
Genomic Scan for Runs of Homozygosity and Selective Signature Analysis to Identify Candidate Genes in Large White Pigs

Abstract
:1. Introduction
2. Results
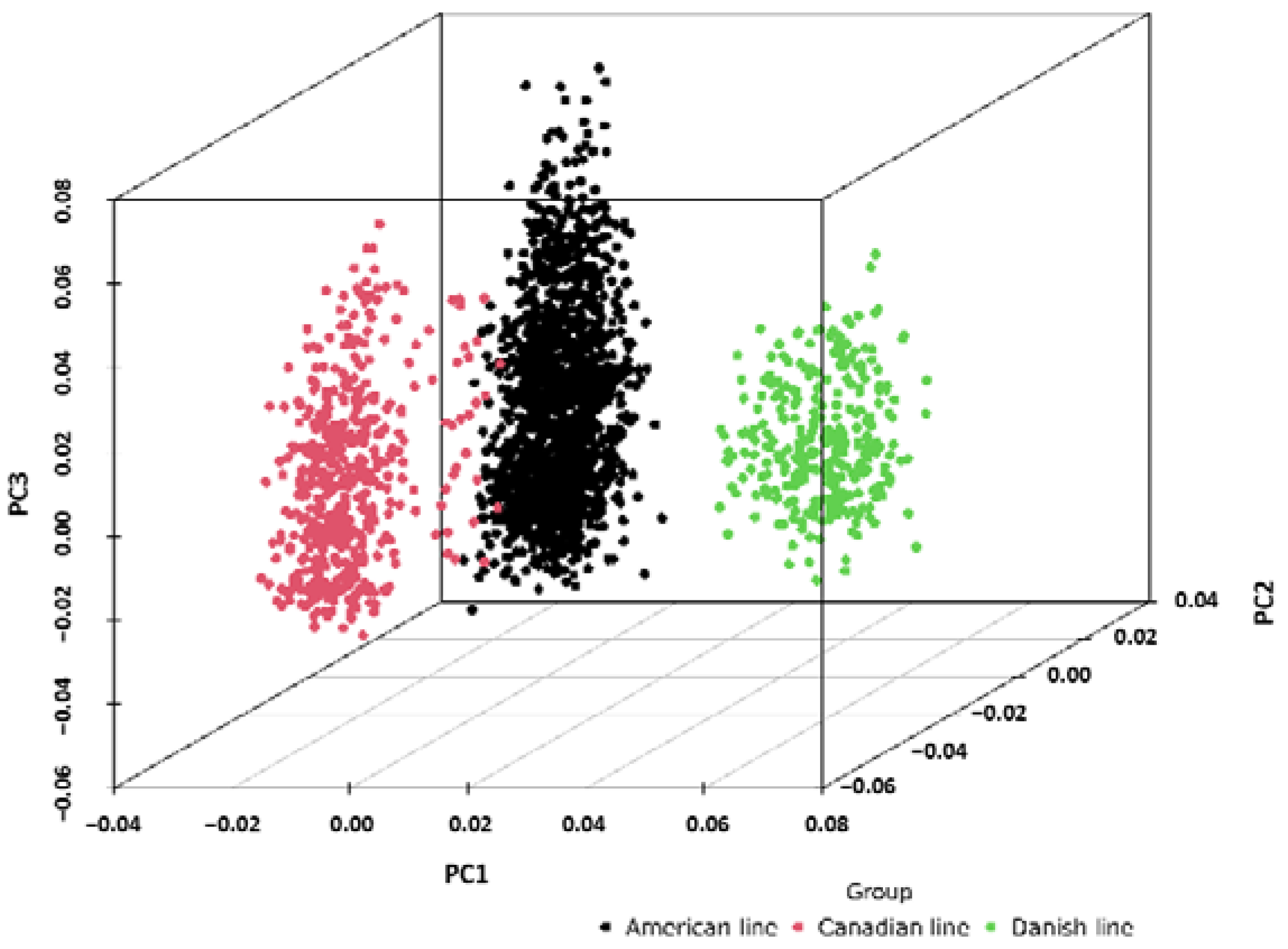
2.1. Population Stratification Assessment
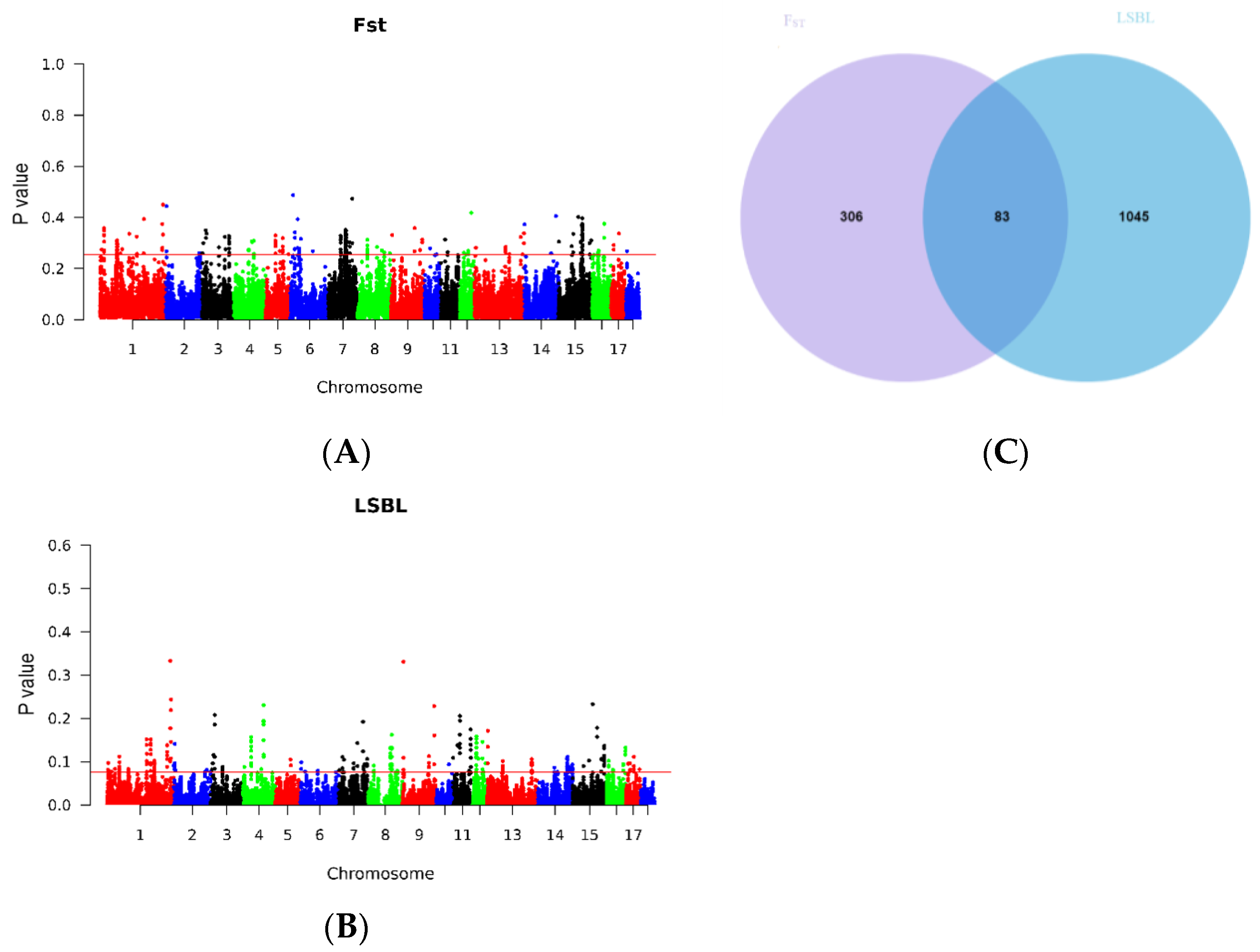
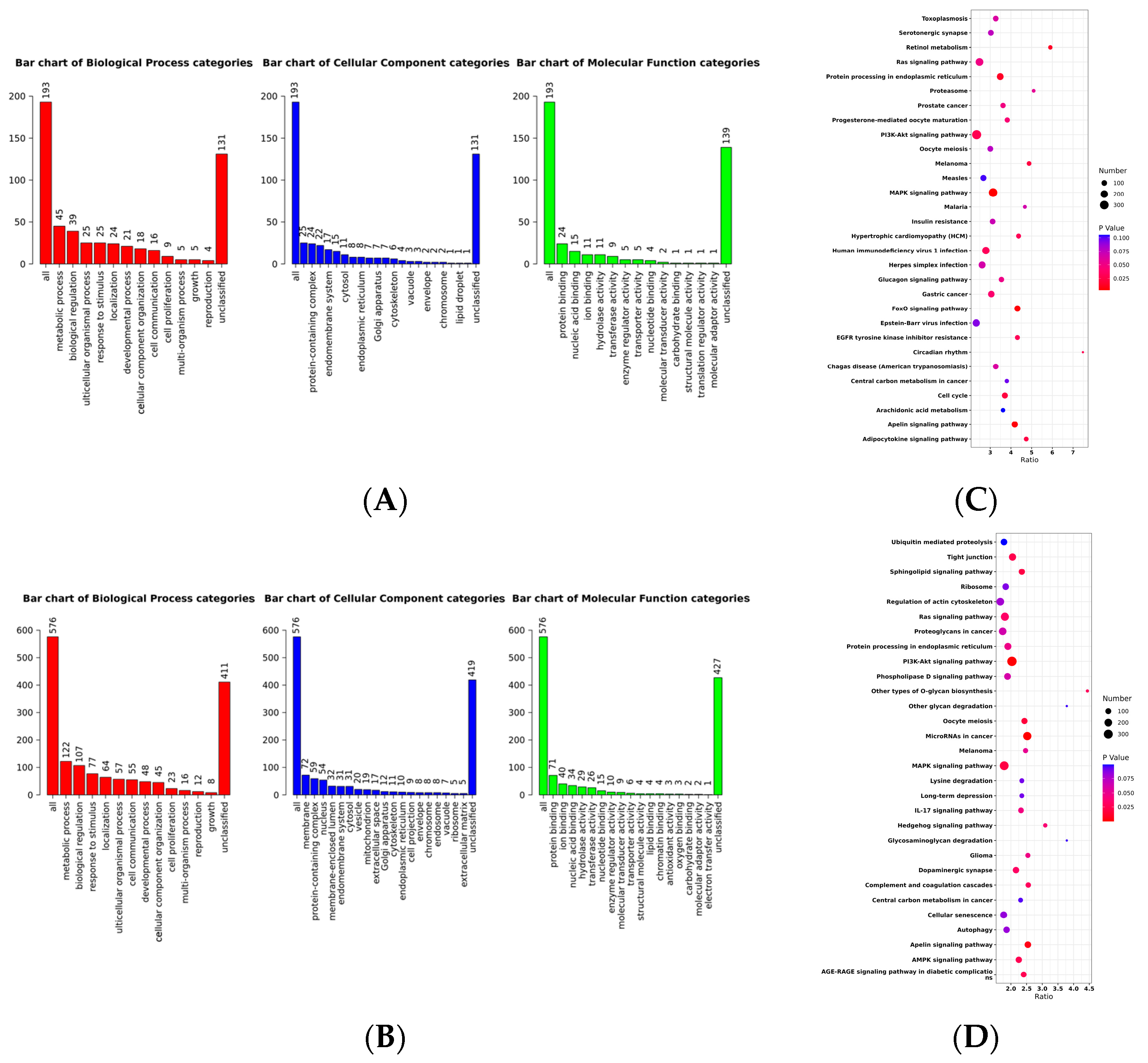
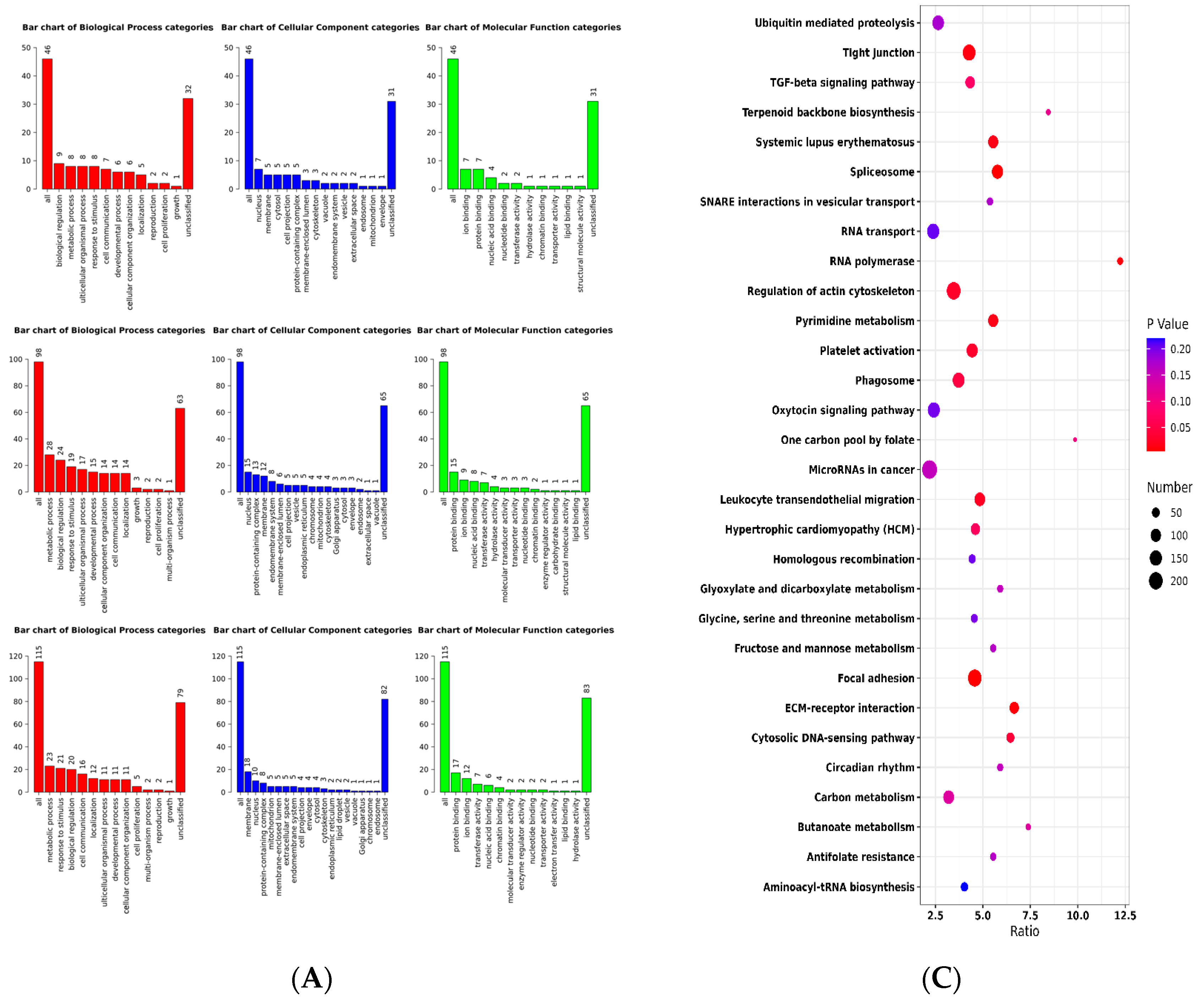
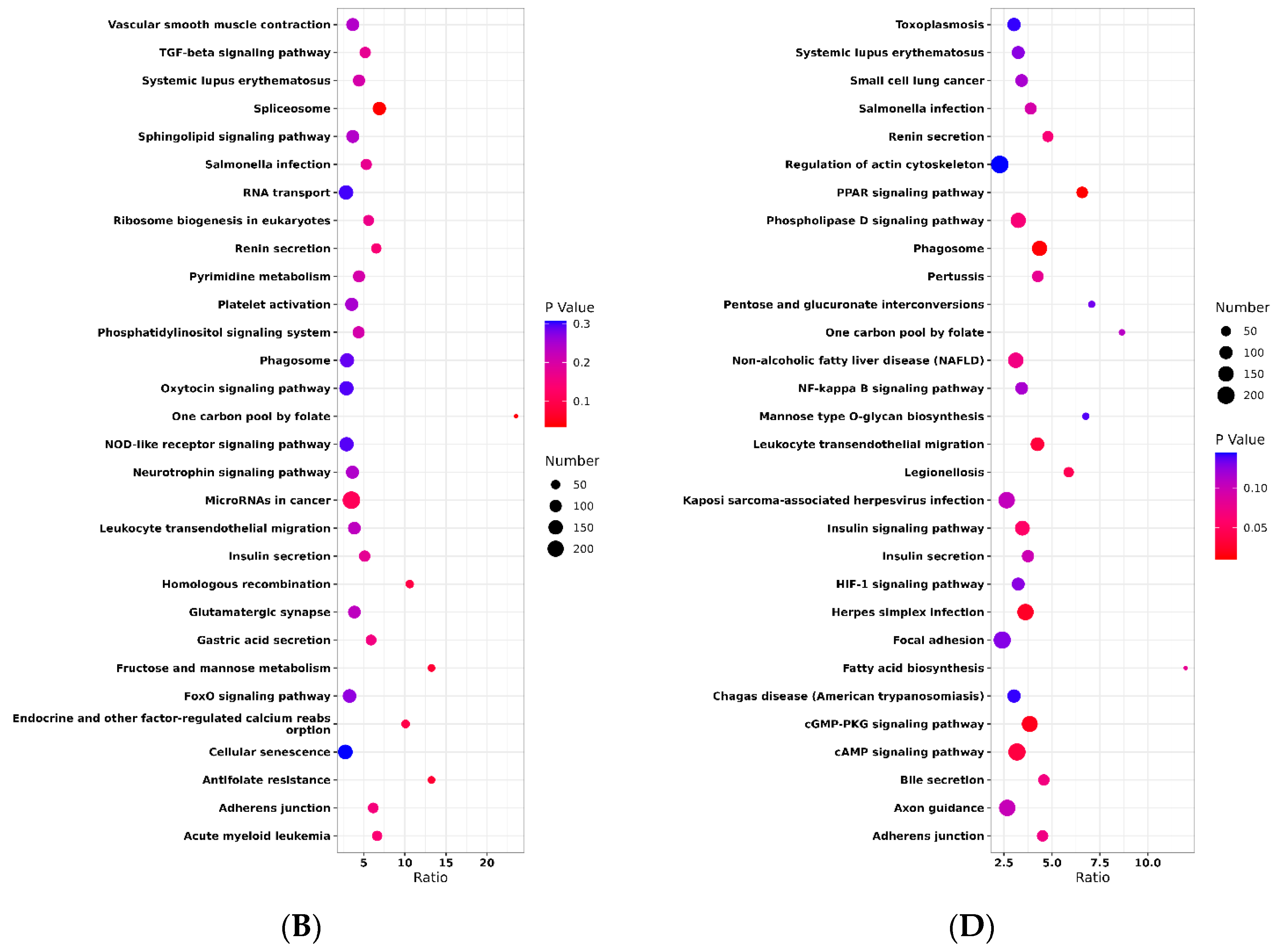
2.2. Selective Signature Analysis
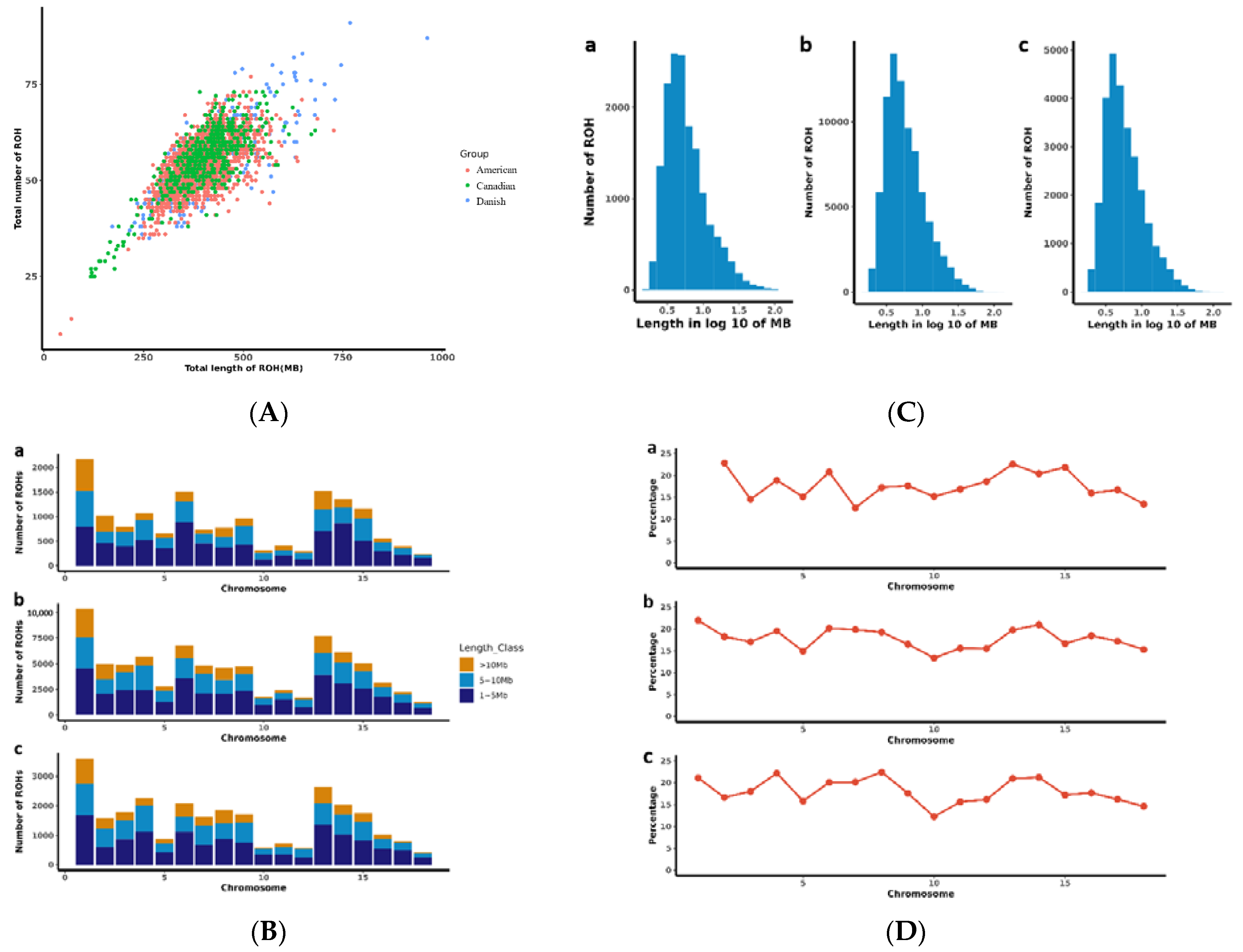
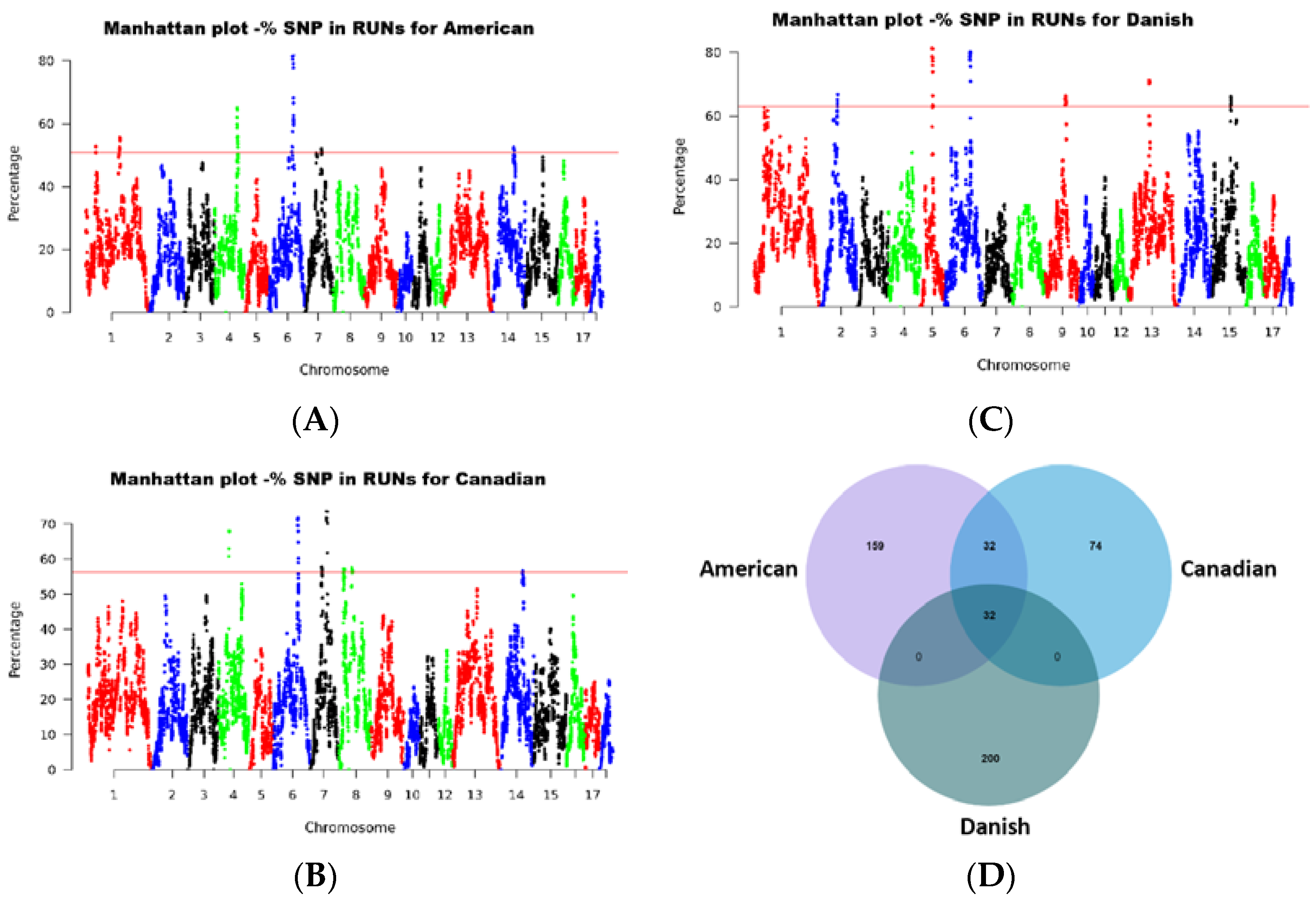
2.3. Runs of Homozygosity Analysis
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. DNA Sampling and Sequencing of DNA
4.3. Population Structure
4.4. Partitioning Heritabilities of Complex Traits Based on Selection Signatures
4.5. Runs of Homozygosity Detection
4.6. Detection of Common Runs of Homozygosity
4.7. Pathway and Functional Analysis
4.8. Gene Annotation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, S.; Zhang, K.; Peng, X.; Zhan, H.; Lu, J.; Xie, S.; Zhao, S.; Li, X.; Ma, Y. Selective sweep analysis reveals extensive parallel selection traits between large white and Duroc pigs. Evol. Appl. 2020, 13, 2807–2820. [Google Scholar] [CrossRef] [PubMed]
- Ai, H.; Yang, B.; Li, J.; Xie, X.; Chen, H.; Ren, J. Population history and genomic signatures for high-altitude adaptation in Tibetan pigs. BMC Genom. 2014, 15, 834. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, S.; Tang, Z.; Li, K.; Rothschild, M.F.; Liu, B.; Fan, B. Genome-wide scans to detect positive selection in Large White and Tongcheng pigs. Anim. Genet. 2014, 45, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, P.; Zhou, W.; Ma, X.; Wang, X.; Xu, Y.; Jiang, N.; Zhao, M.; Zhou, T.; Yin, Y.J.; et al. Genome data uncover conservation status, historical relatedness and candidate genes under selection in Chinese indigenous pigs in the Taihu Lake region. Front. Genet. 2020, 11, 591. [Google Scholar] [CrossRef]
