dijk, W.; Marmontel, O.; Nony, S.; Chatelain, C.; et al. APOB CRISPR-Cas9 Engineering in Hypobetalipoproteinemia: A Promising Tool for Functional Studies of Novel Variants. Int. J. Mol. Sci. 2022, 23, 4281. [Google Scholar] [CrossRef]
- Walker, A.F.; Graham, S.; Maple-Brown, L.; Egede, L.E.; Campbell, J.A.; Walker, R.J.; Wade, A.N.; Mbanya, J.C.; Long, J.A.; Yajnik, C.; et al. Interventions to address global inequity in diabetes: International progress. Lancet 2023, 402, 250–264. [Google Scholar] [CrossRef]
- Stitt, A.W.; Curtis, T.M.; Chen, M.; Medina, R.J.; McKay, G.J.; Jenkins, A.; Gardiner, T.A.; Lyons, T.J.; Hammes, H.P.; Simó, R.; et al. The progress in understanding and treatment of diabetic retinopathy. Prog. Retin. Eye Res. 2016, 51, 156–186. [Google Scholar] [CrossRef]
- Benati, D.; Patrizi, C.; Recchia, A. Gene editing prospects for treating inherited retinal diseases. J. Med. Genet. 2020, 57, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Bilanges, B.; Posor, Y.; Vanhaesebroeck, B. PI3K isoforms in cell signalling and vesicle trafficking. Nat. Rev. Mol. Cell Biol. 2019, 20, 515–534. [Google Scholar] [CrossRef] [PubMed]
- Toker, A.; Dibble, C.C. PI 3-Kinase Signaling: AKTing up inside the Cell. Mol. Cell 2018, 71, 875–876. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.Y.; Ma, G.E.; Qi, H.; Dong, L.J.; Chen, F.; Wang, Y.; Mao, X.X.; Guo, X.Q.; Cui, J.; Matsubara, J.A.; et al. Genome Editing of Pik3cd Impedes Abnormal Retinal Angiogenesis. Hum. Gene Ther. 2023, 34, 30–41. [Google Scholar] [CrossRef]
- Wu, W.Y.; Xu, H.Z.; Meng, Z.S.; Zhu, J.X.; Xiong, S.Q.; Xia, X.B.; Lei, H.T. Axl Is Essential for in-vitro Angiogenesis Induced by Vitreous From Patients With Proliferative Diabetic Retinopathy. Front. Med. 2021, 8, 787150. [Google Scholar] [CrossRef]
- Ao, H.; Li, H.; Zhao, X.; Liu, B.; Lu, L. TXNIP positively regulates the autophagy and apoptosis in the rat müller cell of diabetic retinopathy. Life Sci. 2021, 267, 118988. [Google Scholar] [CrossRef]
- Li, H.; Li, T.; Wang, H.; He, X.; Li, Y.; Wen, S.; Peng, R.; Nie, Y.; Lu, Y.; Yang, H.; et al. Diabetes Promotes Retinal Vascular Endothelial Cell Injury by Inducing CCN1 Expression. Front. Cardiovasc. Med. 2021, 8, 689318. [Google Scholar] [CrossRef]
- Li, A.; Lee, C.M.; Hurley, A.E.; Jarrett, K.E.; De Giorgi, M.; Lu, W.; Balderrama, K.S.; Doerfler, A.M.; Deshmukh, H.; Ray, A.; et al. A Self-Deleting AAV-CRISPR System for In Vivo Genome Editing. Mol. Ther. Methods Clin. Dev. 2019, 12, 111–122. [Google Scholar] [CrossRef]
- Nelson, C.E.; Wu, Y.; Gemberling, M.P.; Oliver, M.L.; Waller, M.A.; Bohning, J.D.; Robinson-Hamm, J.N.; Bulaklak, K.; Castellanos Rivera, R.M.; Collier, J.H.; et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019, 25, 427–432. [Google Scholar] [CrossRef]
- Shen, W.; Liu, S.; Ou, L. rAAV immunogenicity, toxicity, and durability in 255 clinical trials: A meta-analysis. Front. Immunol. 2022, 13, 1001263. [Google Scholar] [CrossRef]
- Puentes-Tellez, M.A.; Sánchez, O.F.; Rojas-Rodriguez, F.; Benincore-Flórez, E.; Barbosa, H.; Alméciga Díaz, C.J. Evaluation of HIV-1 derived lentiviral vectors as transductors of Mucopolysaccharidosis type IV a fibroblasts. Gene 2021, 780, 145527. [Google Scholar] [CrossRef] [PubMed]
