
Proteomic Profiling of Chemotherapy Responses in FOLFOX-Resistant Colorectal Cancer Cells
 , ,
, ,
Abstract
:1. Introduction
2. Results
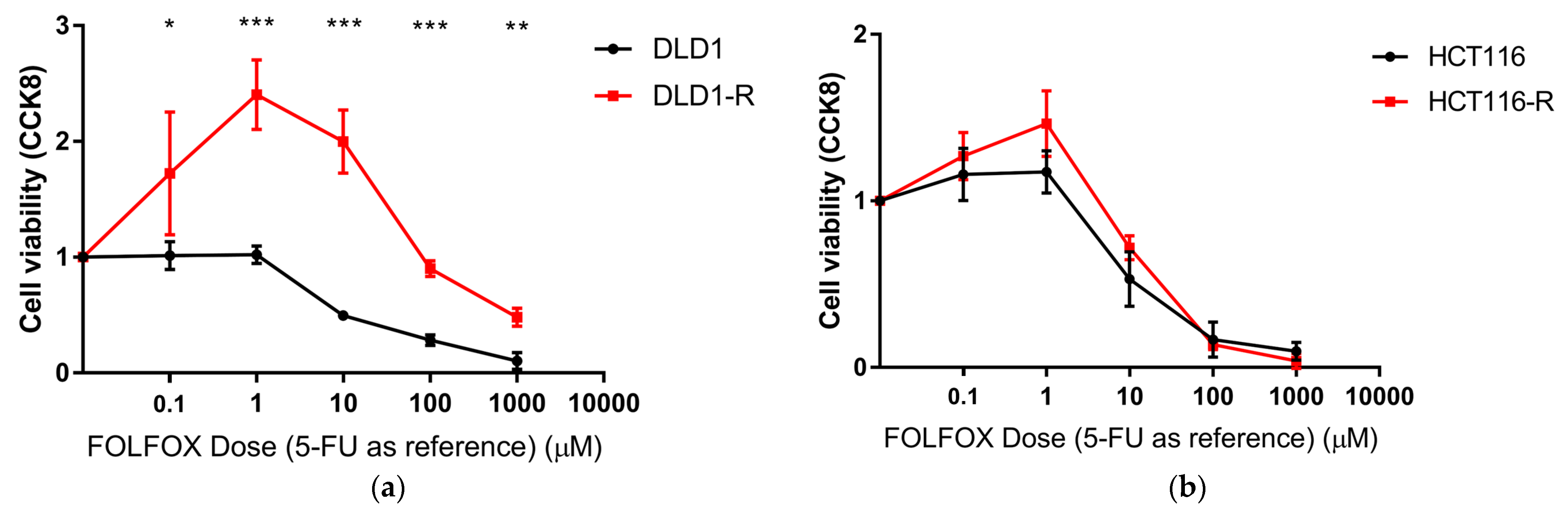
2.1. Verification of Chemoresistance of Cells
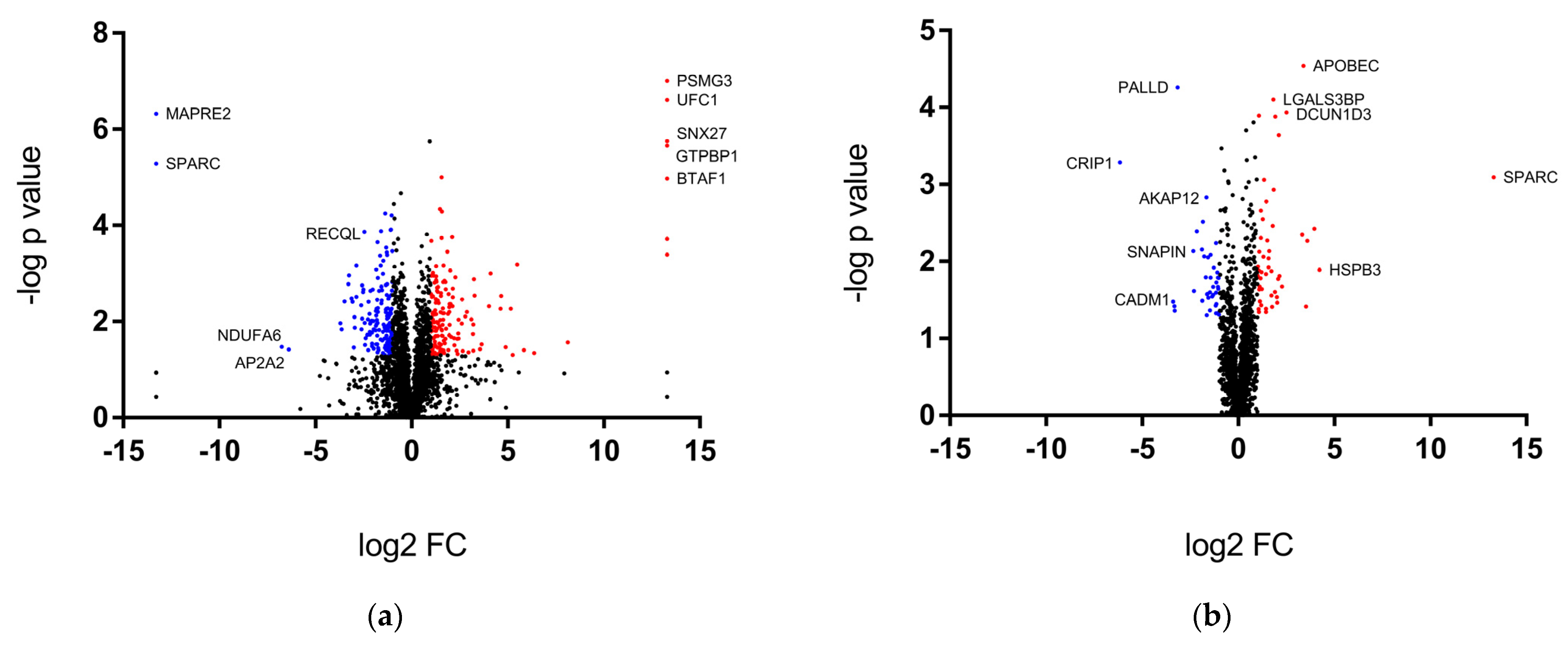
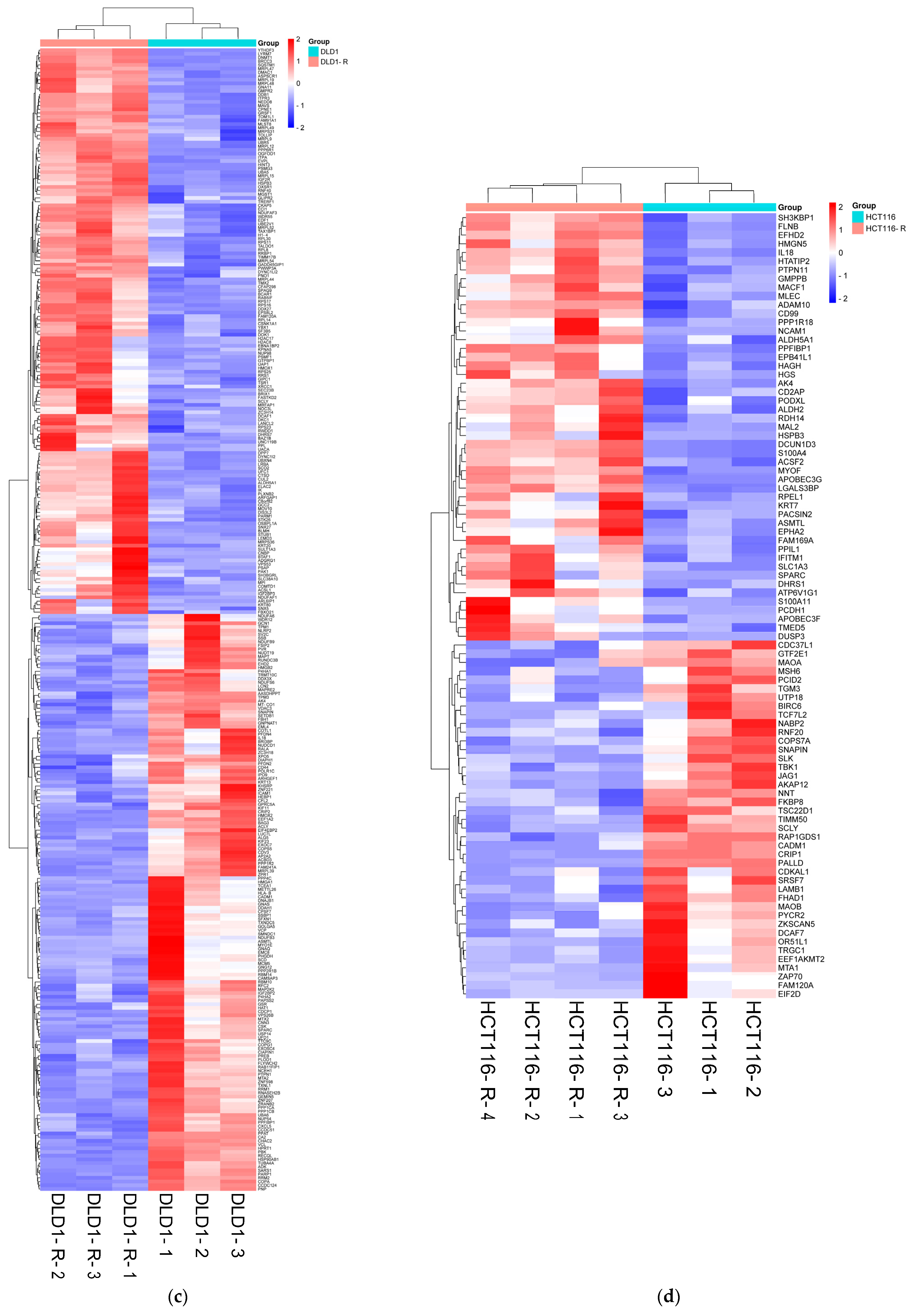
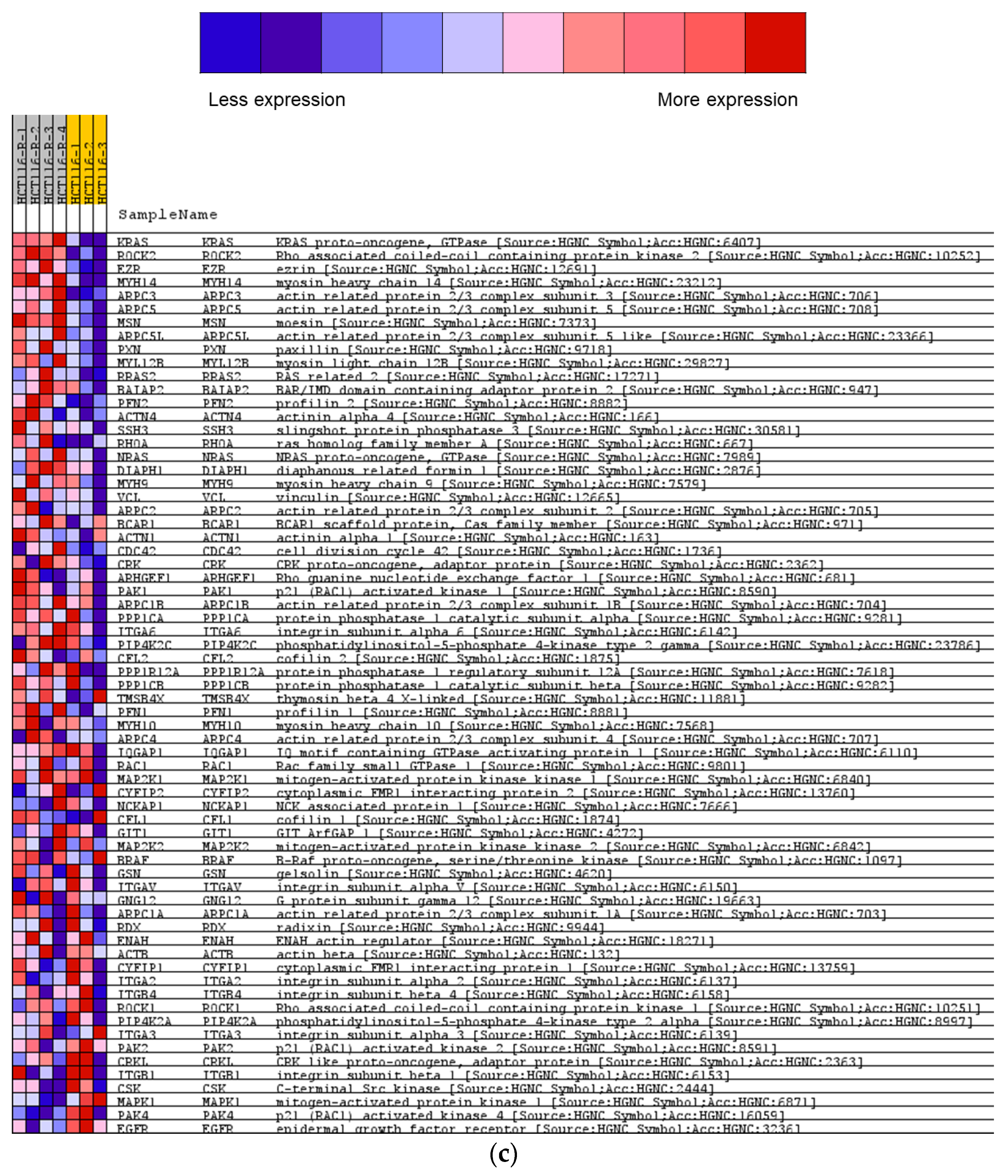
2.2. Overview of Proteomics Data
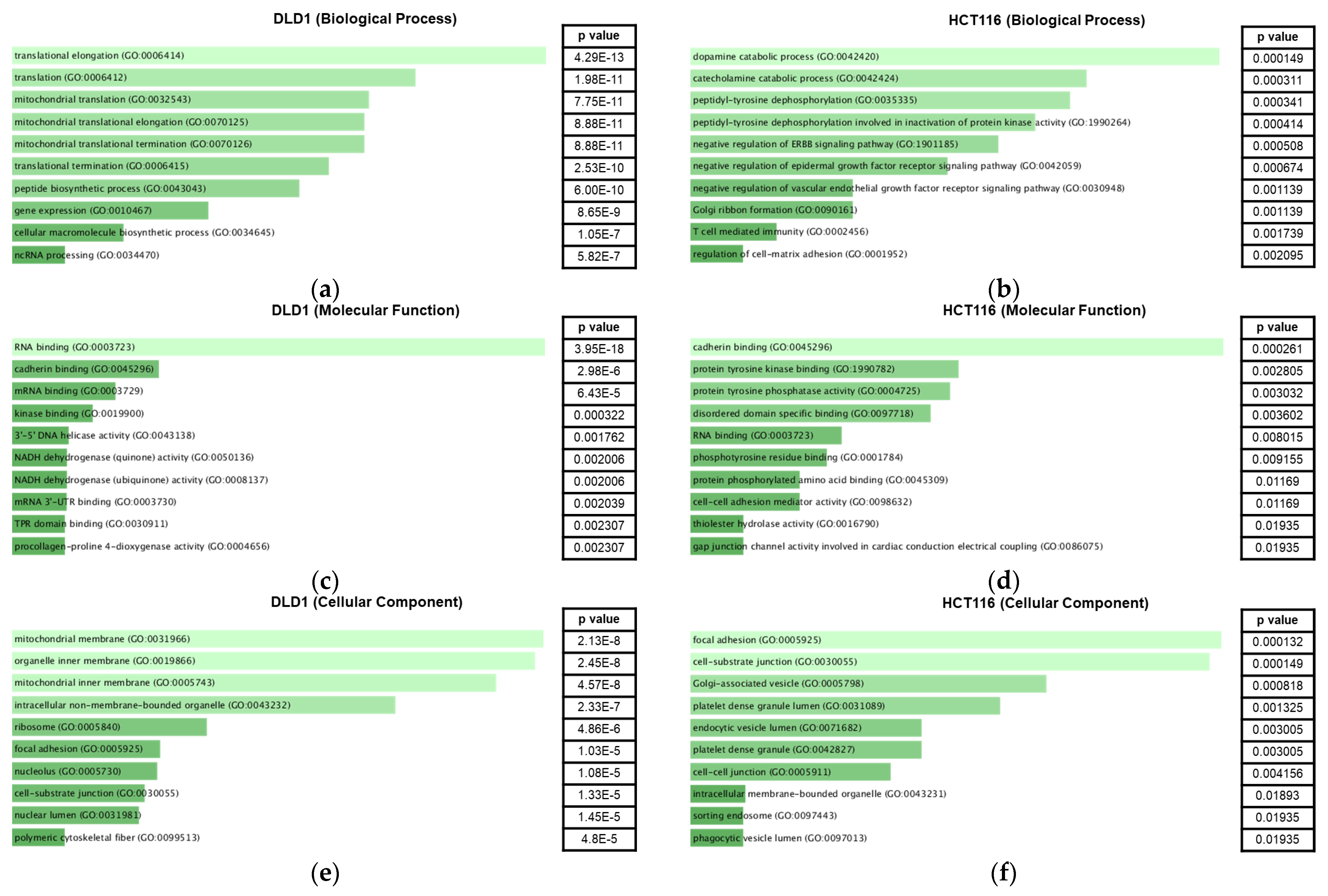
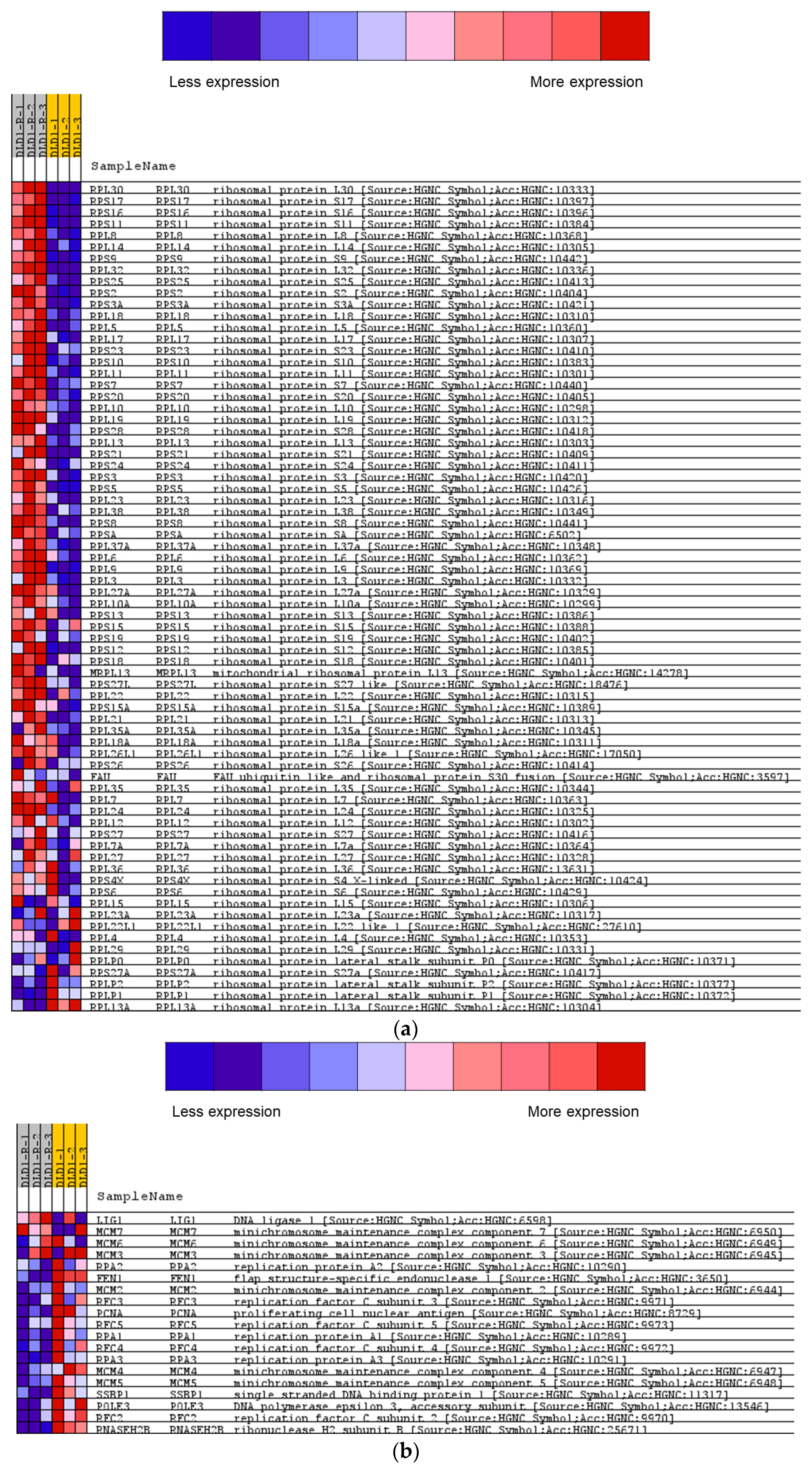
2.3. Gene Ontology Analysis of Significantly Enriched Proteins
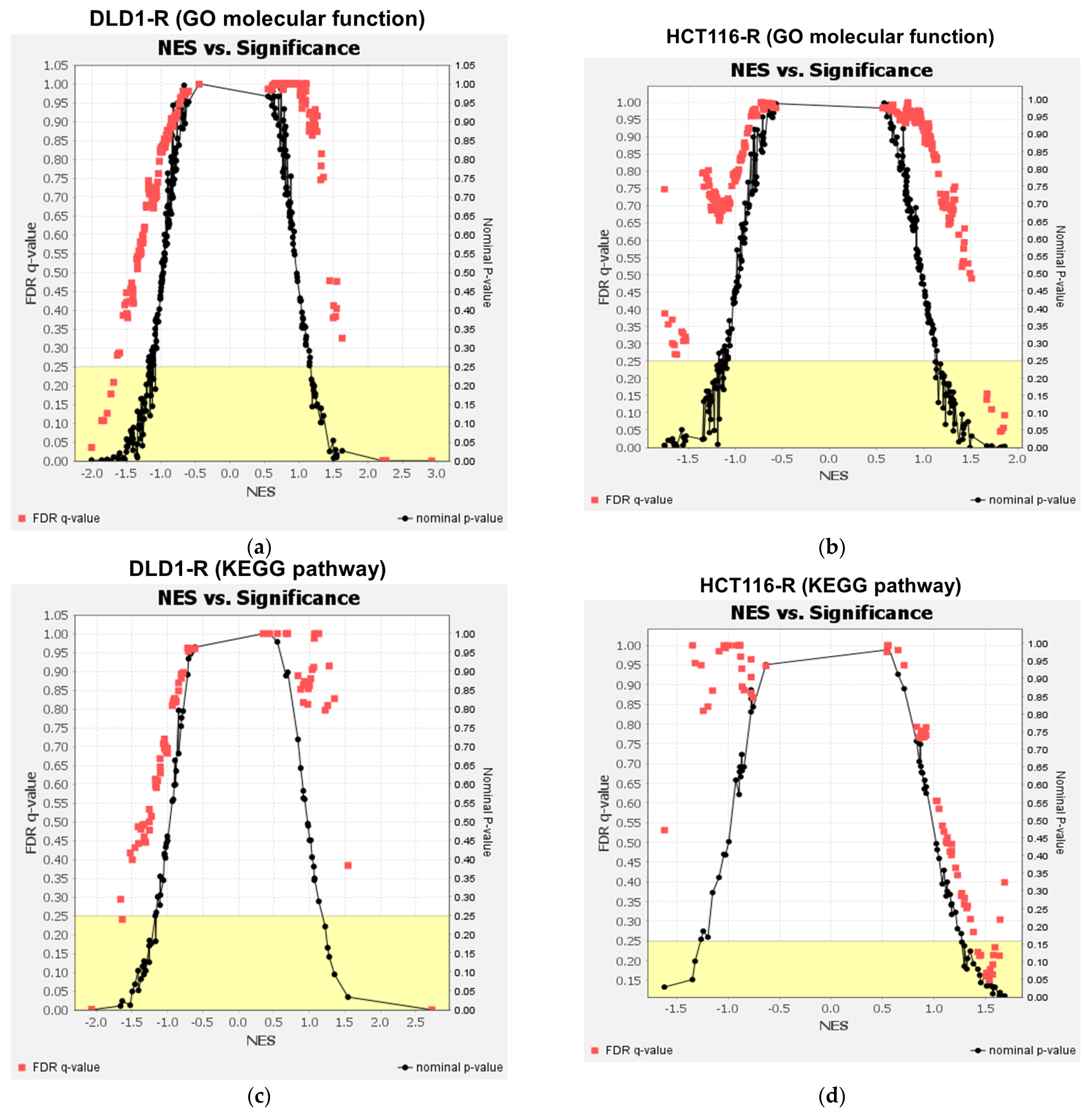
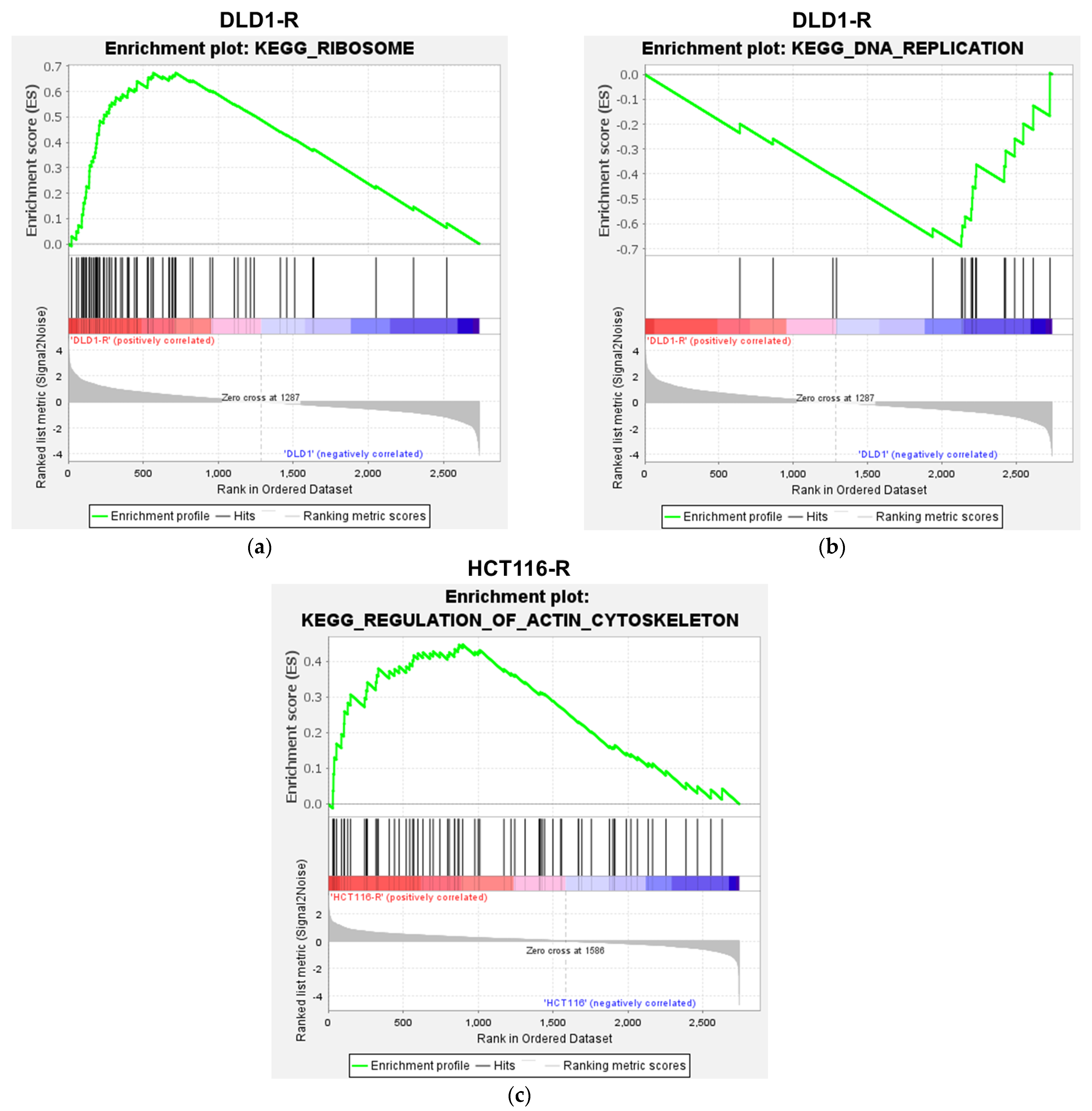
2.4. Gene Set Enrichment Analysis of the Proteomic Data
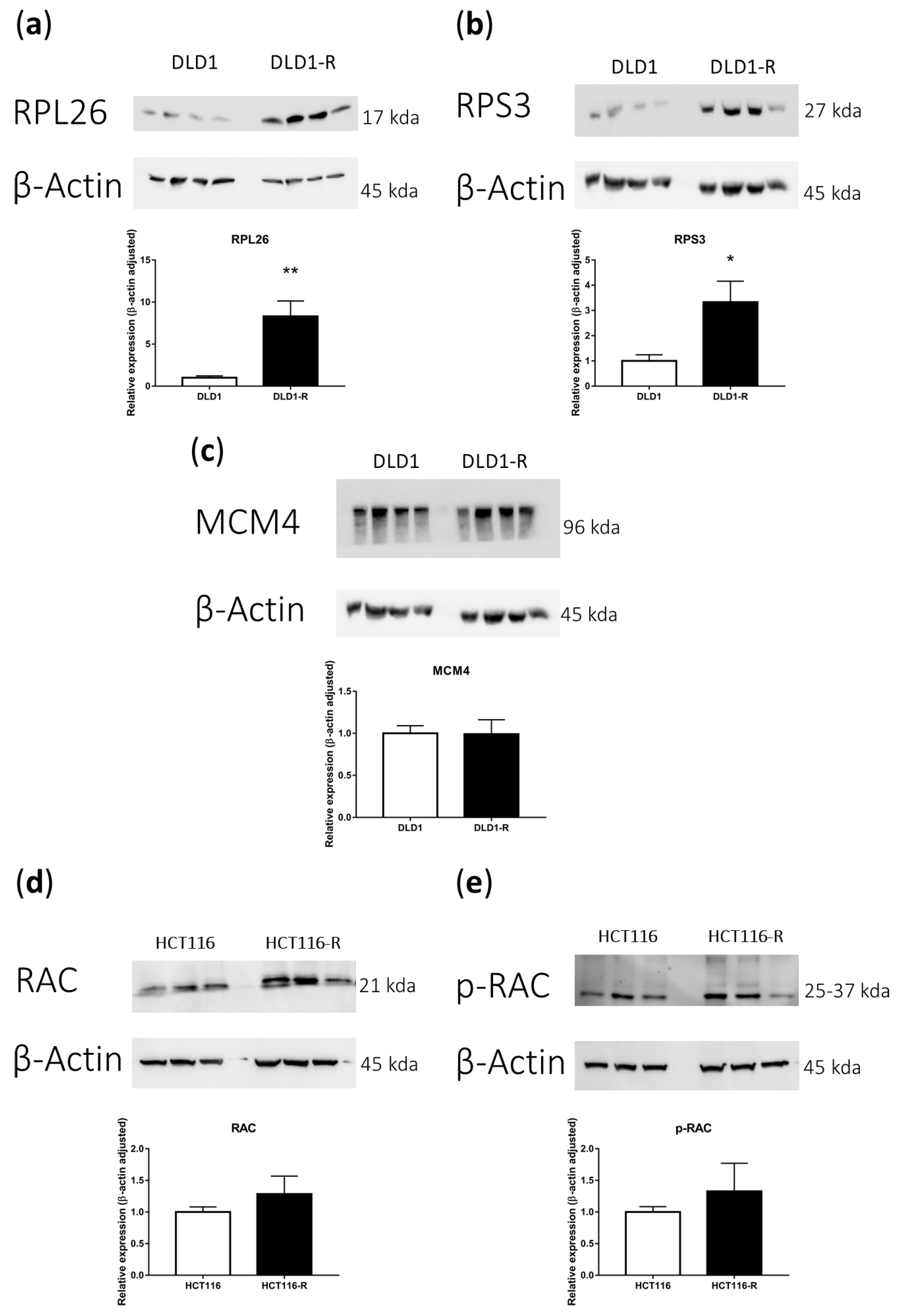
2.5. Western Blot Verification of Selected Pathways
3. Discussion
4. Materials and Methods
4.1. Cell Line and Culture Condition
4.2. Development of Chemoresistance
4.3. MS Sample Preparation for Proteomics Analysis
4.4. LC-MS/MS Analysis
4.5. MS Data Analysis
4.6. Western Blotting
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


References
- Morgan, E.; Arnold, M.; Gini, A.; Lorenzoni, V.; Cabasag, C.J.; Laversanne, M.; Vignat, J.; Ferlay, J.; Murphy, N.; Bray, F. Global burden of colorectal cancer in 2020 and 2040: Incidence and mortality estimates from GLOBOCAN. Gut 2023, 72, 338. [Google Scholar] [CrossRef] [PubMed]
