
Natural Compound Boldine Lessens Myotonic Dystrophy Type 1 Phenotypes in DM1 Drosophila Models, Patient-Derived Cell Lines, and HSALR Mice

 and
and 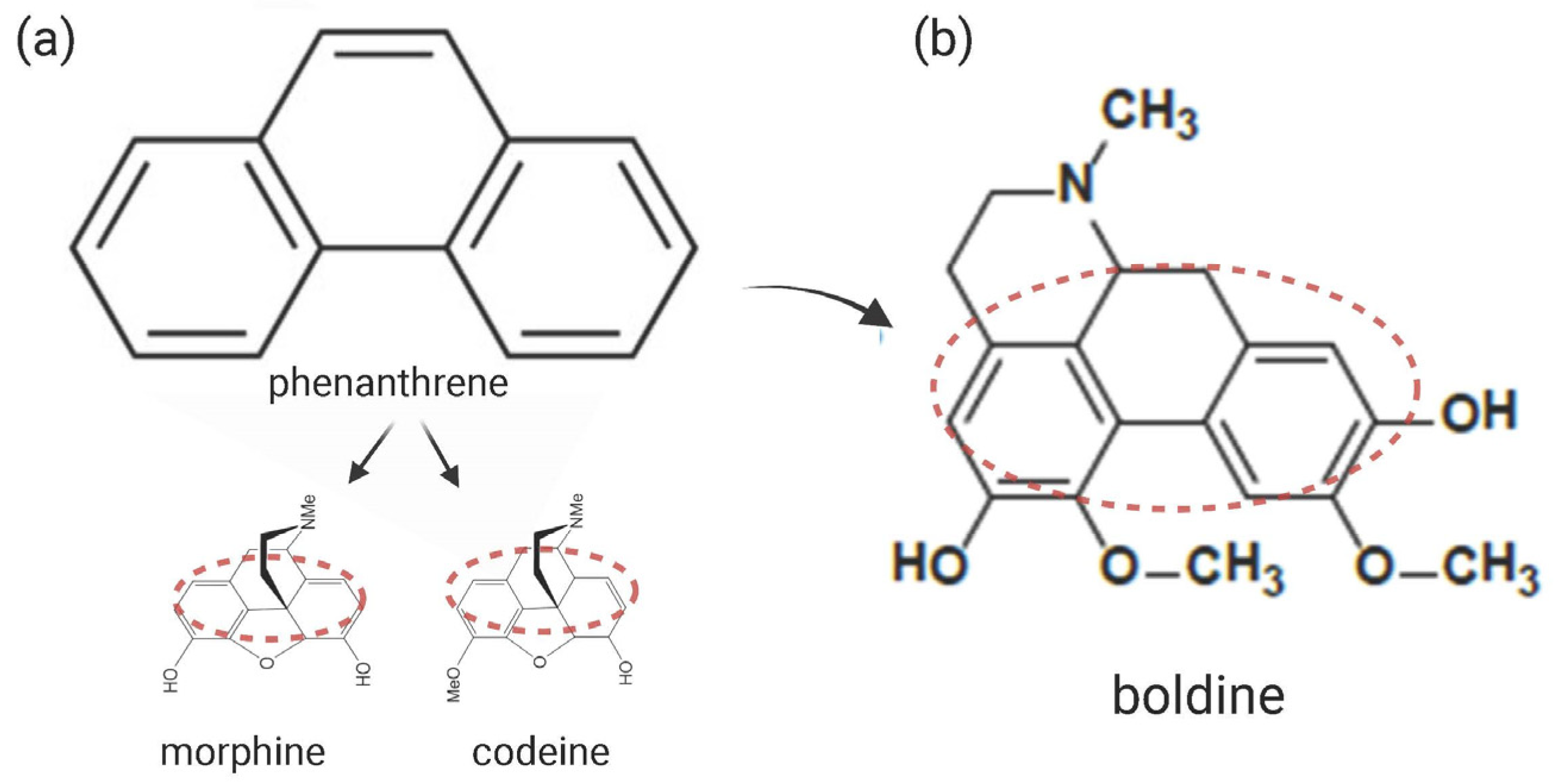{kind=link}
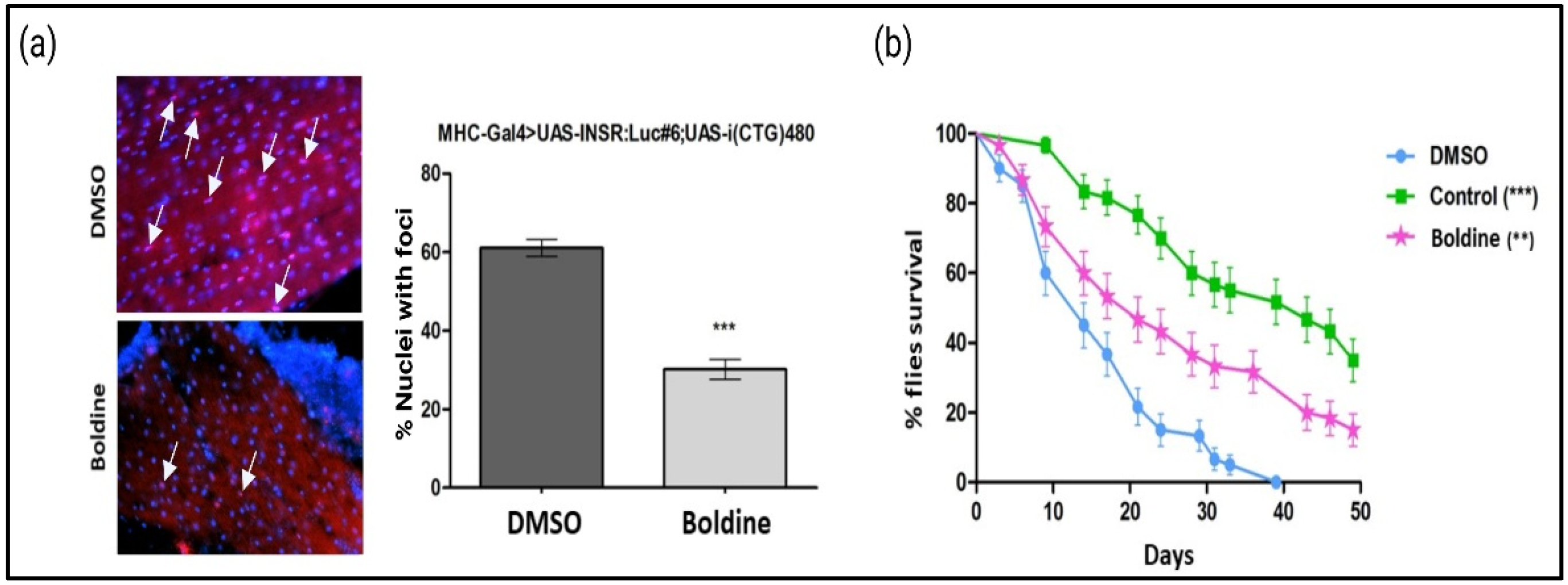{kind=link}
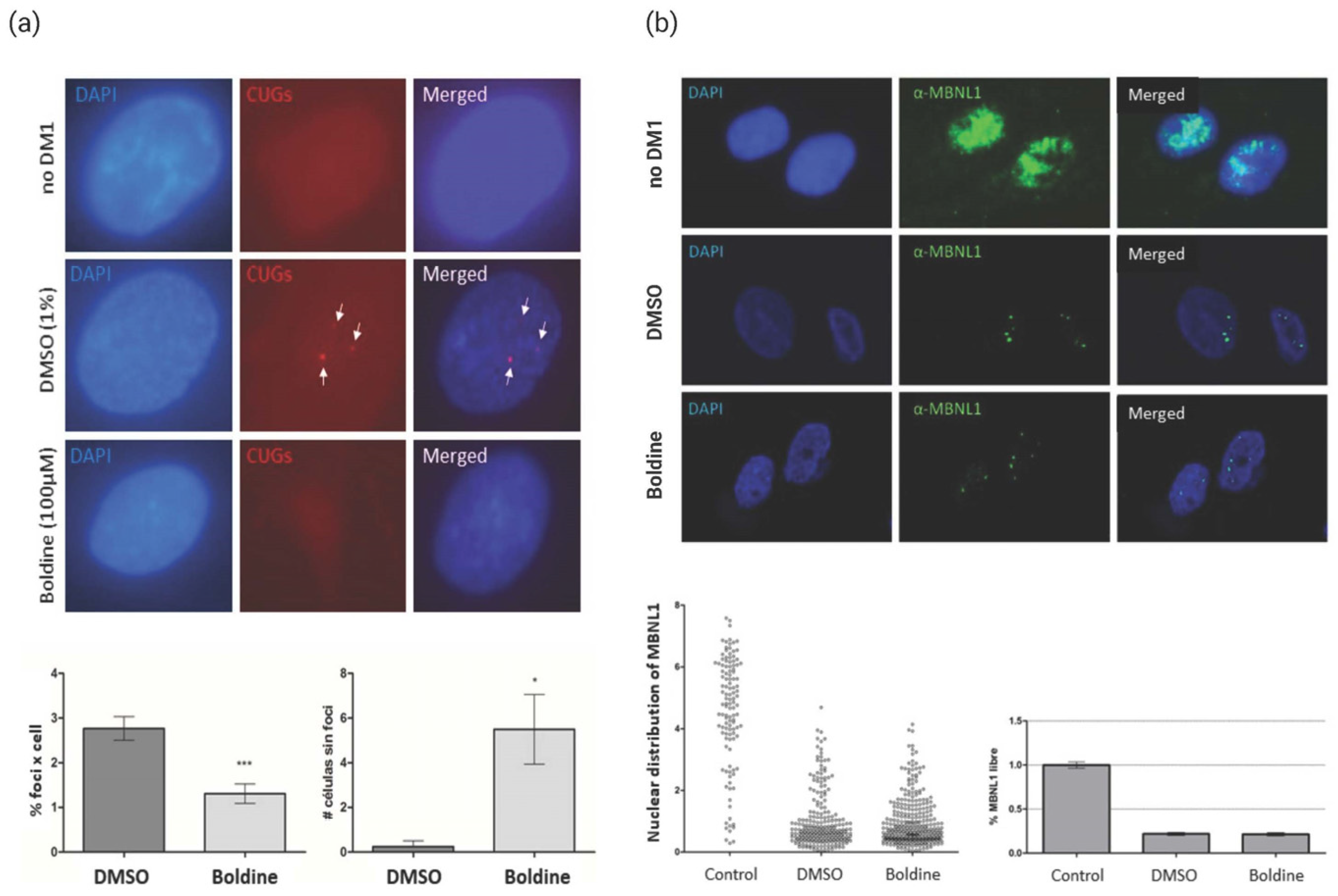{kind=link}
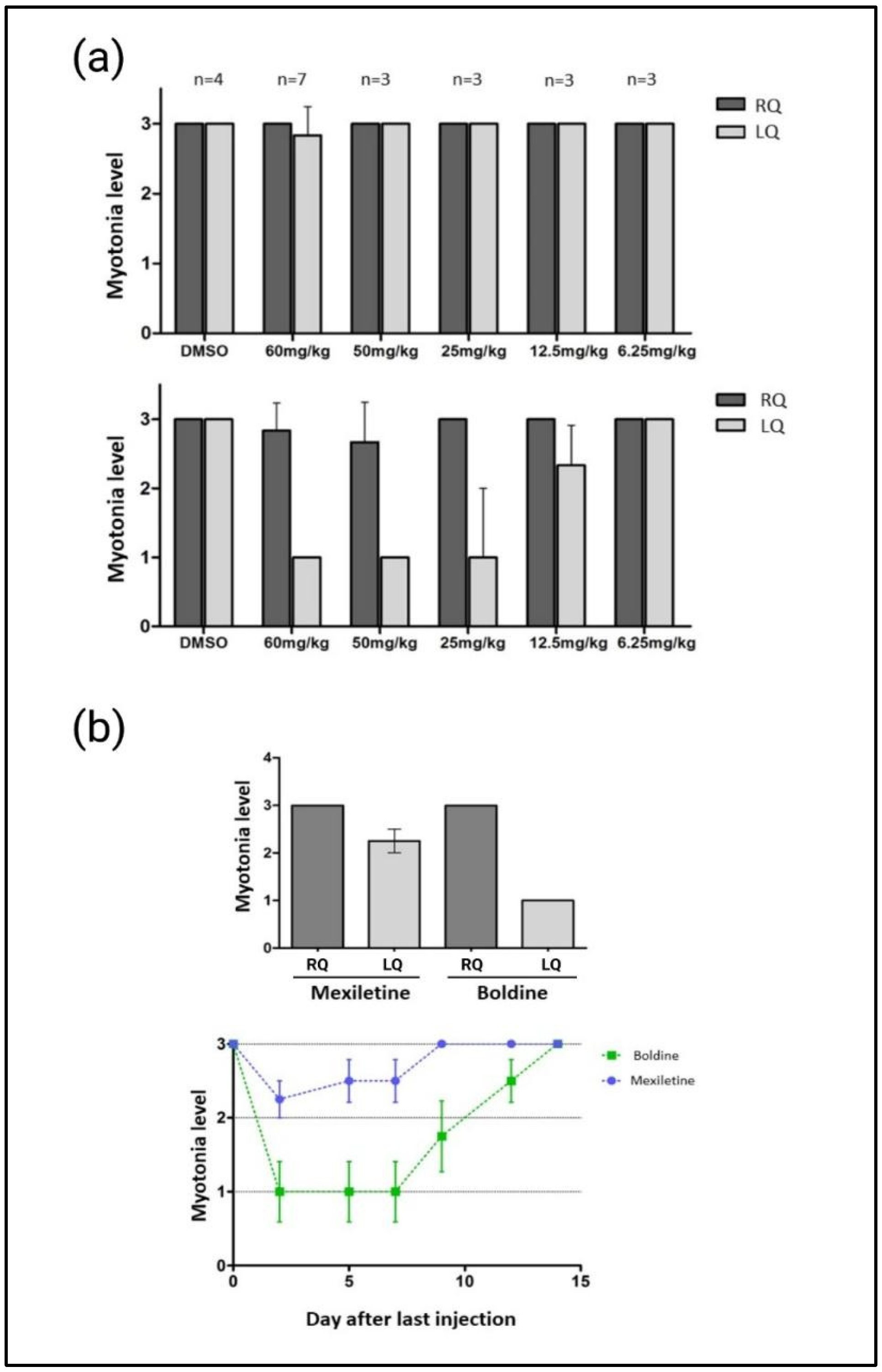{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Boldine Improves DM1 Phenotypes in Flies
2.2. Activity of Boldine in DM1 Human Cell Lines
2.3. Validation of the Boldine Activity in a Murine Model of DM1
2.4. Study of the Mechanism of Action of Boldine
3. Discussion
4. Materials and Methods
4.1. Drosophila Stocks
4.2. Drosophila-Based Experimental Approaches
- 8-point test: With the Biomek FXP pipetting robot, 5 μL of boldine was dispensed, with a final concentration of 12.5 μM, in a row of eight consecutive wells of a 96-well plate containing 250 µL Drosophila culture medium. Two replicated plates were prepared. The control (DMSO 0.25%) was dispensed in the first row of each plate. Each well was seeded with one L1 larva of genotype MHC-Gal4>UAS INSR:Luc#6; UAS-i(CTG)480, using a COPAS embryo dispenser. The plates were incubated at 25 °C for 2 weeks, after which they were kept