
Step-by-Step Double-Trouble OBAIRH and DMD Diagnosis in a One-Year-Old Boy

Abstract
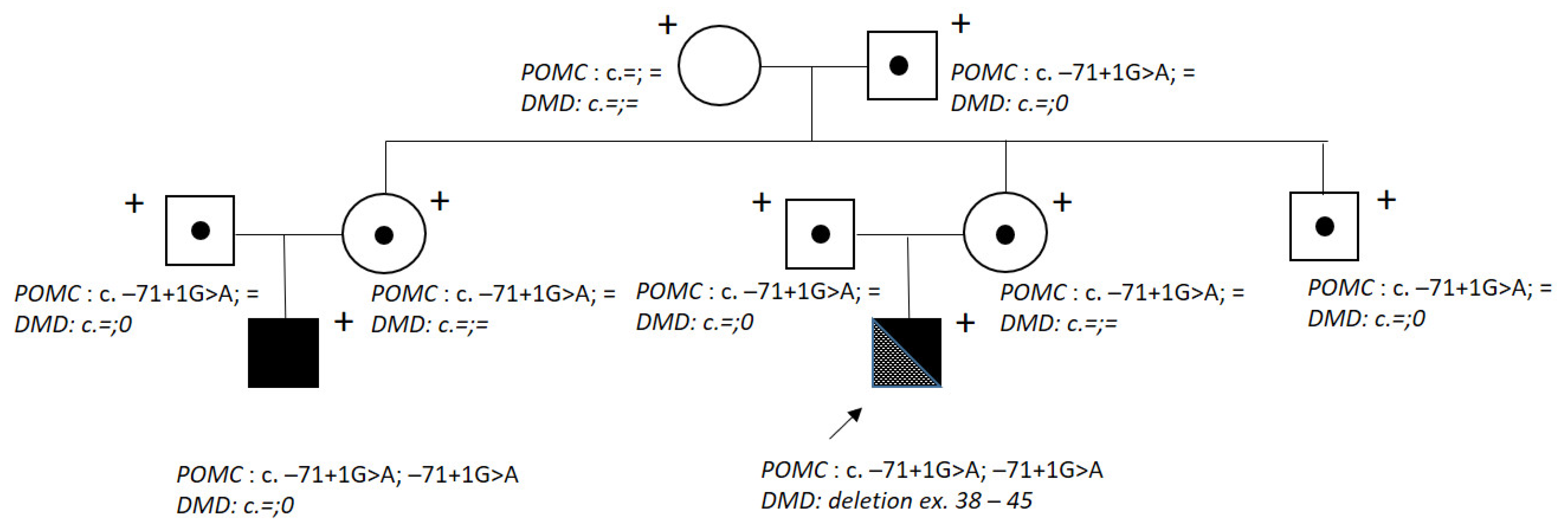
:1. Introduction
2. Results
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khudiakova, E.S. Components of Ethnic Identity of Tatars of Perm Territory (According to Spontaneous Texts). Compon. Ethn. Identity Tatars Perm Territ. 2020, 6, 190–205. [Google Scholar] [CrossRef]
- Rosstat—All-Russian Population Census. 2020. Available online: https://rosstat.gov.ru/vpn_popul (accessed on 23 June 2023).
- Weber, F.J.; Latshang, T.D.; Blum, M.R.; Kohler, M.; Wertli, M.M. Prognostic Factors, Disease Course, and Treatment Efficacy in Duchenne Muscular Dystrophy: A Systematic Review and Meta-Analysis. Muscle Nerve 2022, 66, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Zhang, J.; Shi, K.; Liu, Z. Drug Development Progress in Duchenne Muscular Dystrophy. Front. Pharmacol. 2022, 13, 950651. [Google Scholar] [CrossRef] [PubMed]
- Graves, L.E.; Khouri, J.M.; Kristidis, P.; Verge, C.F. Proopiomelanocortin Deficiency Diagnosed in Infancy in Two Boys and a Review of the Known Cases. J. Paediatr. Child Health 2021, 57, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Viakhireva, I.; Kalinchenko, N.; Vasilyev, E.; Chistousova, G.V.; Filatova, A.; Marakhonov, A.; Rubtsov, P.M.; Skoblov, M.; Tiulpakov, A. A Founder Mutation in the POMC 5′-UTR Causes Proopiomelanocortin Deficiency Through Splicing-Mediated Decrease of MRNA. J. Clin. Endocrinol. Metab. 2022, 107, e3654–e3660. [Google Scholar] [CrossRef] [PubMed]
- Sergeyeva, A.; Gordeuk, V.R.; Tokarev, Y.N.; Sokol, L.; Prchal, J.F.; Prchal, J.T. Congenital Polycythemia in Chuvashia. Blood 1997, 89, 2148–2154. [Google Scholar] [CrossRef] [PubMed]
- Bliznetz, E.A.; Tverskaya, S.M.; Zinchenko, R.A.; Abrukova, A.V.; Savaskina, E.N.; Nikulin, M.V.; Kirillov, A.G.; Ginter, E.K.; Polyakov, A.V. Genetic Analysis of Autosomal Recessive Osteopetrosis in Chuvashiya: The Unique Splice Site Mutation in TCIRG1 Gene Spread by the Founder Effect. Eur. J. Hum. Genet. 2009, 17, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Gordeuk, V.R.; Sergueeva, A.I.; Miasnikova, G.Y.; Okhotin, D.; Voloshin, Y.; Choyke, P.L.; Butman, J.A.; Jedlickova, K.; Prchal, J.T.; Polyakova, L.A. Congenital Disorder of Oxygen Sensing: Association of the Homozygous Chuvash Polycythemia VHL Mutation with Thrombosis and Vascular Abnormalities but Not Tumors. Blood 2004, 103, 3924–3932. [Google Scholar] [CrossRef] [PubMed]
- Gundorova, P.; Zinchenko, R.A.; Kuznetsova, I.A.; Bliznetz, E.A.; Stepanova, A.A.; Polyakov, A.V. Molecular-Genetic Causes for the High Frequency of Phenylketonuria in the Population from the North Caucasus. PLoS ONE 2018, 13, e0201489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galeeva, N.M.; Voevoda, M.I.; Spiridonova, M.G.; Stepanov, V.A.; Poliakov, A.V. Population frequency and age of c.806C > T mutation in CYB5R3 gene as cause of recessive congenital methemoglobinemia in Yakutia. Genetika 2013, 49, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Zinchenko, R.A.; Ginter, E.K.; Marakhonov, A.V.; Petrova, N.V.; Kadyshev, V.V.; Vasilyeva, T.P.; Alexandrova, O.U.; Polyakov, A.V.; Kutsev, S.I. Epidemiology of Rare Hereditary Diseases in the European Part of Russia: Point and Cumulative Prevalence. Front. Genet. 