- Aguiar Junior, S.; Oliveira, M.M.d.; Silva, D.R.M.e.; Mello, C.A.L.d.; Calsavara, V.F.; Curado, M.P. Survival of Patients with Colorectal Cancer in a Cancer Center. Arq. De Gastroenterol. 2020, 57, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, S.; Liu, Y.; Zhang, C.; Li, H.; Lai, B. Metastatic patterns and survival outcomes in patients with stage IV colon cancer: A population-based analysis. Cancer Med. 2020, 9, 361–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolpin, B.M.; Meyerhardt, J.A.; Mamon, H.J.; Mayer, R.J. Adjuvant treatment of colorectal cancer. CA A Cancer J. Clin. 2007, 57, 168–185. [Google Scholar] [CrossRef] [Green Version]
- Skarkova, V.; Kralova, V.; Vitovcova, B.; Rudolf, E. Selected Aspects of Chemoresistance Mechanisms in Colorectal Carcinoma-A Focus on Epithelial-to-Mesenchymal Transition, Autophagy, and Apoptosis. Cells 2019, 8, 234. [Google Scholar] [CrossRef] [Green Version]
- Gustavsson, B.; Carlsson, G.; Machover, D.; Petrelli, N.; Roth, A.; Schmoll, H.J.; Tveit, K.M.; Gibson, F. A review of the evolution of systemic chemotherapy in the management of colorectal cancer. Clin. Color. Cancer 2015, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Jensen, N.F.; Stenvang, J.; Beck, M.K.; Hanáková, B.; Belling, K.C.; Do, K.N.; Viuff, B.; Nygård, S.B.; Gupta, R.; Rasmussen, M.H.; et al. Establishment and characterization of models of chemotherapy resistance in colorectal cancer: Towards a predictive signature of chemoresistance. Mol. Oncol. 2015, 9, 1169–1185. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Shen, X.; Chen, G.; Du, J. Drug Resistance in Colorectal Cancer: From Mechanism to Clinic. Cancers 2022, 14, 2928. [Google Scholar] [CrossRef]