- Gibson, J.; Morton, N.E.; Collins, A. Extended tracts of homozygosity in outbred human populations. Hum. Mol. Genet. 2006, 15, 789–795. [Google Scholar] [CrossRef]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef]
- Robinson, J.A.; Räikkönen, J.; Vucetich, L.M.; Vucetich, J.A.; Peterson, R.O.; Lohmueller, K.E.; Wayne, R.K. Genomic signatures of extensive inbreeding in Isle Royale wolves, a population on the threshold of extinction. Sci. Adv. 2019, 5, eaau0757. [Google Scholar] [CrossRef]
- Peripolli, E.; Munari, D.; Silva, M.; Lima, A.; Irgang, R.; Baldi, F. Runs of homozygosity: Current knowledge and applications in livestock. Anim. Genet. 2017, 48, 255–271. [Google Scholar] [CrossRef]
- Broman, K.W.; Weber, J.L. Long homozygous chromosomal segments in reference families from the centre d’Etude du polymorphisme humain. Am. J. Hum. Genet. 1999, 65, 1493–1500. [Google Scholar] [CrossRef]
- Howie, B.N.; Donnelly, P.; Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009, 5, e1000529. [Google Scholar] [CrossRef]
- Wu, F.; Sun, H.; Lu, S.; Gou, X.; Yan, D.; Xu, Z.; Zhang, Z.; Qadri, Q.R.; Zhang, Z.; Wang, Z. Genetic diversity and selection signatures within Diannan small-ear pigs revealed by next-generation sequencing. Front. Genet. 2020, 11, 733. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yang, J.; Li, G.; Ding, R.; Zhuang, Z.; Ruan, D.; Wu, J.; Yang, H.; Zheng, E.; Cai, G.; et al. Identification of homozygous regions with adverse effects on the five economic traits of Duroc pigs. Front. Vet. Sci. 2022, 9, 855933. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Sun, H.; Zhang, Z.; Zhao, Q.; Olasege, B.S.; Li, Q.; Yue, Y.; Ma, P.; Zhang, X.; Wang, Q.; et al. Assessment of autozygosity derived from runs of homozygosity in Jinhua pigs disclosed by sequencing data. Front. Genet. 2019, 10, 274. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef]