frozen at −20 °C until the day of data reading. For the quantification of luciferase activity, each adult fly present in each of the wells was homogenized in 150 µL of 1X buffer from the Luciferase Assay Kit System (Promega; Madison, WI, USA). Of each homogenate, 50 µL was transferred to white 96-well plates (Nunc; Roskilde, Denmark), where 10 µL of the luciferase reagent was added per well using the Envision Multilabel Reader dispenser. Luciferase levels were measured using this same plate reader. The differences between the two groups were calculated using a two-tailed Student t-test and p = 0.05.
- Quantification of transcription levels for the luciferase reporter and UAS/Gal4 system: Sampling plates were prepared following the method described in the previous section. To study the effect of boldine on luciferase expression, 3 L1 larvae of the MHC Gal4;UAS-Luc genotype were seeded in each well, while for the effect of boldine on the system Gal4/UAS, 3 L1 larvae of the Mhc-Gal4>UAS-INSR:Luc#6 genotype were sown. The methodology described in [19] was followed to quantify the individuals and subsequently determine luciferase activity.
- Detection of ribonuclear foci in Drosophila: 500 µL of Drosophila culture medium was dispensed in glass vials of 5 mL capacity (Fisher; Tucson, AZ, USA) with the Biomek FXP pipetting robot. Boldine was dispensed at a final concentration of 12.5 µM in 3 vials, while DMSO was dispensed (0.25%) in another 3 vials. In each vial, 10 L1 larvae of the MHC Gal4>UAS-INSR:Luc#6;UAS-i(CTG)480 genotype were seeded using the COPAS embryo dispenser. The vials were incubated at 25 °C for 2 weeks, after which time they were frozen and kept at −20 °C until the day of data reading. The thoraxes of the adults to be analyzed were fixed overnight in paraformaldehyde at 4% in PBS at 4 °C. Subsequently, they were kept in a 30% sucrose solution in PBS for 2 days. After incubation time, the thoraxes were embedded in OCT, frozen in liquid nitrogen and maintained at a temperature of −80 °C until processing. For a further evaluation of the flies, 15 µm cross-sections were obtained with the Leica CM 1510S cryotome (Wetzlar, Germany). Fluorescence in situ hybridization (FISH) was then performed. The slides with the thorax sections were washed three times for 5 min each with 1× PBS. Fresh acetylation buffer was added to the slides (580 µL of 0.1 M triethalonamine and 125 µL of 0.25% (v/v) acetic anhydride in 50 mL of water). After 