- Kwon, Y.W.; Jo, H.S.; Bae, S.; Seo, Y.; Song, P.; Song, M.; Yoon, J.H. Application of Proteomics in Cancer: Recent Trends and Approaches for Biomarkers Discovery. Front. Med. 2021, 8, 747333. [Google Scholar] [CrossRef]
- Cantor, D.I.; Cheruku, H.R.; Westacott, J.; Shin, J.S.; Mohamedali, A.; Ahn, S.B. Proteomic investigations into resistance in colorectal cancer. Expert Rev. Proteom. 2020, 17, 49–65. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirabelli, P.; Coppola, L.; Salvatore, M. Cancer Cell Lines Are Useful Model Systems for Medical Research. Cancers 2019, 11, 1098. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Mouradov, D.; Wang, X.; Jorissen, R.N.; Chambers, M.C.; Zimmerman, L.J.; Vasaikar, S.; Love, C.G.; Li, S.; Lowes, K.; et al. Colorectal Cancer Cell Line Proteomes Are Representative of Primary Tumors and Predict Drug Sensitivity. Gastroenterology 2017, 153, 1082–1095. [Google Scholar] [CrossRef] [PubMed]
- Lindhorst, P.H.; Hummon, A.B. Proteomics of Colorectal Cancer: Tumors, Organoids, and Cell Cultures-A Minireview. Front. Mol. Biosci. 2020, 7, 604492. [Google Scholar] [CrossRef] [PubMed]
- Elhamamsy, A.R.; Metge, B.J.; Alsheikh, H.A.; Shevde, L.A.; Samant, R.S. Ribosome Biogenesis: A Central Player in Cancer Metastasis and Therapeutic Resistance. Cancer Res. 2022, 82, 2344–2353. [Google Scholar] [CrossRef]