- Ge, J.; Liu, K.; Niu, W.; Chen, M.; Wang, M.; Xue, Y.; Gao, C.; Ma, P.X.; Lei, B. Gold and gold-silver alloy nanoparticles enhance the myogenic differentiation of myoblasts through p38 MAPK signaling pathway and promote in vivo skeletal muscle regeneration. Biomaterials 2018, 175, 19–29. [Google Scholar] [CrossRef]
- Dong, S.; Han, Y.; Zhang, J.; Ye, Y.; Duan, M.; Wang, K.; Wei, M.; Chamba, Y.; Shang, P. Haplotypes within the regulatory region of MYL4 are associated with pig muscle fiber size. Gene 2023, 850, 146934. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Yang, Y.; Fan, X.; Liang, G.; Wang, Z.; Li, J.; Wang, L.; Chen, Y.; Adetula, A.A.; Tang, Y.; et al. circRNAome profiling reveals circFgfr2 regulates myogenesis and muscle regeneration via a feedback loop. J. Cachex-Sarcopenia Muscle 2022, 13, 696–712. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, W.; Wang, R.; Lei, C.; Zhou, R.; Tang, Z.; Li, K. Wnt antagonist, secreted frizzled-related protein 1, is involved in prenatal skeletal muscle development and is a target of miRNA-1/206 in pigs. BMC Mol. Biol. 2015, 16, 4. [Google Scholar] [CrossRef]
- Comuzzie, A.G.; Cole, S.A.; Laston, S.L.; Voruganti, V.S.; Haack, K.; Gibbs, R.A.; Butte, N.F. Novel genetic loci identified for the pathophysiology of childhood obesity in the Hispanic population. PLoS ONE 2012, 7, e51954. [Google Scholar] [CrossRef]
- Li, J.; Wu, J.; Jian, Y.; Zhuang, Z.; Qiu, Y.; Huang, R.; Lu, P.; Guan, X.; Huang, X.; Li, S.; et al. Genome-Wide Association Studies Revealed Significant QTLs and Candidate Genes Associated with Backfat and Loin Muscle Area in Pigs Using Imputation-Based Whole Genome Sequencing Data. Animals 2022, 12, 2911. [Google Scholar] [CrossRef]
- Dey, B.K.; Gagan, J.; Dutta, A. miR-206 and -486 Induce Myoblast Differentiation by Downregulating Pax7. Mol. Cell. Biol. 2011, 31, 203–214. [Google Scholar] [CrossRef]
