
Memantine Improves the Disturbed Glutamine and γ-Amino Butyric Acid Homeostasis in the Brain of Rats Subjected to Experimental Autoimmune Encephalomyelitis

Abstract
:1. Introduction
2. Results
2.1. The Effect of Memantine on the Clinical Course of EAE
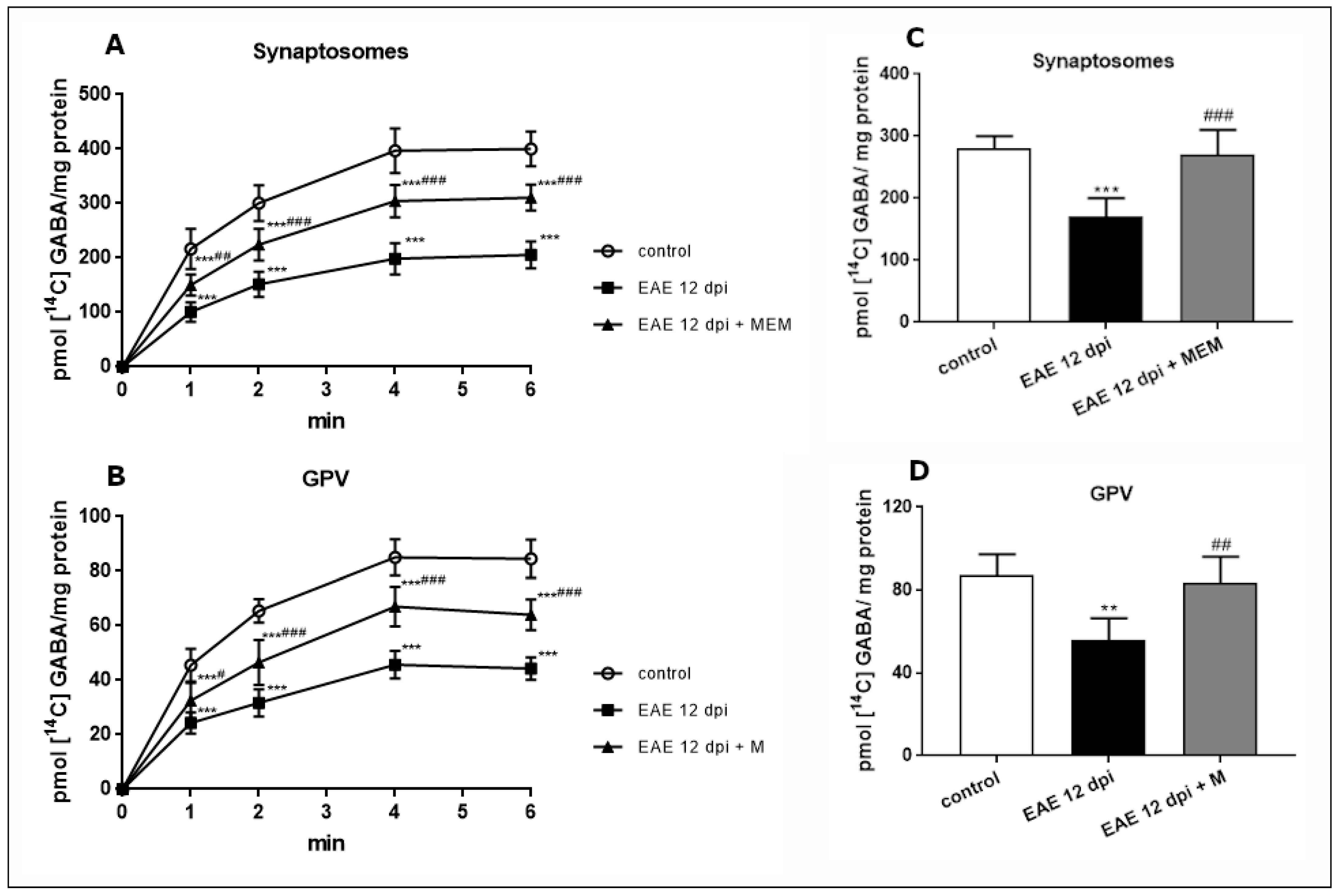
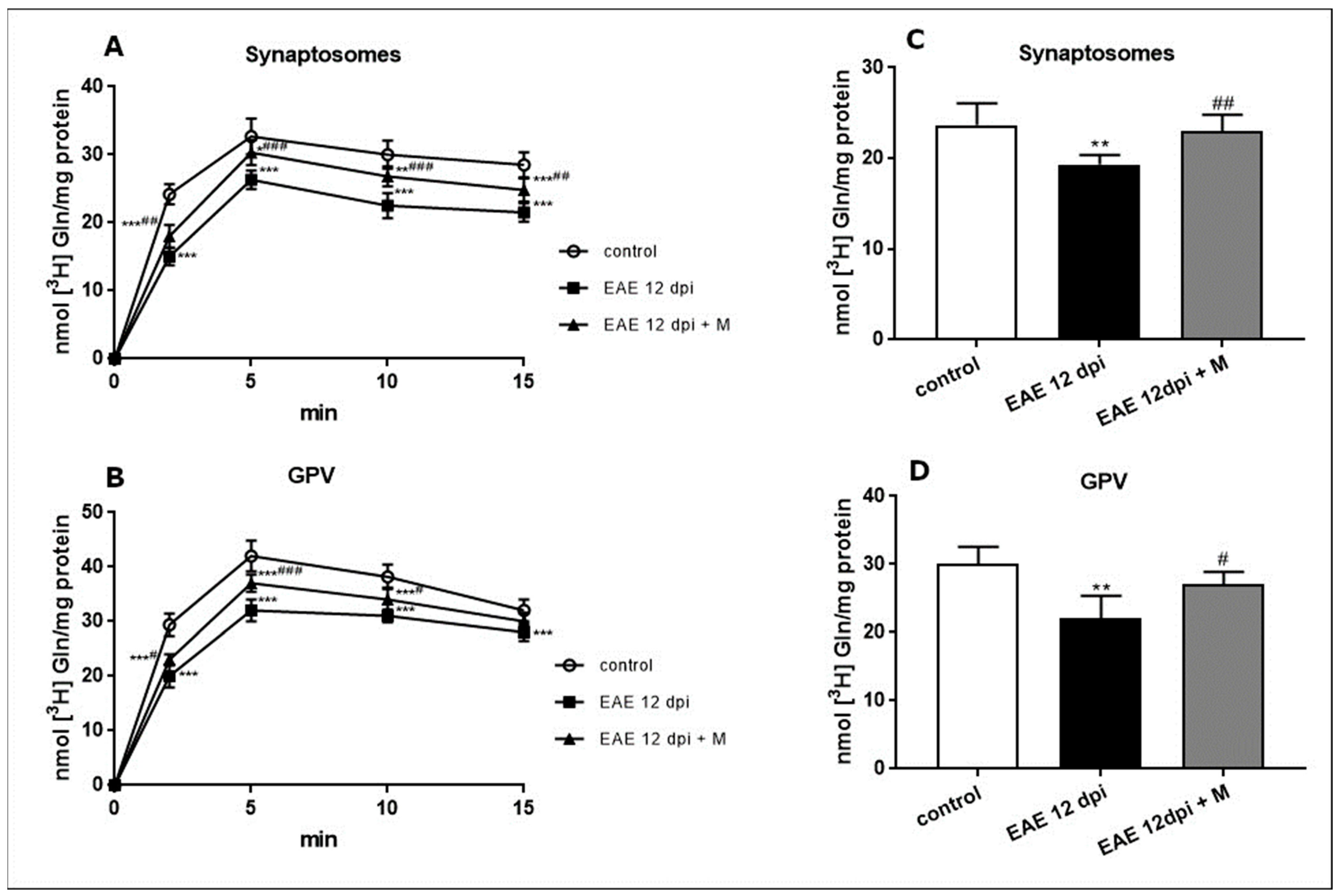
2.2. Functional Changes in GABA and Gln Amino Acids Transport during the Course of EAE and the Effect of Memantine
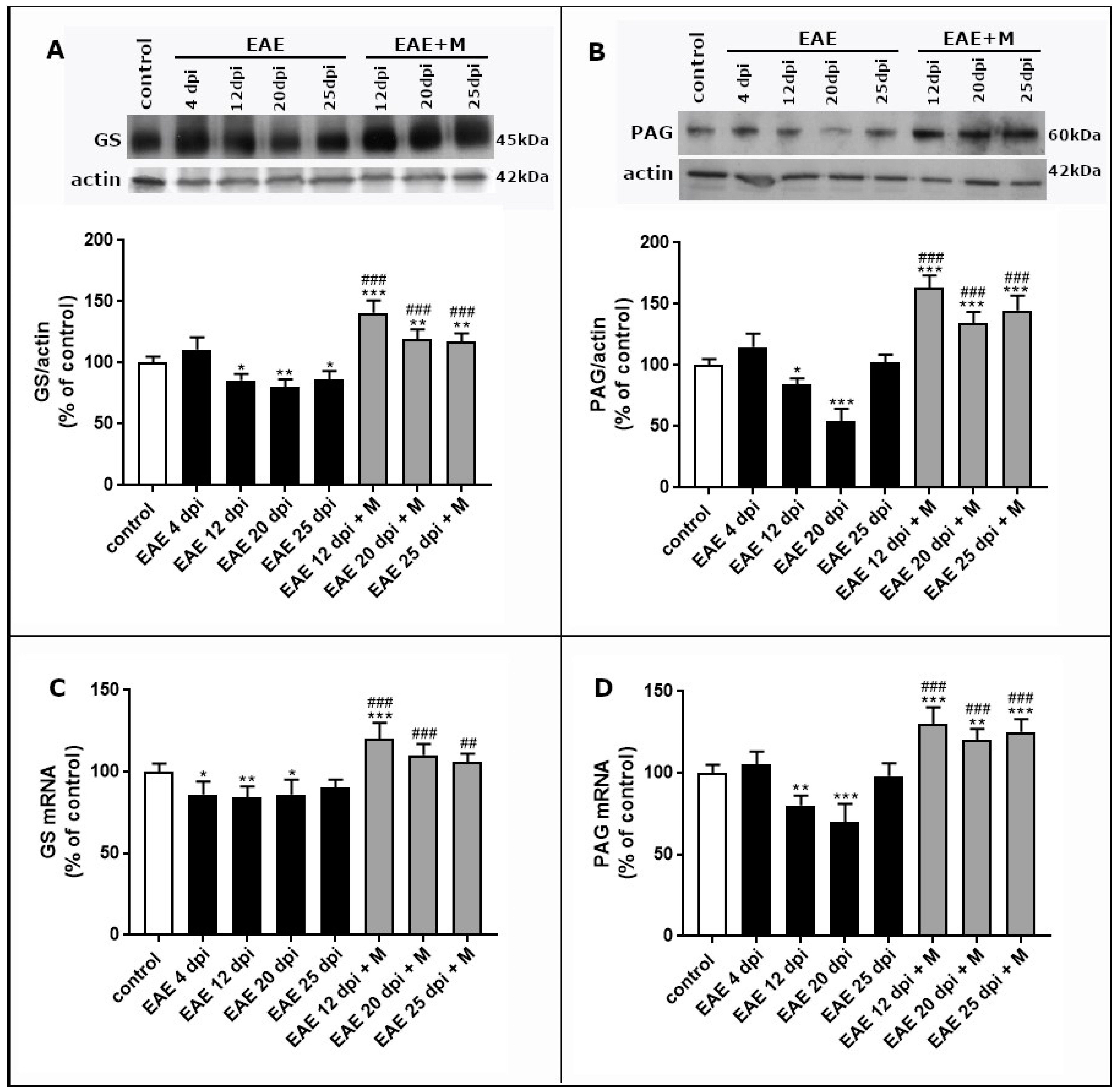
2.3. Changes in Expression of Selected Enzymes Involved in Glutamate–Glutamine Cycle and the Effect of Memantine
2.3.1. The Effect of Memantine on the Expression of GS during the Course of EAE
2.3.2. The Effect of Memantine on the Expression of PAG during the Course of EAE
2.4. The Effect of Memantine on the Activity of Selected Enzymes Involved in Glu Metabolism
3. Discussion
3.1. Changes in Metabolism of Amino Acids Involved in Neuron-Astrocyte Crosstalk during EAE
3.2. The Protective Effect of Memantine on Disturbed Homeostasis of Amino Acids Involved in Neuron–Astrocyte Crosstalk
4. Materials and Methods
4.1. Animal Model
4.2. Experimental Groups and Tissue Processing
4.3. Preparation of Synaptosomes
4.4. Preparation of GPV Fraction
4.5. Transport of Amino Acid Neurotransmitters in Brain Synaptosomes and GPV Fractions
4.6. Western Blot Analysis
4.7. qPCR Analysis
4.8. Measurement of GS Activity
4.9. Measurement of PAG Activity
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lassmann, H. Mechanisms of Demyelination and Tissue Destruction in Multiple Sclerosis. Clin. Neurol. Neurosurg. 2002, 104, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Cuzner, M.L.; Hayes, G.M.; Newcombe, J.; Woodroofe, M.N. The Nature of Inflammatory Components during Demyelination in Multiple Sclerosis. J. Neuroimmunol. 1988, 20, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Ponomarev, E.D.; Shriver, L.P.; Maresz, K.; Dittel, B.N. Microglial Cell Activation and Proliferation Precedes the Onset of CNS Autoimmunity. J. Neurosci. Res. 2005, 81, 374–389. [Google Scholar] [CrossRef]