10 min of incubation, they were washed three times (each wash for 5 min) with 1× PBS and prehybridized for 30 min with the hybridization solution (10 mL deionized formamide, 12 µL 5 M NaCl, 400 µL 1 M Tris-HCl, pH = 8, and 20 µL 0.5 EDTA, pH = 8, 2 g Dextran sulfate, 400 µL Denhart’s 50× solution, 1 mL of herring sperm (10 mg/mL), and H2O, to a final volume of 20 mL). The Cy3-labelled probe 5′CAGCAGCAGCAGCAGCA3′-Cy3 (Merck, Darmstadt, Germany;) was used after heating it to 65 °C. Dissolved in hybridization buffer (1/100) was added to the slides for 5 min. The probe was allowed to hybridize at 37 °C overnight in a humid chamber, in darkness. The next day, the slides were washed with 2× SSC (2 washes of 15 min) and PBS (3 washes of 5 min duration), keeping the preparations at 32 °C. Finally, the slides were mounted with Vectastain and the images were taken using a Leica DM2500 optical microscope with a 40× objective. We analyzed the images of at least three different fields for each individual and a minimum of three individuals per group. The number of foci per cell was quantified using the Photoshop image analysis program (vCS4-11.0). Between groups differences were analyzed using a two-tailed t-student test and p = 0.05.