- Cho, I.-C.; Park, H.-B.; Ahn, J.S.; Han, S.-H.; Lee, J.-B.; Lim, H.-T.; Yoo, C.-K.; Jung, E.-J.; Kim, D.-H.; Sun, W.-S. A functional regulatory variant of MYH3 influences muscle fiber-type composition and intramuscular fat content in pigs. PLoS Genet. 2019, 15, e1008279. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Chen, L.; Meng, W.; Yang, K.; Wei, S.; Wei, W.; Chen, J.; Zhang, L. C/EBPα promotes porcine pre-adipocyte proliferation and differentiation via mediating MSTRG. 12568.2/FOXO3 trans-activation for STYX. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2022, 1867, 159206. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, S.; Aspesi, A.; Silengo, L.; Altruda, F.; Dianzani, I.; Chiabrando, D. Alteration of heme metabolism in a cellular model of Diamond–Blackfan anemia. Eur. J. Haematol. 2016, 96, 367–374. [Google Scholar] [CrossRef]
- Wang, B.; Hou, L.; Zhou, W.; Liu, H.; Tao, W.; Wu, W.; Niu, P.; Zhang, Z.; Zhou, J.; Li, Q.; et al. Genome-wide association study reveals a quantitative trait locus and two candidate genes on Sus scrofa chromosome 5 affecting intramuscular fat content in Suhuai pigs. Animal 2021, 15, 100341. [Google Scholar] [CrossRef] [PubMed]
- Song, S.-H.; Kim, K.; Jo, E.-K.; Kim, Y.-W.; Kwon, J.-S.; Bae, S.S.; Sung, J.-H.; Park, S.G.; Kim, J.T.; Suh, W.; et al. Fibroblast growth factor 12 is a novel regulator of vascular smooth muscle cell plasticity and fate. Arter. Thromb. Vasc. Biol. 2016, 36, 1928–1936. [Google Scholar] [CrossRef] [PubMed]
- Lundholm, M.; Mayans, S.; Motta, V.; Löfgren-Burström, A.; Danska, J.; Holmberg, D. Variation in the CD3ζ (Cd247) gene correlates with altered T cell activation and is associated with autoimmune diabetes. J. Immunol. 2010, 184, 5537–5544. [Google Scholar] [CrossRef]
- Ceballos, F.C.; Hazelhurst, S.; Ramsay, M. Assessing runs of Homozygosity: A comparison of SNP Array and whole genome sequence low coverage data. BMC Genom. 2018, 19, 106. [Google Scholar] [CrossRef]
