
Unique Gut Microbiome Signatures among Adult Patients with Moderate to Severe Atopic Dermatitis in Southern Chinese
 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Characteristics of Participants
2.2. Distinct Gut Microbiome Composition between AD Patients and Healthy Subjects
2.3. Gut Microbial Composition in Healthy Individuals and AD Patients
2.4. Identification of Gut Microbial Signatures to Differentiate between AD and Healthy Subjects and Related to the Severity of AD
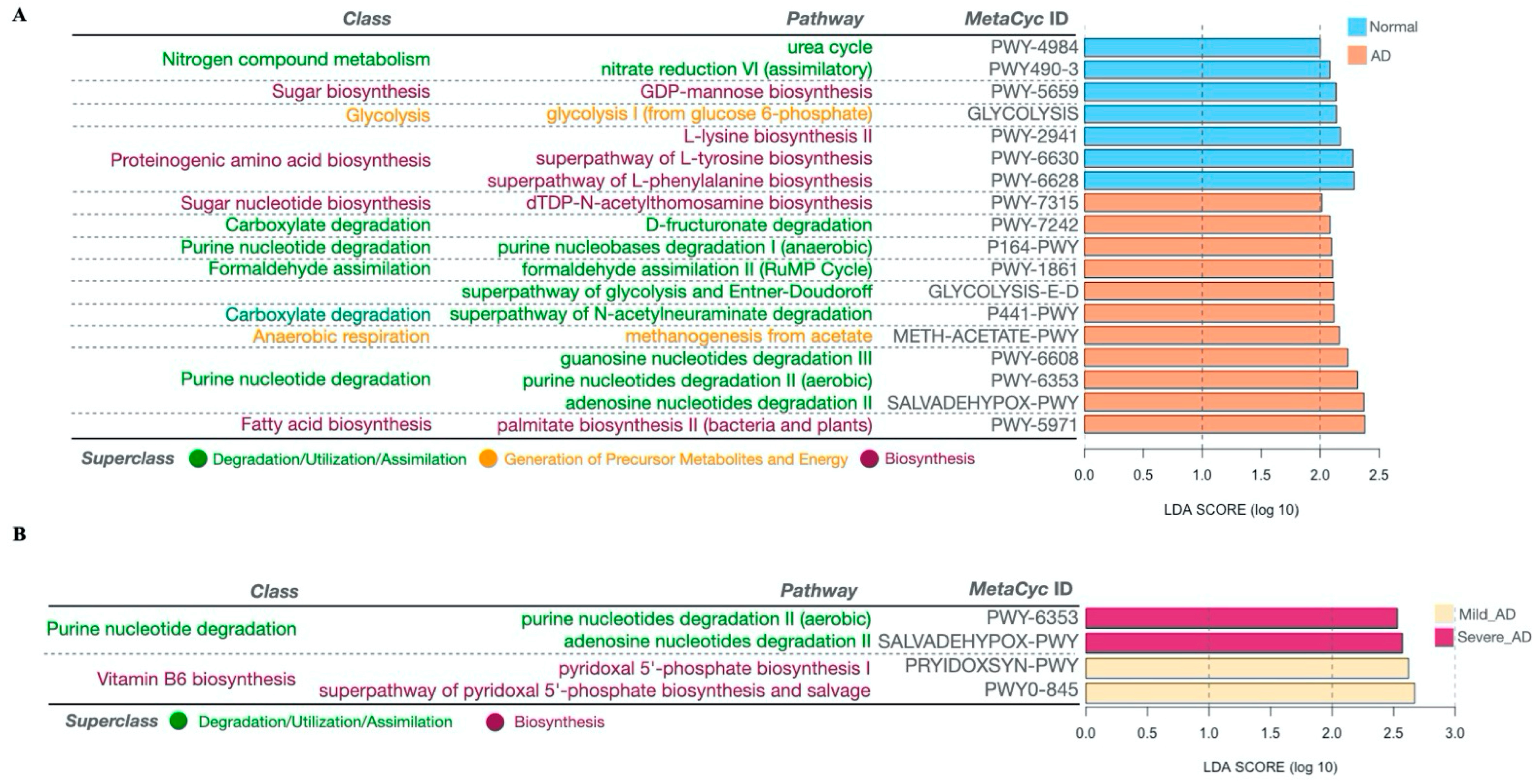
2.5. Significantly Altered MetaCyc Pathways of the Gut Microbiome Associated with AD
3. Discussion
4. Materials and Methods
4.1. Study Design and Participants
4.2. Sample Collection, DNA Extraction, and 16S rRNA Gene Sequencing
4.3. Microbiome Data Analysis
4.4. Functional Profiling Prediction with PICRUSt2
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Langan, S.M.; Irvine, A.D.; Weidinger, S. Atopic dermatitis. Lancet 2020, 396, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Caubet, J.-C.; Eigenmann, P.A. Allergic Triggers in Atopic Dermatitis. Immunol. Allergy Clin. N. Am. 2010, 30, 289–307. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; Lee, E.; Park, Y.M.; Hong, S.-J. Microbiome in the Gut-Skin Axis in Atopic Dermatitis. Allergy Asthma Immunol. Res. 2018, 10, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.A.; Verdi, S.; Maxan, M.-E.; Shin, C.M.; Zierer, J.; Bowyer, R.C.E.; Martin, T.; Williams, F.M.K.; Menni, C.; Bell, J.T.; et al. Gut microbiota associations with common diseases and prescription medications in a population-based cohort. Nat. Commun. 2018, 9, 2655. [Google Scholar] [CrossRef]
- Mahmud, R.; Akter, S.; Tamanna, S.K.; Mazumder, L.; Esti, I.Z.; Banerjee, S.; Akter, S.; Hasan, R.; Acharjee, M.; Hossain, S.; et al. Impact of gut microbiome on skin health: Gut-skin axis observed through the lenses of therapeutics and skin diseases. Gut Microbes 2022, 14, 2096995. [Google Scholar] [CrossRef]
- Galkin, F.; Mamoshina, P.; Aliper, A.; Putin, E.; Moskalev, V.; Gladyshev, V.N.; Zhavoronkov, A. Human Gut Microbiome Aging Clock Based on Taxonomic Profiling and Deep Learning. iScience 2020, 23, 101199. [Google Scholar] [CrossRef]
- Abrahamsson, T.R.; Jakobsson, H.E.; Andersson, A.F.; Björkstén, B.; Engstrand, L.; Jenmalm, M.C. Low diversity of the gut microbiota in infants with atopic eczema. J. Allergy Clin. Immunol. 2012, 129, 434–440.e2. [Google Scholar] [CrossRef]
- Hong, P.-Y.; Lee, B.W.; Aw, M.; Shek, L.P.C.; Yap, G.C.; Chua, K.Y.; Liu, W.-T. Comparative Analysis of Fecal Microbiota in Infants with and without Eczema. PLoS ONE 2010, 5, e9964. [Google Scholar] [CrossRef]