- Kobayashi, T.; Sasaki, Y.; Oshima, Y.; Yamamoto, H.; Mita, H.; Suzuki, H.; Toyota, M.; Tokino, T.; Itoh, F.; Imai, K.; et al. Activation of the ribosomal protein L13 gene in human gastrointestinal cancer. Int. J. Mol. Med. 2006, 18, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Nait Slimane, S.; Marcel, V.; Fenouil, T.; Catez, F.; Saurin, J.C.; Bouvet, P.; Diaz, J.J.; Mertani, H.C. Ribosome Biogenesis Alterations in Colorectal Cancer. Cells 2020, 9, 2361. [Google Scholar] [CrossRef]
- Xie, W.; Qu, L.; Meng, L.; Liu, C.; Wu, J.; Shou, C. PES1 regulates sensitivity of colorectal cancer cells to anticancer drugs. Biochem. Biophys Res. Commun. 2013, 431, 460–465. [Google Scholar] [CrossRef]
- Xie, W.; Feng, Q.; Su, Y.; Dong, B.; Wu, J.; Meng, L.; Qu, L.; Shou, C. Transcriptional regulation of PES1 expression by c-Jun in colon cancer. PLoS ONE 2012, 7, e42253. [Google Scholar] [CrossRef] [Green Version]
- Charitou, T.; Srihari, S.; Lynn, M.A.; Jarboui, M.A.; Fasterius, E.; Moldovan, M.; Shirasawa, S.; Tsunoda, T.; Ueffing, M.; Xie, J.; et al. Transcriptional and metabolic rewiring of colorectal cancer cells expressing the oncogenic KRAS(G13D) mutation. Br. J. Cancer 2019, 121, 37–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, A.; Deng, S.; Gopal, V.; Yap, K.C.; Halim, C.E.; Lye, M.L.; Ong, M.S.; Tan, T.Z.; Sethi, G.; Hooi, S.C.; et al. Cytoskeletal Dynamics in Epithelial-Mesenchymal Transition: Insights into Therapeutic Targets for Cancer Metastasis. Cancers 2021, 13, 1882. [Google Scholar] [CrossRef] [PubMed]