- Schiavo, G.; Bovo, S.; Bertolini, F.; Dall’Olio, S.; Costa, L.N.; Tinarelli, S.; Gallo, M.; Fontanesi, L. Runs of homozygosity islands in Italian cosmopolitan and autochthonous pig breeds identify selection signatures in the porcine genome. Livest. Sci. 2020, 240, 104219. [Google Scholar] [CrossRef]
- Reglődi, D.; Cseh, S.; Somoskői, B.; Fülöp, B.D.; Szentléleky, E.; Szegeczki, V.; Kovacs, A.; Varga, A.; Kiss, P.; Hashimoto, H.; et al. Disturbed spermatogenic signaling in pituitary adenylate cyclase activating polypeptide-deficient mice. Reproduction 2018, 155, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.A.; Leon, S.; Madara, J.C.; Schafer, D.; Fergani, C.; Maguire, C.A.; Verstegen, A.M.; Brengle, E.; Kong, D.; Herbison, A.E.; et al. PACAP neurons in the ventral premammillary nucleus regulate reproductive function in the female mouse. eLife 2018, 7, e35960. [Google Scholar] [CrossRef]
- Avasthi, P.; Scheel, J.F.; Ying, G.; Frederick, J.M.; Baehr, W.; Wolfrum, U. Germline deletion of Cetn1 causes infertility in male mice. J. Cell Sci. 2013, 126, 3204–3213. [Google Scholar] [CrossRef]
- Wang, X.; Chinnam, M.; Wang, J.; Wang, Y.; Zhang, X.; Marcon, E.; Moens, P.; Goodrich, D.W. Thoc1 deficiency compromises gene expression necessary for normal testis development in the mouse. Mol. Cell. Biol. 2009, 29, 2794–2803. [Google Scholar] [CrossRef] [PubMed]
- Crimmins, S.; Sutovsky, M.; Chen, P.-C.; Huffman, A.; Wheeler, C.; Swing, D.A.; Roth, K.; Wilson, J.; Sutovsky, P.; Wilson, S. Transgenic rescue of ataxia mice reveals a male-specific sterility defect. Dev. Biol. 2009, 325, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Padua, M.B.; Jiang, T.; Morse, D.A.; Fox, S.C.; Hatch, H.M.; Tevosian, S.G. Combined loss of the GATA4 and GATA6 transcription factors in male mice disrupts testicular development and confers adrenal-like function in the testes. Endocrinology 2015, 156, 1873–1886. [Google Scholar] [CrossRef] [PubMed]