- Ismail, I.H.; Oppedisano, F.; Joseph, S.J.; Boyle, R.J.; Licciardi, P.V.; Robins-Browne, R.M.; Tang, M.L. Reduced gut microbial diversity in early life is associated with later development of eczema but not atopy in high-risk infants. Pediatr. Allergy Immunol. 2012, 23, 674–681. [Google Scholar] [CrossRef]
- Laursen, M.F.; Zachariassen, G.; Bahl, M.I.; Bergström, A.; Høst, A.; Michaelsen, K.F.; Licht, T.R. Having older siblings is associated with gut microbiota development during early childhood. BMC Microbiol. 2015, 15, 154. [Google Scholar] [CrossRef]
- Lee, E.; Lee, S.-Y.; Kang, M.-J.; Kim, K.; Won, S.; Kim, B.-J.; Choi, K.Y.; Kim, B.-S.; Cho, H.-J.; Kim, Y.; et al. Clostridia in the gut and onset of atopic dermatitis via eosinophilic inflammation. Ann. Allergy Asthma Immunol. 2016, 117, 91–92.e91. [Google Scholar] [CrossRef] [PubMed]
- West, C.E.; Rydén, P.; Lundin, D.; Engstrand, L.; Tulic, M.K.; Prescott, S.L. Gut microbiome and innate immune response patterns in IgE-associated eczema. Clin. Exp. Allergy 2015, 45, 1419–1429. [Google Scholar] [CrossRef] [PubMed]
- Gore, C.; Munro, K.; Lay, C.; Bibiloni, R.; Morris, J.; Woodcock, A.; Custovic, A.; Tannock, G.W. Bifidobacterium pseudocatenulatum is associated with atopic eczema: A nested case-control study investigating the fecal microbiota of infants. J. Allergy Clin. Immunol. 2008, 121, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Penders, J.; Gerhold, K.; Stobberingh, E.E.; Thijs, C.; Zimmermann, K.; Lau, S.; Hamelmann, E. Establishment of the intestinal microbiota and its role for atopic dermatitis in early childhood. J. Allergy Clin. Immunol. 2013, 132, 601–607.e608. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Yoo, Y.; Hwang, J.; Na, Y.-C.; Kim, H.S. Faecalibacterium prausnitzii subspecies–level dysbiosis in the human gut microbiome underlying atopic dermatitis. J. Allergy Clin. Immunol. 2016, 137, 852–860. [Google Scholar] [CrossRef]
- Eichenfield, L.F.; Hanifin, J.M.; Beck, L.A.; Lemanske, R.F.; Sampson, H.A.; Weiss, S.T.; Leung, D.Y. Atopic Dermatitis and Asthma: Parallels in the Evolution of Treatment. Pediatrics 2003, 111, 608–616. [Google Scholar] [CrossRef]
- Eller, E.; Kjaer, H.F.; Høst, A.; Andersen, K.E.; Bindslev-Jensen, C. Food allergy and food sensitization in early childhood: Results from the DARC cohort. Allergy 2009, 64, 1023–1029. [Google Scholar] [CrossRef]
- Schäfer, T. The impact of allergy on atopic eczema from data from epidemiological studies. Curr. Opin. Allergy Clin. Immunol. 2008, 8, 418–422. [Google Scholar] [CrossRef]
- Fuiano, N.; Fusilli, S.; Incorvaia, C. House dust mite-related allergic diseases: Role of skin prick test, atopy patch test, and RAST in the diagnosis of different manifestations of allergy. Eur. J. Pediatr. 2010, 169, 819–824. [Google Scholar] [CrossRef]