- Kubo, S.; Mitani, K. A new hybrid system capable of efficient lentiviral vector production and stable gene transfer mediated by a single helper-dependent adenoviral vector. J. Virol. 2003, 77, 2964–2971. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Ortinski, P.I.; O’Donovan, B.; Dong, X.; Kantor, B. Integrase-Deficient Lentiviral Vector as an All-in-One Platform for Highly Efficient CRISPR/Cas9-Mediated Gene Editing. Mol. Ther. Methods Clin. Dev. 2017, 5, 153–164. [Google Scholar] [CrossRef]
- Uchida, N.; Drysdale, C.M.; Nassehi, T.; Gamer, J.; Yapundich, M.; DiNicola, J.; Shibata, Y.; Hinds, M.; Gudmundsdottir, B.; Haro-Mora, J.J.; et al. Cas9 protein delivery non-integrating lentiviral vectors for gene correction in sickle cell disease. Mol. Ther. Methods Clin. Dev. 2021, 21, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Farzanehpour, M.; Miri, A.; Alvanegh, A.G.; Gouvarchinghaleh, H.E. Viral Vectors, Exosomes, and Vexosomes: Potential armamentarium for delivering CRISPR/Cas to cancer cells. Biochem. Pharmacol. 2023, 212, 115555. [Google Scholar] [CrossRef] [PubMed]
- Alptekin, A.; Parvin, M.; Chowdhury, H.I.; Rashid, M.H.; Arbab, A.S. Engineered exosomes for studies in tumor immunology. Immunol. Rev. 2022, 312, 76–102. [Google Scholar] [CrossRef]
- Wan, T.; Zhong, J.; Pan, Q.; Zhou, T.; Ping, Y.; Liu, X. Exosome-mediated delivery of Cas9 ribonucleoprotein complexes for tissue-specific gene therapy of liver diseases. Sci. Adv. 2022, 8, eabp9435. [Google Scholar] [CrossRef]
- Alghuthaymi, M.A.; Ahmad, A.; Khan, Z.; Khan, S.H.; Ahmed, F.K.; Faiz, S.; Nepovimova, E.; Kuča, K.; Abd-Elsalam, K.A. Exosome/Liposome-like Nanoparticles: New Carriers for CRISPR Genome Editing in Plants. Int. J. Mol. Sci. 2021, 22, 7456. [Google Scholar] [CrossRef]
- Li, L.; Hu, S.; Chen, X. Non-viral delivery systems for CRISPR/Cas9-based genome editing: Challenges and opportunities. Biomaterials 2018, 171, 207–218. [Google Scholar] [CrossRef]
- Mout, R.; Ray, M.; Yesilbag Tonga, G.; Lee, Y.W.; Tay, T.; Sasaki, K.; Rotello, V.M. Direct Cytosolic Delivery of CRISPR/Cas9-Ribonucleoprotein for Efficient Gene Editing. ACS Nano 2017, 11, 2452–2458. [Google Scholar] [CrossRef] [PubMed]
- Kazemian, P.; Yu, S.Y.; Thomson, S.B.; Birkenshaw, A.; Leavitt, B.R.; Ross, C.J.D. Lipid-Nanoparticle-Based Delivery of CRISPR/Cas9 Genome-Editing Components. Mol. Pharm. 2022, 19, 1669–1686. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.H.; Cai, H.; Steinmetz, N.F. Viral nanoparticles for drug delivery, imaging, immunotherapy, and theranostic applications. Adv. Drug Deliv. Rev. 2020, 156, 214–235. [Google Scholar] [CrossRef]
- Donaldson, B.; Lateef, Z.; Walker, G.F.; Young, S.L.; Ward, V.K. Virus-like particle vaccines: Immunology and formulation for clinical translation. Expert Rev. Vaccines 2018, 17, 833–849. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, C.; Zheng, Y.; Zhao, Y.; Wang, Y.; Hao, J.; Zhao, X.; Yi, K.; Shi, L.; Kang, C.; et al. Virus-like nanoparticle as a co-delivery system to enhance efficacy of CRISPR/Cas9-based cancer immunotherapy. Biomaterials 2020, 258, 120275. [Google Scholar] [CrossRef]
- Kumar, D.; Saini, N.; Jain, N.; Sareen, R.; Pandit, V. Gold nanoparticles: An era in bionanotechnology. Expert Opin. Drug Deliv. 2013, 10, 397–409. [Google Scholar] [CrossRef]