- De Tomasi, L.; David, P.; Humbert, C.; Silbermann, F.; Arrondel, C.; Tores, F.; Fouquet, S.; Desgrange, A.; Niel, O.; Bole-Feysot, C.; et al. Mutations in GREB1L cause bilateral kidney agenesis in humans and mice. Am. J. Hum. Genet. 2017, 101, 803–814. [Google Scholar] [CrossRef]
- Niu, N.; Liu, Q.; Hou, X.; Liu, X.; Wang, L.; Zhao, F.; Gao, H.; Shi, L.; Wang, L.; Zhang, L. Genome-wide association study revealed ABCD4 on SSC7 and GREB1L and MIB1 on SSC6 as crucial candidate genes for rib number in Beijing Black pigs. Anim. Genet. 2022, 53, 690–695. [Google Scholar] [CrossRef]
- Feng, Y.; Niu, L.; Wei, W.; Zhang, W.; Li, X.; Cao, J.; Zhao, S. A feedback circuit between miR-133 and the ERK1/2 pathway involving an exquisite mechanism for regulating myoblast proliferation and differentiation. Cell Death Dis. 2013, 4, e934. [Google Scholar] [CrossRef]
- Baas, D.; Caussanel-Boude, S.; Guiraud, A.; Calhabeu, F.; Delaune, E.; Pilot, F.; Chopin, E.; Machuca-Gayet, I.; Vernay, A.; Bertrand, S.; et al. CKIP-1 regulates mammalian and zebrafish myoblast fusion. J. Cell Sci. 2012, 125, 3790–3800. [Google Scholar] [CrossRef]
- Chen, Y.; Rui, B.-B.; Tang, L.-Y.; Hu, C.-M. Lipin family proteins-key regulators in lipid metabolism. Ann. Nutr. Metab. 2015, 66, 10–18. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Waskom, M.L. Seaborn: Statistical data visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Shriver, M.D.; Kennedy, G.C.; Parra, E.J.; Lawson, H.A.; Sonpar, V.; Huang, J.; Akey, J.M.; Jones, K.W. The genomic distribution of population substructure in four populations using 8525 autosomal SNPs. Hum. Genom. 2004, 1, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Lencz, T.; Lambert, C.; DeRosse, P.; Burdick, K.E.; Morgan, T.V.; Kane, J.M.; Kucherlapati, R.; Malhotra, A.K. Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc. Natl. Acad. Sci. USA 2007, 104, 19942–19947. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SSC (Sus Scrofa Chromosome) | Position (Mb) | Distance (bp) * | Genes |
|---|---|---|---|
| 1 | 182.24–182.36 | Upstream 117,606 | STYX |