- Pónyai, G.; Hidvégi, B.; Németh, I.; Sas, A.; Temesvári, E.; Kárpáti, S. Contact and aeroallergens in adulthood atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2008, 22, 1346–1355. [Google Scholar] [CrossRef]
- Hon, K.-L.E.; Leung, T.-F.; Ching, G.; Chow, C.-M.; Luk, V.; Ko, W.-S.F.; Ng, P.-C. Patterns of food and aeroallergen sensitization in childhood eczema. Acta Paediatr. 2008, 97, 1734–1737. [Google Scholar] [CrossRef] [PubMed]
- Kvenshagen, B.; Jacobsen, M.; Halvorsen, R. Atopic dermatitis in premature and term children. Arch. Dis. Child. 2009, 94, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.J.; Heine, R.G.; Hosking, C.S. The diagnostic value of skin prick testing in children with food allergy. Pediatr. Allergy Immunol. 2004, 15, 435–441. [Google Scholar] [CrossRef]
- Monnot, G.C.; Wegrecki, M.; Cheng, T.-Y.; Chen, Y.-L.; Sallee, B.N.; Chakravarthy, R.; Karantza, I.M.; Tin, S.Y.; Khaleel, A.E.; Monga, I.; et al. Staphylococcal phosphatidylglycerol antigens activate human T cells via CD1a. Nat. Immunol. 2023, 24, 110–122. [Google Scholar] [CrossRef]
- Caffarelli, C.; Cavagni, G.; Deriu, F.M.; Zanotti, P.; Atherton, D.J. Gastrointestinal symptoms in atopic eczema. Arch. Dis. Child. 1998, 78, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Bisgaard, H.; Li, N.; Bonnelykke, K.; Chawes, B.L.K.; Skov, T.; Paludan-Müller, G.; Stokholm, J.; Smith, B.; Krogfelt, K.A. Reduced diversity of the intestinal microbiota during infancy is associated with increased risk of allergic disease at school age. J. Allergy Clin. Immunol. 2011, 128, 646–652.e5. [Google Scholar] [CrossRef]
- Wang, M.; Karlsson, C.; Olsson, C.; Adlerberth, I.; Wold, A.E.; Strachan, D.P.; Martricardi, P.M.; Åberg, N.; Perkin, M.R.; Tripodi, S.; et al. Reduced diversity in the early fecal microbiota of infants with atopic eczema. J. Allergy Clin. Immunol. 2008, 121, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Li, J.; Wei, C.; Zhao, H.; Wang, T. mbDenoise: Microbiome data denoising using zero-inflated probabilistic principal components analysis. Genome Biol. 2022, 23, 94. [Google Scholar] [CrossRef]
- Jiang, R.; Li, W.V.; Li, J.J. mbImpute: An accurate and robust imputation method for microbiome data. Genome Biol. 2021, 22, 192. [Google Scholar] [CrossRef]
- Gupta, R.S.; Gao, B. Phylogenomic analyses of clostridia and identification of novel protein signatures that are specific to the genus Clostridium sensu stricto (cluster I). Int. J. Syst. Evol. Microbiol. 2009, 59, 285–294. [Google Scholar] [CrossRef]
- Cassir, N.; Benamar, S.; La Scola, B. Clostridium butyricum: From beneficial to a new emerging pathogen. Clin. Microbiol. Infect. 2016, 22, 37–45. [Google Scholar] [CrossRef]