- Longevity test: 15 male newborns with the genotype MHC-Gal4>UAS-i(CTG)480 were placed in tubes containing DMSO (0.25%) or boldine (50 µM) dissolved in 3 mL of the nutrient medium Drosophila. As a positive control, 15 age-matched males from the reference strain were placed in tubes with DMSO (0.25%) dissolved in 3 mL of nutritional medium. Four replicates were performed per compound, with a total of 60 flies finally analyzed. The flies were transferred to a new tube with freshly prepared food and compound in each one every 2–3 days, and the number of dead individuals was quantified during the change. The results were represented by the Kaplan–Meier survival curves, which show the number of survivors according to the time in days. The curves of the compounds were compared using the Kaplan–Meier test with the GraphPad program.
4.3. Binding RNA Assays
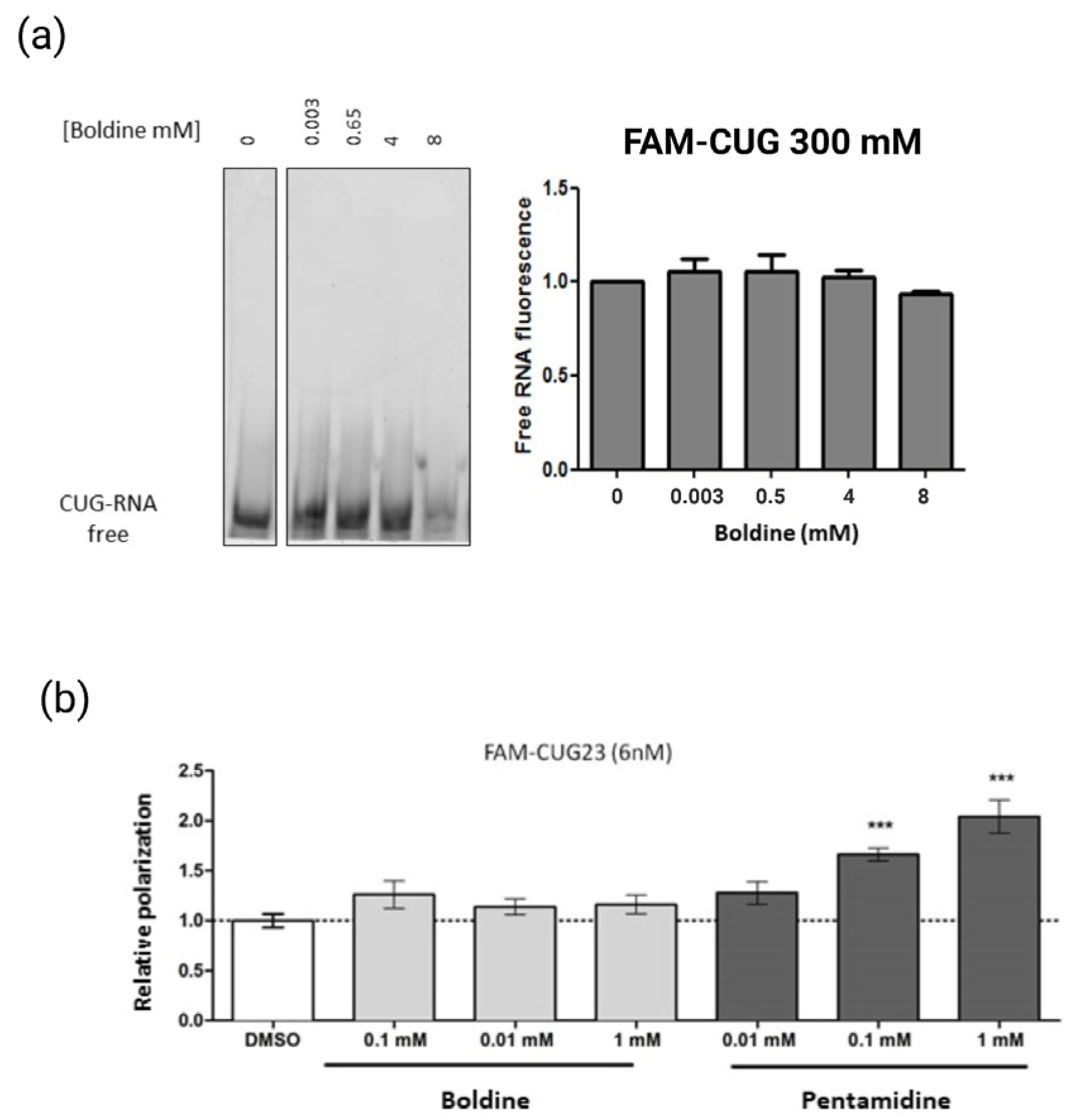
- Gel shift binding assay: To carry out the gel retardation assay, an aliquot of carboxyfluorescein (FAM)–CUG4 RNA (Metabion; Steinkirchen, Germany) was diluted to a concentration of 300 nM in binding buffer (25 mM Tris-HCL pH 7.5, 100 mM NaCl, 5 mM MgCl2, 50 µM, ZnCl2, and 10% glycerol) to a final volume of 10 µL. RNA was incubated with boldine (at concentrations of 0.03 mM, 0.5 mM, 4 mM and 8 mM) or DMSO (control) at 37 °C for 10 min. 5× loading buffer (20% sucrose, 100 mM Tris-Boric (TB) pH 8, 25 mM MgCl2, 0.1% bromophenol blue) and allowed to cool on ice. Subsequently, it was loaded on non-denaturing gels of 8% polyacrylamide without EDTA, previously subjected to a current of 100 V for 30 min. Electrophoresis was carried out in 1× TB supplemented with 5 mM MgCl2 and 50 µM ZnCl2, with a pH of 8.5 at a temperature of 4 °C in Mini-PROTEAN 3 cuvettes (Bio-Rad; Hercules, CA, USA) at 240 V for approximately 30 min. The images of the gels were taken with the Typhoon 9400 fluorescence scanner (GE Healthcare; Chicago, IL, USA), using emission filters BP 520 and SP 526. The intensity of the free RNA band was quantified using the Image J software (NIH). The EC50 value was calculated using linear non-regression analysis.
- Fluorescence polarization assay: FAM-CUG23 (synthetic RNA composed of 23 CUG repeats and conjugated in 5′ with the fluorophore carboxyfluorescein. Metabion) was heated at 70 °C for 10 min and allowed to cool slowly on the bench top. Next, the probe was incubated with boldine or 1% DMSO (control) in lysis buffer (25 mM Tris-HCl pH = 7.5, 100 mM NaCl, 5 mN MgCl2, 50 μM ZnCl2, 10% glycerol, and 0.05% Tween 20) for 20 min on ice and in the dark. To obtain a binding curve of boldine binding to FAM-CUG23 (6 nM), boldine was used at the concentrations of 0.1 mM, 0.01 nM, and 1 mM. As the positive control, the FAM-CUG23 probe (6 nM) was incubated with pentamidine (P0547-Sigma) at various concentrations (0.01 mM, 0.1 mM, 1 mM). All assays were carried out in black 96-well plates, with a total of four replicates per assay and at a final volume of 200 µL per well. The polarization measurements were obtained through