- Espina, C.; Céspedes, M.V.; García-Cabezas, M.A.; Gómez del Pulgar, M.T.; Boluda, A.; Oroz, L.G.; Benitah, S.A.; Cejas, P.; Nistal, M.; Mangues, R.; et al. A critical role for Rac1 in tumor progression of human colorectal adenocarcinoma cells. Am. J. Pathol. 2008, 172, 156–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotelevets, L.; Chastre, E. Rac1 Signaling: From Intestinal Homeostasis to Colorectal Cancer Metastasis. Cancers 2020, 12, 665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, K.; Rao, J.; Zhou, Z.-h.; Yao, X.-h.; Wu, F.; Yang, J.; Yang, L.; Zhang, X.; Cui, Y.-h.; Bian, X.-W.; et al. RAC1-GTP promotes epithelial-mesenchymal transition and invasion of colorectal cancer by activation of STAT3. Lab. Investig. 2018, 98, 989–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustelo, X.R.; Sauzeau, V.; Berenjeno, I.M. GTP-binding proteins of the Rho/Rac family: Regulation, effectors and functions in vivo. BioEssays News Rev. Mol. Cell. Dev. Biol. 2007, 29, 356–370. [Google Scholar] [CrossRef] [Green Version]
- Parri, M.; Chiarugi, P. Rac and Rho GTPases in cancer cell motility control. Cell Commun. Signal. 2010, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Matos, P.; Oliveira, C.; Velho, S.; Gonçalves, V.; da Costa, L.T.; Moyer, M.P.; Seruca, R.; Jordan, P. B-Raf(V600E) cooperates with alternative spliced Rac1b to sustain colorectal cancer cell survival. Gastroenterology 2008, 135, 899–906. [Google Scholar] [CrossRef]
- Alonso-Espinaco, V.; Cuatrecasas, M.; Alonso, V.; Escudero, P.; Marmol, M.; Horndler, C.; Ortego, J.; Gallego, R.; Codony-Servat, J.; Garcia-Albeniz, X.; et al. RAC1b overexpression correlates with poor prognosis in KRAS/BRAF WT metastatic colorectal cancer patients treated with first-line FOLFOX/XELOX chemotherapy. Eur. J. Cancer 2014, 50, 1973–1981. [Google Scholar] [CrossRef]