- García-Fernández, A.; Vivo-Llorca, G.; Sancho, M.; García-Jareño, A.B.; Ramírez-Jiménez, L.; Barber-Cano, E.; Murguía, J.R.; Orzáez, M.; Sancenón, F.; Martínez-Máñez, R. Nanodevices for the Efficient Codelivery of CRISPR-Cas9 Editing Machinery and an Entrapped Cargo: A Proposal for Dual Anti-Inflammatory Therapy. Pharmaceutics 2022, 14, 1495. [Google Scholar] [CrossRef]
- Ho, L.W.C.; Chan, C.K.W.; Han, R.; Lau, Y.F.Y.; Li, H.; Ho, Y.P.; Zhuang, X.; Choi, C.H.J. Mammalian Cells Exocytose Alkylated Gold Nanoparticles via Extracellular Vesicles. ACS Nano 2022, 16, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Lee, C.W.; Chiou, A.; Wei, P.K. Size-dependent endocytosis of gold nanoparticles studied by three-dimensional mapping of plasmonic scattering images. J. Nanobiotechnol. 2010, 8, 33. [Google Scholar] [CrossRef]
- Shahbazi, R.; Sghia-Hughes, G.; Reid, J.L.; Kubek, S.; Haworth, K.G.; Humbert, O.; Kiem, H.P.; Adair, J.E. Targeted homology-directed repair in blood stem and progenitor cells with CRISPR nanoformulations. Nat. Mater. 2019, 18, 1124–1132. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, L.; Zheng, W.; Cong, L.; Guo, Z.; Xie, Y.; Wang, L.; Tang, R.; Feng, Q.; Hamada, Y.; et al. Thermo-triggered Release of CRISPR-Cas9 System by Lipid-Encapsulated Gold Nanoparticles for Tumor Therapy. Angew. Chem. Int. Ed. Engl. 2018, 57, 1491–1496. [Google Scholar] [CrossRef]
- Jhaveri, A.M.; Torchilin, V.P. Multifunctional polymeric micelles for delivery of drugs and siRNA. Front. Pharmacol. 2014, 5, 77. [Google Scholar] [CrossRef]
- Mariano, A.; Lubrano, C.; Bruno, U.; Ausilio, C.; Dinger, N.B.; Santoro, F. Advances in Cell-Conductive Polymer Biointerfaces and Role of the Plasma Membrane. Chem. Rev. 2022, 122, 4552–4580. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.; Lächelt, U.; Bartek, J.; Wagner, E.; Moghimi, S.M. Polyplex Evolution: Understanding Biology, Optimizing Performance. Mol. Ther. 2017, 25, 1476–1490. [Google Scholar] [CrossRef] [PubMed]
- Ryu, N.; Kim, M.A.; Park, D.; Lee, B.; Kim, Y.R.; Kim, K.H.; Baek, J.I.; Kim, W.J.; Lee, K.Y.; Kim, U.K. Effective PEI-mediated delivery of CRISPR-Cas9 complex for targeted gene therapy. Nanomedicine 2018, 14, 2095–2102. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, S.; Uchida, S.; Toh, K.; Tockary, T.A.; Dirisala, A.; Hayashi, K.; Fukushima, S.; Kataoka, K. Co-encapsulation of Cas9 mRNA and guide RNA in polyplex micelles enables genome editing in mouse brain. J. Control. Release 2021, 332, 260–268. [Google Scholar] [CrossRef]
- Liu, X.; Li, G.; Liu, Y.; Zhou, F.; Huang, X.; Li, K. Advances in CRISPR/Cas gene therapy for inborn errors of immunity. Front. Immunol. 2023, 14, 1111777. [Google Scholar] [CrossRef]
- Yin, W.; Chen, Z.; Huang, J.; Ye, H.; Lu, T.; Lu, M.; Rao, Y. Application of CRISPR-Cas9 gene editing technology in crop breeding. Sheng Wu Gong Cheng Xue Bao 2023, 39, 399–424. [Google Scholar] [CrossRef]
- Xu, C.F.; Chen, G.J.; Luo, Y.L.; Zhang, Y.; Zhao, G.; Lu, Z.D.; Czarna, A.; Gu, Z.; Wang, J. Rational designs of in vivo CRISPR-Cas delivery systems. Adv. Drug Deliv. Rev. 2021, 168, 3–29. [Google Scholar] [CrossRef]
- Sun, H.; Zhi, S.; Wu, G.; Wu, G.; Cao, T.; Hao, H.; Zhou, S.; Liang, P.; Huang, J. Cost-effective generation of A-to-G mutant mice by zygote electroporation of adenine base editor ribonucleoproteins. J. Genet Genom. 2020, 47, 337–340. [Google Scholar] [CrossRef]