- Groom, A.J.; Smith, T.; Turski, L. Multiple Sclerosis and Glutamate. Ann. N. Y. Acad. Sci. 2003, 993, 229–275. [Google Scholar] [CrossRef] [PubMed]
- Werner, P.; Pitt, D.; Raine, C.S. Multiple Sclerosis: Altered Glutamate Homeostasis in Lesions Correlates with Oligodendrocyte and Axonal Damage. Ann. Neurol. 2001, 50, 169–180. [Google Scholar] [CrossRef]
- Matute, C.; Alberdi, E.; Domercq, M.; Pérez-Cerdá, F.; Pérez-Samartín, A.; Sánchez-Gómez, M.V. The Link between Excitotoxic Oligodendroglial Death and Demyelinating Diseases. Trends Neurosci. 2001, 24, 224–230. [Google Scholar] [CrossRef]
- Wang, D.; Ayers, M.M.; Catmull, D.V.; Hazelwood, L.J.; Bernard, C.C.A.; Orian, J.M. Astrocyte-Associated Axonal Damage in Pre-Onset Stages of Experimental Autoimmune Encephalomyelitis. Glia 2005, 51, 235–240. [Google Scholar] [CrossRef]
- Sulkowski, G.; Dabrowska-Bouta, B.; Chalimoniuk, M.; Struzyńska, L. Effects of Antagonists of Glutamate Receptors on Pro-Inflammatory Cytokines in the Brain Cortex of Rats Subjected to Experimental Autoimmune Encephalomyelitis. J. Neuroimmunol. 2013, 261, 67–76. [Google Scholar] [CrossRef]
- Sulkowski, G.; Dąbrowska-Bouta, B.; Struzyńska, L. Modulation of Neurological Deficits and Expression of Glutamate Receptors during Experimental Autoimmune Encephalomyelitis after Treatment with Selected Antagonists of Glutamate Receptors. Biomed. Res. Int. 2013, 2013, 186068. [Google Scholar] [CrossRef]
- Sulkowski, G.; Dąbrowska-Bouta, B.; Salińska, E.; Struzyńska, L. Modulation of Glutamate Transport and Receptor Binding by Glutamate Receptor Antagonists in EAE Rat Brain. PLoS ONE 2014, 9, e113954. [Google Scholar] [CrossRef]
- Dąbrowska-bouta, B.; Strużyńska, L.; Sidoryk-węgrzynowicz, M.; Sulkowski, G. Memantine Modulates Oxidative Stress in the Rat Brain Following Experimental Autoimmune Encephalomyelitis. Int. J. Mol. Sci. 2021, 22, 11330. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Calcium and Excitotoxic Neuronal Injury. Ann. N. Y. Acad. Sci. 1994, 747, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Glutamate Neurotoxicity and Diseases of the Nervous System. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.K. Structure-Function Relationship of Transporters in the Glutamate–Glutamine Cycle of the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 1177. [Google Scholar] [CrossRef]