- Lipner, M.B.; Peng, X.L.; Jin, C.; Xu, Y.; Gao, Y.; East, M.P.; Rashid, N.U.; Moffitt, R.A.; Herrera Loeza, S.G.; Morrison, A.B.; et al. Irreversible JNK1-JUN inhibition by JNK-IN-8 sensitizes pancreatic cancer to 5-FU/FOLFOX chemotherapy. JCI Insight 2020, 5, e129905. [Google Scholar] [CrossRef] [Green Version]
- McDermott, M.; Eustace, A.J.; Busschots, S.; Breen, L.; Crown, J.; Clynes, M.; O’Donovan, N.; Stordal, B. In vitro Development of Chemotherapy and Targeted Therapy Drug-Resistant Cancer Cell Lines: A Practical Guide with Case Studies. Front. Oncol. 2014, 4, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam Khan, M.Z.; Tam, S.Y.; Azam, Z.; Law, H.K.W. Proteomic profiling of metabolic proteins as potential biomarkers of radioresponsiveness for colorectal cancer. J. Proteom. 2022, 262, 104600. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2021, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]
- Tam, S.Y.; Wu, V.W.C.; Law, H.K.W. Dynamics of oxygen level-driven regulators in modulating autophagy in colorectal cancer cells. Biochem. Biophys. Res. Commun. 2019, 517, 193–200. [Google Scholar] [CrossRef]
- Tam, S.Y.; Wu, V.W.C.; Law, H.K.W. JNK Pathway Mediates Low Oxygen Level Induced Epithelial–Mesenchymal Transition and Stemness Maintenance in Colorectal Cancer Cells. Cancers 2020, 12, 224. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | GO Molecular Function | Up-Regulated or Down-Regulated | NES | p Value |