- Wang, Y.; Song, J.; Zhai, Y.; Zhang, C.; Gerritsen, J.; Wang, H.; Chen, X.; Li, Y.; Zhao, B.; Zhao, B.; et al. Romboutsia sedimentorum sp. nov., isolated from an alkaline-saline lake sediment and emended description of the genus Romboutsia. Int. J. Syst. Evol. Microbiol. 2015, 65, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Salem, I.; Ramser, A.; Isham, N.; Ghannoum, M.A. The Gut Microbiome as a Major Regulator of the Gut-Skin Axis. Front. Microbiol. 2018, 9, 1459. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, C.A.; Monteleone, G.; McLaughlin, J.T.; Paus, R. The gut-skin axis in health and disease: A paradigm with therapeutic implications. Bioessays 2016, 38, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, Z.; Ma, H.; Ji, S.; Chen, Z.; Cui, Z.; Chen, J.; Tang, S. Dysbiosis and Implication of the Gut Microbiota in Diabetic Retinopathy. Front. Cell. Infect. Microbiol. 2021, 11, 646348. [Google Scholar] [CrossRef]
- Fang, Z.; Lu, W.; Zhao, J.; Zhang, H.; Qian, L.; Wang, Q.; Chen, W. Probiotics modulate the gut microbiota composition and immune responses in patients with atopic dermatitis: A pilot study. Eur. J. Nutr. 2020, 59, 2119–2130. [Google Scholar] [CrossRef]
- Ling, Z.; Li, Z.; Liu, X.; Cheng, Y.; Luo, Y.; Tong, X.; Yuan, L.; Wang, Y.; Sun, J.; Li, L.; et al. Altered Fecal Microbiota Composition Associated with Food Allergy in Infants. Appl. Environ. Microbiol. 2014, 80, 2546–2554. [Google Scholar] [CrossRef]
- Zhu, L.; Xu, F.; Wan, W.; Yu, B.; Tang, L.; Yang, Y.; Du, Y.; Chen, Z.; Xu, H. Gut microbial characteristics of adult patients with allergy rhinitis. Microb. Cell Factories 2020, 19, 171. [Google Scholar] [CrossRef]
- Zheng, H.; Liang, H.; Wang, Y.; Miao, M.; Shi, T.; Yang, F.; Liu, E.; Yuan, W.; Ji, Z.-S.; Li, D.-K. Altered Gut Microbiota Composition Associated with Eczema in Infants. PLoS ONE 2016, 11, e0166026. [Google Scholar] [CrossRef]
- Magnúsdóttir, S.; Ravcheev, D.; de Crécy-Lagard, V.; Thiele, I. Systematic genome assessment of B-vitamin biosynthesis suggests co-operation among gut microbes. Front. Genet. 2015, 6, 148. [Google Scholar] [CrossRef]
- Qian, B.; Shen, S.; Zhang, J.; Jing, P. Effects of Vitamin B6 Deficiency on the Composition and Functional Potential of T Cell Populations. J. Immunol. Res. 2017, 2017, 2197975. [Google Scholar] [CrossRef] [PubMed]
- Hanifin, J.M.; Baghoomian, W.; Grinich, E.; Leshem, Y.A.; Jacobson, M.; Simpson, E.L. The Eczema Area and Severity Index—A Practical Guide. Dermatitis® 2022, 33, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Tofte, S.; Graeber, M.; Cherill, R.; Omoto, M.; Thurston, M.; Hanifin, J. Eczema area and severity index (EASI): A new tool to evaluate atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 1998, 11, S197. [Google Scholar] [CrossRef]