- Laake, J.H.; Slyngstad, T.A.; Haug, F.Š.; Ottersen, O.P. Glutamine from Glial Cells Is Essential for the Maintenance of the Nerve Terminal Pool of Glutamate: Immunogold Evidence from Hippocampal Slice Cultures. J. Neurochem. 1995, 65, 871–881. [Google Scholar] [CrossRef]
- Riepe, R.E.; Norenberg, M.D. Müller cell localization of glutamine synthetase in rat retina. Nature 1977, 286, 654–655. [Google Scholar] [CrossRef]
- Derouiche, A.; Frotscher, M. Astroglial processes around identified glutamatergic synapses contain glutamine synthetase: Evidence for transmitter degradation. Brain Res. 1991, 552, 346–350. [Google Scholar] [CrossRef]
- Danbolt, N.C. Glutamate Uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Pow, D.V.; Robinson, S.R. Glutamate in Some Retinal Neurons Is Derived Solely from Glia. Neuroscience 1994, 60, 355–366. [Google Scholar] [CrossRef]
- Reubi, J.C.; Van Der Berg, C.; Cuénod, M. Glutamine as Precursor for the GABA and Glutamate Trasmitter Pools. Neurosci. Lett. 1978, 10, 171–174. [Google Scholar] [CrossRef]
- Tapia, R.; Meza-Ruíz, G. Differences in Some Properties of Newborn and Adult Brain Glutamate Decarboxylase. J. Neurobiol. 1975, 6, 171–181. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.C.; Sonnewald, U.; Huang, X.; Stevenson, J.; Zielke, H.R. Exogenous Glutamate Concentration Regulates the Metabolic Fate of Glutamate in Astrocytes. J. Neurochem. 1996, 66, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Pascual, J.M.; Carceller, F.; Roda, J.M.; Cerdán, S. Glutamate, Glutamine, and GABA as Substrates for the Neuronal and Glial Compartments after Focal Cerebral Ischemia in Rats. Stroke 1998, 29, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, T.; Shigemoto, R.; Mizuno, N. Metabolism of Glutamate and Ammonia in Astrocyte: An Immunocytochemical