- Uddin, F.; Rudin, C.M.; Sen, T. CRISPR Gene Therapy: Applications, Limitations, and Implications for the Future. Front. Oncol. 2020, 10, 1387. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Marchisio, M.A. CRISPR-associated type V proteins as a tool for controlling mRNA stability in S. cerevisiae synthetic gene circuits. Nucleic Acids Res. 2023, 51, 1473–1487. [Google Scholar] [CrossRef]
- Nie, J.; Han, Y.; Jin, Z.; Hang, W.; Shu, H.; Wen, Z.; Ni, L.; Wang, D.W. Homology-directed repair of an MYBPC3 gene mutation in a rat model of hypertrophic cardiomyopathy. Gene Ther. 2023, 30, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Darrow, J.J. Luxturna: FDA documents reveal the value of a costly gene therapy. Drug Discov. Today 2019, 24, 949–954. [Google Scholar] [CrossRef]
- Maguire, A.M.; Russell, S.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Drack, A.V.; Simonelli, F.; Leroy, B.P.; Reape, K.Z.; High, K.A.; et al. Durability of Voretigene Neparvovec for Biallelic RPE65-Mediated Inherited Retinal Disease: Phase 3 Results at 3 and 4 Years. Ophthalmology 2021, 128, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, J.W.; Mehat, M.S.; Sundaram, V.; Robbie, S.J.; Barker, S.E.; Ripamonti, C.; Georgiadis, A.; Mowat, F.M.; Beattie, S.G.; Gardner, P.J.; et al. Long-term effect of gene therapy on Leber’s congenital amaurosis. N. Engl. J. Med. 2015, 372, 1887–1897. [Google Scholar] [CrossRef]
- Rodgers, G.P.; Dover, G.J.; Uyesaka, N.; Noguchi, C.T.; Schechter, A.N.; Nienhuis, A.W. Augmentation by erythropoietin of the fetal-hemoglobin response to hydroxyurea in sickle cell disease. N. Engl. J. Med. 1993, 328, 73–80. [Google Scholar] [CrossRef]
- Couette, M.; Forté, S.; Oudin Doglioni, D.; Mekontso-Dessap, A.; Calvet, D.; Kuo, K.H.M.; Bartolucci, P. Early Strokes Are Associated with More Global Cognitive Deficits in Adults with Sickle Cell Disease. J. Clin. Med. 2023, 12, 1615. [Google Scholar] [CrossRef]
- Witzigmann, D.; Kulkarni, J.A.; Leung, J.; Chen, S.; Cullis, P.R.; van der Meel, R. Lipid nanoparticle technology for therapeutic gene regulation in the liver. Adv. Drug Deliv. Rev. 2020, 159, 344–363. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Delivery Strategies | Delivery Approach | Limitations | Advantages | Applications | References |
|---|---|---|---|---|---|
| LV | CRISPR/Cas9 and sgRNA | LV vectors are at risk of off-target mutations and have a limited loading capacity of 10 kb bases. | It can deliver CRISPR land to cells in a single transfection and has a high cloning capacity. Low immunogenicity and inexpensive expansion | In vitro | [14,15] |
| AAV | CRISPR/Cas9 | Includes a 4.7 kb fragment that readily integrates into the host’s genome. | AAV capsids are structurally flexible, serotype-diverse, and easily adaptable to suppress the immune response. | In vitro and in vivo | [16,17,18] |
| AdV | CRISPR/Cas9 | Packaging restricted to 8 kb fragments, prone to adverse immune reactions, more challenging to prepare | Lower risk of off-target effects and insertion mutagenesis, together with better clinical outcomes | In vivo | [19,20] |
| VLP | RNA | Limited clinical translation, instability, and insufficient support for widespread use | Excellent biosecurity, low immune response, and flexibility | In vivo | [21,22] |