| 2 | 2.79–2.91 | Upstream 74,339 | SHANK2 |
| 4 | 83.49–83.61 | Upstream 4395 | CD247 |
| 5 | 71.79–71.91 | Upstream 10,224 | LRRK2 |
| 9 | 130.54–130.66 | Upstream 1660 | FLVCR1 |
| 12 | 16.79–16.91 | Downstream 14,536 | MYL4 |
| 54.85–55.35 | Upstream 49,706 | MYH3 | |
| 13 | 129.24–129.36 | Upstream 83 | FGF12 |
| 14 | 131.14–131.216 | Upstream 41,712 | FGFR2 |
| 17 | 10.44–10.56 | Downstream 3575 | SFRP1 |
| 10.64–10.76 | Upstream 118,817 | miR-486 |
| Groups | Chromosome | Start (bp) | End (bp) | Length (bp) | Number of SNPs | pigQTLdb |
|---|---|---|---|---|---|---|
| American | 1 | 43,045,542 | 43,357,270 | 311,728 | 5 | - |
| 146,085,059 | 148,974,102 | 2,889,043 | 42 | S: 145,869,313 E: 173,242,773 Average daily gain | ||
| 4 | 98,912,988 | 102,212,696 | 3,299,708 | 51 | - | |
| 6 | 102,107,540 | 102,337,903 | 230,363 | 4 | - | |
| 102,717,110 | 102,796,136 | 79,026 | 2 | - | ||
| 102,917,556 | 104,370,915 | 1,453,359 | 23 | - | ||
| 104,981,850 | 110,109,305 | 5,127,455 | 121 | - | ||
| 7 | 72,338,896 | 74,402,073 | 2,063,177 | 42 | S: 72,215,870 E: 87,765,126 Fat area percentage in carcass | |
| 14 | 98,912,988 | 102,212,696 | 3,299,708 | 43 | - | |
| Canadian | 4 | 44,727,463 | 48,379,816 | 3,652,353 | 54 | S: 44,723,094 E: 91,039,884 Ham weight |
| 6 | 105,047,268 | 107,701,419 | 2,654,151 | 64 | - | |
| 7 | 50,032,121 | 52,080,918 | 2,048,797 | 31 | - | |
| 70,691,786 | 74,260,534 | 3,568,748 | 66 | S: 70,292,251 E: 83,677,435 Teat number | ||
| 8 | 22,032,465 | 23,585,036 | 1,552,571 | 29 | - | |
| 24,568,718 | 25,096,226 | 527,508 | 2 | S: 24,414,300 E: 25,683,843 Umbilical hernia | ||
| 57,175,682 | 58,758,032 | 1,582,350 | 35 | S: 56,966,700 E: 67,491,976 Hematocrit | ||
| 14 | 92,778,821 | 94,149,712 | 1,370,891 | 39 | - | |
| Danish | 2 | 71,619,091 | 74,328,649 | 2,709,558 | 33 | S: 71,416,758 E: 128,795,277 Leaf fat weight |
| 5 | 51,535,641 | 54,004,977 | 2,469,336 | 52 | - | |