Study. Brain Res. 1988, 457, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A. Paradigm Shift in NMDA Receptor Antagonist Drug Development: Molecular Mechanism of Uncompetitive Inhibition by Memantine in the Treatment of Alzheimer’s Disease and Other Neurologic Disorders. J. Alzheimer’s Dis. 2004, 6, S61–S74. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A. Pharmacological and Functional Characterization of Astrocytic GABA Transport: A Short Review. Neurochem. Res. 2000, 25, 1241–1244. [Google Scholar] [CrossRef]
- Ishibashi, M.; Egawa, K.; Fukuda, A. Diverse Actions of Astrocytes in GABAergic Signaling. Int. J. Mol. Sci. 2019, 20, 2964. [Google Scholar] [CrossRef]
- Booth, R.F.G.; Clark, J.B. A Rapid Method for the Preparation of Relatively Pure Metabolically Competent Synaptosomes from Rat Brain. Biochem. J. 1978, 176, 365–370. [Google Scholar] [CrossRef]
- Daniels, K.K.; Vickroy, T.W. Simultaneous Isolation of Glial and Neuronal Fractions from Rat Brain Homogenates: Comparison of High-Affinity L-Glutamate Transport Properties. Neurochem. Res. 1998, 23, 103–113. [Google Scholar] [CrossRef]
- Nakamura, Y.; Iga, K.; Shibata, T.; Shudo, M.; Kataoka, K. Glial Plasmalemmal Vesicles: A Subcellular Fraction from Rat Hippocampal Homogenate Distinct from Synaptosomes. Glia 1993, 9, 48–56. [Google Scholar] [CrossRef]
- Struzyńska, L. The Protective Role of Astroglia in the Early Period of Experimental Lead Toxicity in the Rat. Acta Neurobiol. Exp. 2000, 60, 167–173. [Google Scholar]
- Grygorowicz, T.; Wełniak-Kamińska, M.; Struzyńska, L. Early P2X7R-Related Astrogliosis in Autoimmune Encephalomyelitis. Mol. Cell. Neurosci. 2016, 74, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Castegna, A.; Palmieri, L.; Spera, I.; Porcelli, V.; Palmieri, F.; Fabis-Pedrini, M.J.; Kean, R.B.; Barkhouse, D.A.; Curtis, M.T.; Hooper, D.C. Oxidative Stress and Reduced Glutamine Synthetase Activity in the Absence of Inflammation in the Cortex of Mice with Experimental Allergic Encephalomyelitis. Neuroscience 2011, 185, 97–105. [Google Scholar] [CrossRef]