an Envision plate reader (Envision 2104, Perkin Elmer; Waltham, MA, USA), using the excitation filter FP480 and emission filter FP535. The millipolarization (mP) values were calculated for each of the compounds following the formula (mP = 1000 × (S − G × P)/(S + G × P)), where S and P were the counts relative to the parallel (S) and perpendicular (P) planes of the arousal, and the G (grid factor) was an equipment-dependent factor. The results were normalized to the values of the negative control (DMSO). Between-group differences were analyzed using a two-tailed Student t-test and p = 0.05.
4.4. Mouse Strains
4.5. Mouse Experimental Approaches
- Treatment routes: In order to test the activity of small molecules in mice, and characterize some of their pharmacokinetic properties, the compounds were administered in different ways. For intramuscular administration, the compounds were injected into the quadricep muscles of the right and left hind legs, using 1 mL Hamilton syringes at a volume of 10 µL. For intraperitoneal administration, 50 µL of the compounds were injected into the right lower quadrant of the mouse abdomen, using a 24-gauge short-bevel needle and a 1.2 mL syringe. An oroesophageal tube was introduced into the animal’s esophagus (2–3 cm), and a 1.2 mL syringe was used for intragastric administration. The compounds were administered by trained staff of the SCIE animal facility (University of Valencia, Burjasot, Spain).
- Detection of ribonuclear foci in HSALR mice: The dissection of the quadricep muscles of both hind legs was carried out at 4 °C. Immediately after the animals were sacrificed, half of each quadricep muscle was placed in a cryotome cast, immersed in OCT, and stored at a temperature of −80 °C until further processing. During its processing, 6 µm sections were obtained with the Leica CM 1510S cryotome. Fluorescence in situ hybridization (FISH) was performed for 6 µm thick quadricep sections, fixed (73% ethanol, 25% acid acetic acid, and 2% formaldehyde) for 30 min at 4 °C and pre-hybridized for 10 min with the pre-hybridization buffer (30% formamide, 2× SSC) at room temperature. The hybridization was performed in a dark chamber at 37 °C for 2 h, using the buffer hybridization (30% formamide, 2× SSC, 0.02% bovine serum albumin, tRNA of yeast (1 mg/mL), and 2 mM sodium metavanadate) and 2 ng/µL of the probe marked Cy3-5′CAGCAGCAGCAGCAGCA3′-Cy3 (Sigma). After hybridization, the samples were washed with the pre-hybridization buffer for 30 min at 45 °C, followed by a second wash with SSC05X for 30 min at the same temperature. The samples were mounted with the Vectashield solution with DAPI. The number of foci per nucleus were quantified using a Leica DM2500 optical microscope with a 63× objective, whereby the number of foci present in three different fields was noted, in each of which at least 25 cells were counted. The number of foci obtained was divided by the number of the counted cells. The between-group differences were analyzed using a two-tailed Student t-test and p = 0.05.