2021, 12, 678957. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne Muscular Dystrophy. Nat. Rev. Dis. Prim. 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Guiraud, S.; Chen, H.; Burns, D.T.; Davies, K.E. Advances in Genetic Therapeutic Strategies for Duchenne Muscular Dystrophy. Exp. Physiol. 2015, 100, 1458–1467. [Google Scholar] [CrossRef] [Green Version]
- Donkervoort, S.; Schindler, A.; Tesi-Rocha, C.; Schreiber, A.; Leach, M.E.; Dastgir, J.; Hu, Y.; Mankodi, A.; Wagner, K.R.; Friedman, N.R.; et al. ‘Double Trouble’: Diagnostic Challenges in Duchenne Muscular Dystrophy in Patients with an Additional Hereditary Skeletal Dysplasia. Neuromuscul. Disord. 2013, 23, 955–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vondracek, P.; Hermanova, M.; Sedlackova, J.; Fajkusova, L.; Stary, D.; Michenkova, A.; Gaillyova, R.; Seeman, P.; Mazanec, R. Charcot-Marie-Tooth Neuropathy Type 1A Combined with Duchenne Muscular Dystrophy. Eur. J. Neurol. 2007, 14, 1182–1185. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cui, F.; Chen, D.; Pu, C.; Chen, Z.; Yang, F.; Wu, H.; Huang, X. Coexistence of Peripheral Myelin Protein 22 and Dystrophin Mutations in a Chinese Boy: CMT Type 1A Comorbid with DMD. Muscle Nerve 2013, 48, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Rudnik-Schöneborn, S.; Weis, J.; Kress, W.; Häusler, M.; Zerres, K. Becker’s Muscular Dystrophy Aggravating Facioscapulohumeral Muscular Dystrophy—Double Trouble as an Explanation for an Atypical Phenotype. Neuromuscul. Disord. 2008, 18, 881–885. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Patient’s Age | Five Months | One Year | |
|---|---|---|---|
| Index (Unit) | Reference Values | ||
| AST (MU/L) | <40 | 374 MU/L | 228 MU/L |
| ALT (MU/L) | <40 | 424 MU/L | 197 MU/L |
| CK (U/L) | 24–190 | 14,333 U/L | 10,005 U/L |
| CK-MB (U/L) | 0–24 | 261 U/L | 253 U/L |
| LDH (U/L) | 225–937 | 1594 U/L | 1633 U/L |
| Cortisol levels (nmol/L) | 101.2–535.7 | <13.8 nmol/L | <13.8 nmol/L |
| Glucose levels (mmol/L) | 3.5–4.5 | 3.79 nmol/L | 4.1 nmol/L |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shchagina, O.; Kurilova, V.; Zinina, E.; Porubov, V.; Efishova, S.; Polyakov, A. Step-by-Step Double-Trouble OBAIRH and DMD Diagnosis in a One-Year-Old Boy. Int. J. Mol. Sci. 2023, 24, 12357. https://doi.org/10.3390/ijms241512357
Shchagina O, Kurilova V, Zinina E, Porubov V, Efishova S, Polyakov A. Step-by-Step Double-Trouble OBAIRH and DMD Diagnosis in a One-Year-Old Boy. International Journal of Molecular Sciences. 2023; 24(15):12357. https://doi.org/10.3390/ijms241512357
Chicago/Turabian StyleShchagina, Olga, Vera Kurilova, Elena Zinina, Vyacheslav Porubov, Svetlana Efishova, and Aleksander Polyakov. 2023. "Step-by-Step Double-Trouble OBAIRH and DMD Diagnosis in a One-Year-Old Boy" International Journal of Molecular Sciences 24, no. 15: 12357. https://doi.org/10.3390/ijms241512357