- Hardin-Pouzet, H.; Krakowski, M.; Bourbonnière, L.; Didier-Bazes, M.; Tran, E.; Owens, T. Glutamate Metabolism Is Down-Regulated in Astrocytes during Experimental Allergic Encephalomyelitis. Glia 1997, 20, 79–85. [Google Scholar] [CrossRef]
- Schousboe, A.; Bak, L.K.; Waagepetersen, H.S. Astrocytic Control of Biosynthesis and Turnover of the Neurotransmitters Glutamate and GABA. Front. Endocrinol. 2013, 4, 102. [Google Scholar] [CrossRef]
- Ziemińska, E.; Hilgier, W.; Waagepetersen, H.S.; Hertz, L.; Sonnewald, U.; Schousboe, A.; Albrecht, J. Analysis of Glutamine Accumulation in Rat Brain Mitochondria in the Presence of a Glutamine Uptake Inhibitor, Histidine, Reveals Glutamine Pools with a Distinct Access to Deamidation. Neurochem. Res. 2004, 29, 2121–2123. [Google Scholar] [CrossRef] [PubMed]
- Gonsette, R.E. Neurodegeneration in Multiple Sclerosis: The Role of Oxidative Stress and Excitotoxicity. J. Neurol. Sci. 2008, 274, 48–53. [Google Scholar] [CrossRef]
- Liu, J.; Feng, X.; Wang, Y.; Xia, X.; Zheng, J.C. Astrocytes: GABAceptive and GABAergic Cells in the Brain. Front. Cell. Neurosci. 2022, 16, 892497. [Google Scholar] [CrossRef]
- Dienel, G.A. Astrocytic energetics during excitatory neurotransmission: What are contributions of glutamate oxidation and glycolysis? Neurochem. Int. 2013, 63, 244–258. [Google Scholar] [CrossRef]
- Dienel, G.A.; Schousboe, A.; McKenna, M.C.; Rothman, D.L. A tribute to Leif Hertz: The historical context of his pioneering studies of the roles of astrocytes in brain energy metabolism, neurotransmission, cognitive functions, and pharmacology identifies important, unresolved topics for future studies. J. Neurochem. 2023. [Google Scholar] [CrossRef]
- Chen, H.S.V.; Lipton, S.A. The Chemical Biology of Clinically Tolerated NMDA Receptor Antagonists. J. Neurochem. 2006, 97, 1611–1626. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka, K.; Chuchmacz, J.; Wójtowicz, P.; Szymański, P. Memantine in Neurological Disorders—Schizophrenia and Depression. J. Mol. Med. 2021, 99, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Turalde, C.; Espiritu, A.; Anlacan, V. Memantine for Multiple Sclerosis: A Systematic Review and Meta-Analysis of Randomized Trials. Front. Neurol. 2021, 11, 574748. [Google Scholar] [CrossRef] [PubMed]