- Evaluation of alternative splicing via semi-quantitative RT-PCR: The other half of each muscle quadricep was frozen in liquid nitrogen and stored at a temperature of −80 °C, after which RNA was extracted from the samples. Approximately 40 mg of the muscle was homogenized in 1 mL of TriReagent (Sigma). The homogenates were left for 5 min at room temperature before 200 µL of chloroform was added to each sample. The mixture was stirred and left for 5 min at room temperature. The tubes were then centrifuged for 15 min at 12,000× g and 4 °C. The aqueous phase was transferred to a new tube, where 500 µL of isopropanol and 3 µg of glycogen (GlicoBlue™ from Ambion; Austin, TX, USA) were added after 10 min at room temperature; the samples were centrifuged again for 10 min. The supernatant was removed, and the pellet was washed with 1 mL of 75% EtOH. After another centrifugation for 5 min at 7500× g and 4 °C, the supernatant was removed, and the pellet was dried at room temperature. Once dry, it was resuspended in RNase-free water, previously heated to 60 °C. The amount of RNA obtained was quantified by measuring its absorbance at 260 nm using a spectrophotometer (Eppendorf BioPhotometer; Hamburg, Germany). All RNA samples were diluted to a final concentration of 0.5 µg/µL and stored at −80 °C. The genomic DNA present in the extractions was removed by the digestion of 2 µg of RNA with DNase I (Fermentas; Waltham, MA, USA), at a total volume of 8 µL (RNA 0.5 µg/µL, 1 µL of 10× DNase buffer, 1.5 µL Dnase, and H2O, with a final volume of 8 µL). The digestion was performed at 37 °C for 30 min. DNase I was inactivated through the addition of 1 µL of 25 mM EDTA, followed by 10 min at 65 °C. For the mold for cDNA synthesis, 5 µL of the DNase I digestion was added to the 8 µL Mix 1 (1 µL 10 mM dNTPs, 1 µL hexamers (Invitrogen; Waltham, MA, USA), and 5 µL H2O). To denature the DNA, the tubes were left at 65 °C for 5 min. Immediately afterward, the tubes were placed on ice and 7 µL of Mix 2 (4 µL of buffer) was added (5× Superscript, 2 µL 0.1 M DTT, 1 µL RNase Inhibitor (Invitrogen)) to each. Finally, 1 µL of the enzyme Superscript TMII reverse transcriptase (2U, Invitrogen) was added. To carry out the retrotranscription reaction, the mixture was heated to 25 °C for 10 min in a thermocycler (Mastercycler Eppendorf), followed by 50 min at 42 °C and 15 min at 70 °C. For the controls, the same reactions were performed either without the RT enzyme in the reaction mix or without the RNA template. The cDNA obtained was stored at a temperature of −20 °C. For the detection of the Serca, Clcn1, and Gadph transcripts, 2 µL of the cDNA obtained in the previous section together with 0.25 µL of the GoTaq polymerase enzyme (Promega) was used as a template for the PCR reaction, to which 10 µL of Flexi buffer was added (GoTaq, 3 µL Mg2+, 1 µL dNTPS (10 mM), 1 µL forward primer (Serca e21F/Clce6F/Gadph F)) (10 mM), 1 µL of reverse primer (Serca e23R/Clc e8R/Gadph R)(10 mM), and DNase-free H2O, to a final volume of 50 µL. The following program was used for the amplification: 95 °C for 10 min, 25 or 27 cycles consisting of 30 s at 95 °C, 30 s at 58 °C, 1 min at 72 °C, and a last cycle of 5 min at 72 °C. The PCR products were resolved on 2.5% agarose gel. The expected amplicon in the case of the transcribed Serca was of two bands (an upper one of 300 bp and a lower one of 225 bp), while the expected amplicon for the Clcn1 transcripts was of a higher band of 450 bp and 350 bp in the case of the inclusion of exon 7, and 350 bp if exon 7 was excluded. The intensity of the bands was quantified with the Image J program (NIH), normalizing the exon 22 inclusion percentages of Serca and exclusion of exon 7 of the Clcn1 gene with the values of the Gadph gene. The between-group differences were analyzed using a two-tailed Student t-test and p = 0.05. The designed primers and amplification temperatures for the reactions are detailed in Table S1.