|---|---|---|---|---|
| DLD1-R | Structural constituent of ribosome | Up-regulated | 2.93 | <0.001 |
| Structural molecule activity | Up-regulated | 2.26 | <0.001 | |
| rRNA binding | Up-regulated | 2.22 | <0.001 | |
| ATP-dependent activity acting on DNA | Down-regulated | −1.69 | 0.01 | |
| Electron transfer activity | Down-regulated | −1.72 | <0.001 | |
| Histone deacetylase binding | Down-regulated | −1.78 | 0.004 | |
| Glycosyltransferase activity | Down-regulated | −1.83 | <0.001 | |
| Disordered domain-specific binding | Down-regulated | −1.86 | 0.002 | |
| DNA helicase activity | Down-regulated | −2.01 | 0.002 | |
| HCT116-R | Oxidoreductase activity acting on the aldehyde or oxo group of donors | Up-regulated | 1.86 | 0.003 |
| Actin binding | Up-regulated | 1.85 | <0.001 | |
| Oxidoreductase activity acting on the aldehyde or oxo group of donors NAD or NADP as acceptor | Up-regulated | 1.83 | 0.002 | |
| Actin filament binding | Up-regulated | 1.81 | <0.001 | |
| Hydrolase activity acting on carbon nitrogen but not peptide bonds | Up-regulated | 1.72 | 0.005 | |
| Calcium ion binding | Up-regulated | 1.67 | 0.001 | |
| Ubiquitin-like protein binding | Up-regulated | 1.67 | 0.005 |
| Cell Type | GO Molecular Function | Up-Regulated or Down-Regulated | NES | p Value |
|---|---|---|---|---|
| DLD1-R | Ribosome | Up-regulated | 2.72 | <0.001 |
| Dilated cardiomyopathy | Down-regulated | −1.64 | 0.023 | |
| DNA replication | Down-regulated | −2.07 | <0.001 | |