- Kerschensteiner, M.; Stadelmann, C.; Buddeberg, B.S.; Merkler, D.; Bareyre, F.M.; Anthony, D.C.; Linington, C.; Brück, W.; Schwab, M.E. Targeting Experimental Autoimmune Encephalomyelitis Lesions to a Predetermined Axonal Tract System Allows for Refined Behavioral Testing in an Animal Model of Multiple Sclerosis. Am. J. Pathol. 2004, 164, 1455–1469. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.; Weissert, R.; Diem, R.; Storch, M.K.; De Graaf, K.L.; Kramer, B.; Bähr, M. Acute Neuronal Apoptosis in a Rat Model of Multiple Sclerosis. J. Neurosci. 2001, 21, 6214–6220. [Google Scholar] [CrossRef]
- Ohgoh, M.; Hanada, T.; Smith, T.; Hashimoto, T.; Ueno, M.; Yamanishi, Y.; Watanabe, M.; Nishizawa, Y. Altered Expression of Glutamate Transporters in Experimental Autoimmune Encephalomyelitis. J. Neuroimmunol. 2002, 125, 170–178. [Google Scholar] [CrossRef]
- Sulkowski, G.; Bubko, I.; Struzynska, L.; Januszewski, S.; Walski, M.; Rafalowska, U. Astrocytic Response in the Rodent Model of Global Cerebral Ischemia and during Reperfusion. Exp. Toxicol. Pathol. 2002, 54, 31–38. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein Measurement with the Folin Phenol Reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Divac, I.; Fonnum, F.; Storm-Mathisen, J. High Affinity Uptake of Glutamate in Terminals of Corticostriatal Axons. Nature 1977, 266, 377–378. [Google Scholar] [CrossRef]
- Martin, D.L.; Smith, A.A. Ions and the Transport of Gamma-Aminobutyric Acid by Synaptosomes. J. Neurochem. 1972, 19, 841–855. [Google Scholar] [CrossRef]
- Troeger, M.B.; Rafalowska, U.; Erecińska, M. Effect of Oleate on Neurotransmitter Transport and Other Plasma Membrane Functions in Rat Brain Synaptosomes. J. Neurochem. 1984, 42, 1735–1742. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Chomczynski, P.; Sacchi, N. Single-Step Method of RNA Isolation by Acid Guanidinium Thiocyanate-Phenol-Chloroform Extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Pishak, M.R.; Phillips, A.T. A Modified Radioisotopic Assay for Measuring Glutamine Synthetase Activity in Tissue Extracts. Anal. Biochem. 1979, 94, 82–88. [Google Scholar] [CrossRef]