- Hanifin, J.M.; Thurston, M.; Omoto, M.; Cherill, R.; Tofte, S.J.; Graeber, M.; The Easi Evaluator Group. The eczema area and severity index (EASI): Assessment of reliability in atopic dermatitis. Exp. Dermatol. 2001, 10, 11–18. [Google Scholar] [CrossRef]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackermann, G.; et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Kazutaka, K.; Misakwa, K.; Kei-ichi, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2′s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Oliver, F. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J. Biotechnol. 2017, 261, 169–176. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Liu, C.; Cui, Y.; Li, X.; Yao, M. microeco: An R package for data mining in microbial community ecology. FEMS Microbiol. Ecol. 2021, 97, fiaa255. [Google Scholar] [CrossRef]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbø, M.; Åse; Knight, R.T.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | AD (n = 104) | Normal (n = 130) | p Value 1 | |||
|---|---|---|---|---|---|---|
| n | n | |||||
| Age | 0.216 | |||||
| Sex 2 | Female | 50 | 41 | 0.839 | ||
| Male | 29 | 27 | ||||
| Allergy 2 | Yes | 31 | 10 | 0.0002 | ||
| No | 32 | 50 | ||||
| Not sure | 16 | 8 | ||||
| Food allergy 2 | Yes | 12 | 5 | 0.128 | ||
| No | 65 | 63 | ||||
| Not sure | 2 | 0 | ||||
| Non-food allergy 2 | Yes | 17 | 5 | 0.030 | ||
| No | 62 | 63 | ||||
| Diarrhea 2 | Yes | 17 | 18 | 0.611 | ||
| No | 62 | 50 | ||||
| Constipation 2 | Yes | 26 | 30 | 0.221 | ||
| No | 53 | 38 | ||||
| BMI 2 | 0.709 | |||||
| Overweight (BMI > 25) 2 | Yes | 21 | 19 | 0.983 | ||
| No | 52 | 44 | ||||
| Not sure | 6 | 5 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Hou, J.; Tsui, J.C.-C.; Wang, L.; Zhou, J.; Chan, U.K.; Lo, C.J.Y.; Siu, P.L.K.; Loo, S.K.F.; Tsui, S.K.W. Unique Gut Microbiome Signatures among Adult Patients with Moderate to Severe Atopic Dermatitis in Southern Chinese. Int. J. Mol. Sci. 2023, 24, 12856. https://doi.org/10.3390/ijms241612856
Wang Y, Hou J, Tsui JC-C, Wang L, Zhou J, Chan UK, Lo CJY, Siu PLK, Loo SKF, Tsui SKW. Unique Gut Microbiome Signatures among Adult Patients with Moderate to Severe Atopic Dermatitis in Southern Chinese. International Journal of Molecular Sciences. 2023; 24(16):12856. https://doi.org/10.3390/ijms241612856
Chicago/Turabian StyleWang, Yiwei, Jinpao Hou, Joseph Chi-Ching Tsui, Lin Wang, Junwei Zhou, Un Kei Chan, Claudia Jun Yi Lo, Pui Ling Kella Siu, Steven King Fan Loo, and Stephen Kwok Wing Tsui. 2023. "Unique Gut Microbiome Signatures among Adult Patients with Moderate to Severe Atopic Dermatitis in Southern Chinese" International Journal of Molecular Sciences 24, no. 16: 12856. https://doi.org/10.3390/ijms241612856