| Lipo | CRISPR/Cas9 DNA | High storage and transport requirements, limited DNA concentration at delivery | High load efficiency, editing security, efficiency, and specificity | In vivo | [23,24] |
| Exosome | CRISPR/Cas9 DNA | Complex preparation, extreme storage and transport conditions, and susceptibility to degradation | Natural targeting ability, reduced immune response, and excellent biosecurity | In vivo | [25,26] |
| Polymer-based | CRISPR/Cas9 sgRNA | Possible to aggregate, destabilize, and be eliminated from the organism. | Small size, controlled release, biodegradable, lower immunogenicity | In vivo | [27] |
| Inorganic nanoparticles | CRISPR/Cas9 | Slow degradation in vivo, simple hepatic accumulation, and specific toxicity in vivo | Small size, sizeable small size, high effectiveness, delayed controlled release, targeted action, and the ability to escape an organelle called | In vivo | [28,29] |
| Gene Editing Tools | Off-Target Risk | Improvement | Gene Type | Clinical Application | Reference |
|---|---|---|---|---|---|
| ZFN | High | Optimization of DNA structural and catalytic domains using the modular structure of ZFNs | DNA | Hemophilia B and β-Thalassemia proceeded to clinical stages I and II, respectively. | [30,31,32] |
| TALEN | High | High-throughput solid-phase assembly, connection-independent cloning, and “Golden Gate” molecular cloning are just a few examples. | DNA | Clinical Phase I in HPV-related cervical intraepithelial neoplasia | [33,34] |
| CRISPR/Cas9 | Moderate | Improved targeting to the interior of the nucleus and increased mRNA stability | DNA | β-Thalassemia clinical Phase II | [35] |
| CRISPR/Cas13 | Low | Figuring out whether an RNA substrate binding site exists at the catalytic site of the Cas13 protein | single-stranded RNA | Proceed to preclinical studies | [36,37] |
| BE | Moderate | Enhancing their sequence preferences and coming up with methods to efficiently assess off-targeting | DNA | Numerous studies have laid the groundwork for conducting clinical | [38] |
| PE | low | It enhanced PE in various cells and organisms to evaluate off-target effects across the genome. | pegRNA | No clinical studies have been conducted at this time | [8] |
| TwinPE | Low | The effectiveness of gene editing is significantly boosted by adding two pegRNAs on top of PE. | Paired pegRNA | No clinical studies have been conducted at this time | [39] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, X.; Zhang, M.; Li, G.; Zhang, S.; Zhang, J.; Fu, X.; Sun, F. Applications and Research Advances in the Delivery of CRISPR/Cas9 Systems for the Treatment of Inherited Diseases. Int. J. Mol. Sci. 2023, 24, 13202. https://doi.org/10.3390/ijms241713202
Lu X, Zhang M, Li G, Zhang S, Zhang J, Fu X, Sun F. Applications and Research Advances in the Delivery of CRISPR/Cas9 Systems for the Treatment of Inherited Diseases. International Journal of Molecular Sciences. 2023; 24(17):13202. https://doi.org/10.3390/ijms241713202
Chicago/Turabian StyleLu, Xinyue, Miaomiao Zhang, Ge Li, Shixin Zhang, Jingbo Zhang, Xiaoge Fu, and Fengying Sun. 2023. "Applications and Research Advances in the Delivery of CRISPR/Cas9 Systems for the Treatment of Inherited Diseases" International Journal of Molecular Sciences 24, no. 17: 13202. https://doi.org/10.3390/ijms241713202