- Shapiro, R.A.; Morehouse, R.F.; Curthoys, N.P. Inhibition by Glutamate of Phosphate-Dependent Glutaminase of Rat Kidney. Biochem. J. 1982, 207, 561–566. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | EAE | EAE + Memantine | ||||||
|---|---|---|---|---|---|---|---|---|
| 4 d.p.i. | 12 d.p.i. | 20 d.p.i. | 25 d.p.i. | 12 d.p.i. | 20 d.p.i. | 25 d.p.i. | ||
| Animals with severe EAE (%) | - | 0 | 100 | 0 | 0 | 73 | 0 | 0 |
| Inductive phase (days) | - | - | 9 ± 2 | - | - | 11 ± 1 # | - | - |
| Average score | - | 0.2 ± 0.3 | 4.1 ± 0.2 | 2.2 ± 0.6 | 0.4 ± 0.5 | 2.4 ± 0.6 ## | 1.2 ± 0.6 ## | 0.2 ± 0.3 |
| Cumulative CI (score) | - | 2.5 | 61.5 | 33.5 | 6.5 | 36.0 ## | 17.5 ## | 2.5 ## |
| Number of animals | 15 | 15 | 15 | 15 | 15 | 15 | 15 | 15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dąbrowska-Bouta, B.; Strużyńska, L.; Sidoryk-Węgrzynowicz, M.; Sulkowski, G. Memantine Improves the Disturbed Glutamine and γ-Amino Butyric Acid Homeostasis in the Brain of Rats Subjected to Experimental Autoimmune Encephalomyelitis. Int. J. Mol. Sci. 2023, 24, 13149. https://doi.org/10.3390/ijms241713149
Dąbrowska-Bouta B, Strużyńska L, Sidoryk-Węgrzynowicz M, Sulkowski G. Memantine Improves the Disturbed Glutamine and γ-Amino Butyric Acid Homeostasis in the Brain of Rats Subjected to Experimental Autoimmune Encephalomyelitis. International Journal of Molecular Sciences. 2023; 24(17):13149. https://doi.org/10.3390/ijms241713149
Chicago/Turabian StyleDąbrowska-Bouta, Beata, Lidia Strużyńska, Marta Sidoryk-Węgrzynowicz, and Grzegorz Sulkowski. 2023. "Memantine Improves the Disturbed Glutamine and γ-Amino Butyric Acid Homeostasis in the Brain of Rats Subjected to Experimental Autoimmune Encephalomyelitis" International Journal of Molecular Sciences 24, no. 17: 13149. https://doi.org/10.3390/ijms241713149