| 54,091,881 | 54,253,706 | 161,825 | 5 | S: 54,354,525 E: 54,411,945 uterine horn length | ||
| 6 | 105,105,811 | 107,369,304 | 2,263,493 | 57 | - | |
| 9 | 83,483,921 | 88,168,152 | 4,684,231 | 111 | S: 80,796,751 E: 97,479,874 Immunoglobulin G level | |
| 13 | 86,541,183 | 88,583,284 | 2,042,101 | 48 | S: 86,471,446 E: 118,227,339 Lean meat percentage | |
| 15 | 76,452,937 | 76,592,155 | 139,218 | 5 | S: 76,167,178 E: 76,761,699 Intramuscular fat content | |
| 77,669,772 | 79,049,130 | 1,379,358 | 26 | S: 77,173,290 E: 90,664,324 Drip loss |
| SSC (Sus Scrofa Chromosome) | Start (bp) | End (bp) | Distance(bp) * | Genes |
|---|---|---|---|---|
| American line | ||||
| 4 | 98,912,988 | 102,212,696 | Upstream 11,008 | PLEKHO1 |
| 6 | 102,917,556 | 104,370,915 | Upstream 770,187 | LPIN2 |
| Meta-analysis | ||||
| 6 | 105,105,811 | 107,369,304 | Upstream 308,820 | ADCYAP1 |
| Upstream 634,739 | CETN1 | |||
| Upstream 934,231 | THOC1 | |||
| Upstream 982,113 | USP14 | |||
| Upstream 1,314,132 | GREB1L | |||
| Upstream 1,893,085 | miR-133 | |||
| Upstream 2,177,038 | GATA6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, C.; Wang, Y.; Zhou, P.; Shi, H.; Ma, X.; Yin, Z.; Liu, Y. Genomic Scan for Runs of Homozygosity and Selective Signature Analysis to Identify Candidate Genes in Large White Pigs. Int. J. Mol. Sci. 2023, 24, 12914. https://doi.org/10.3390/ijms241612914
Yin C, Wang Y, Zhou P, Shi H, Ma X, Yin Z, Liu Y. Genomic Scan for Runs of Homozygosity and Selective Signature Analysis to Identify Candidate Genes in Large White Pigs. International Journal of Molecular Sciences. 2023; 24(16):12914. https://doi.org/10.3390/ijms241612914
Chicago/Turabian StyleYin, Chang, Yuwei Wang, Peng Zhou, Haoran Shi, Xinyu Ma, Zongjun Yin, and Yang Liu. 2023. "Genomic Scan for Runs of Homozygosity and Selective Signature Analysis to Identify Candidate Genes in Large White Pigs" International Journal of Molecular Sciences 24, no. 16: 12914. https://doi.org/10.3390/ijms241612914