| HCT116-R | Regulation of actin cytoskeleton | Up-regulated | 1.63 | 0.004 |
| Valine leucine and isoleucine degradation | Up-regulated | 1.59 | 0.028 | |
| Leukocyte transendothelial migration | Up-regulated | 1.58 | 0.027 | |
| Endocytosis | Up-regulated | 1.57 | 0.009 | |
| Propanoate metabolism | Up-regulated | 1.57 | 0.029 | |
| Vasopressin-regulated water reabsorption | Up-regulated | 1.54 | 0.042 | |
| Tight junction | Up-regulated | 1.54 | 0.035 | |
| Pyruvate metabolism | Up-regulated | 1.54 | 0.032 | |
| Lysine degradation | Up-regulated | 1.51 | 0.031 | |
| Fc-gamma-R-mediated phagocytosis | Up-regulated | 1.45 | 0.04 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tam, S.-Y.; Islam Khan, M.Z.; Chen, J.-Y.; Yip, J.H.-Y.; Yan, H.-Y.; Tam, T.-Y.; Law, H.K.-W. Proteomic Profiling of Chemotherapy Responses in FOLFOX-Resistant Colorectal Cancer Cells. Int. J. Mol. Sci. 2023, 24, 9899. https://doi.org/10.3390/ijms24129899
Tam S-Y, Islam Khan MZ, Chen J-Y, Yip JH-Y, Yan H-Y, Tam T-Y, Law HK-W. Proteomic Profiling of Chemotherapy Responses in FOLFOX-Resistant Colorectal Cancer Cells. International Journal of Molecular Sciences. 2023; 24(12):9899. https://doi.org/10.3390/ijms24129899
Chicago/Turabian StyleTam, Shing-Yau, Md Zahirul Islam Khan, Ju-Yu Chen, Jerica Hiu-Yui Yip, Hong-Yiu Yan, Tsz-Yan Tam, and Helen Ka-Wai Law. 2023. "Proteomic Profiling of Chemotherapy Responses in FOLFOX-Resistant Colorectal Cancer Cells" International Journal of Molecular Sciences 24, no. 12: 9899. https://doi.org/10.3390/ijms24129899