- Quantification of myotonia levels via electromyography (EMG): All animals were immobilized with anesthesia during electromyograms. The measurements were obtained with a 30 G concentric needle electrode using the TECA TD-20 MK II EMG/EP electromyograph. Ten measurements were carried out in the quadriceps of both legs of each individual. Myotonic discharges were classified according to the following scale: 0, no myotonia; 1, myotonic discharges in under 50% of insertions; 2, myotonic discharges in over 50% of insertions; and 3, myotonic discharges in practically all the insertions (>90%) [26].
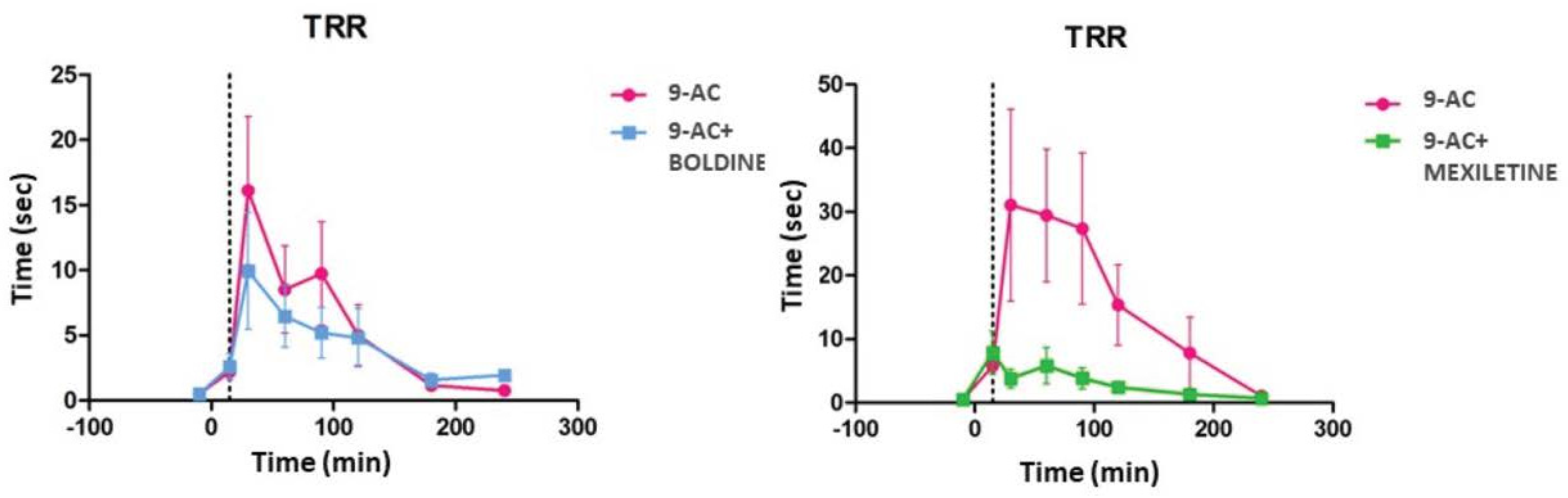
- Quantification of repositioning response time (TRR) in a mouse model of chemically induced myotonia in mouse: On each test day, we prepared a 2.4 g/L solution of anthracene-9-carboxylic acid (9-AC, Sigma) in water and 0.3% bicarbonate. The volume of the solution required to obtain a 60 mg/kg concentration of 9-AC, as previously published [33], was administered intraperitoneally to each CD-1 mouse included in this study. The compounds boldine and mexiletine (Sigma) were previously dissolved directly in saline and administered via an intragastric route at a concentration of 10 mg/kg. TRR was measured and quantified as the time taken for a mouse to roll over on all fours after being placed in a supine position. TRR was determined for each mouse at 10, 30, 60, 120, 180, and 240 min after the 9-AC administration, and calculated as the average of 10 measurements made at one-minute intervals. The between-group differences were calculated using a one-way ANOVA analysis of variance pathway, followed by an unpaired Student’s t-test.
4.6. Patient-Derived and Other Cell Lines
4.7. Cell Experimental Approaches
- Detection of ribonuclear foci: Fibroblast cells were seeded at a density of 10 cells/mL in 24-well plates on 12 mm circular coverslips with the growth medium. After 24 h, the fibroblast proliferation medium was replaced by means of transdifferentiation into myoblasts (DMEM supplemented with 2% of horse serum, penicillin (50 IU/mL), streptomycin (50 µg/mL), 100 µg/mL of Apotransferrin, 10 µg/mL insulin, and 2 µg/mL doxycycline), with a final volume of 500 µL per well. For the muscle cells to attach on the coverslip, they were left on the plate for 24 h. After this time, the compounds to be tested were added and left to act for another 24 h. Subsequently, the cells were fixed for 15 min with 4% PFA at room temperature room, washed 3 times with 1× PBS and stored in 70% EtOH at a temperature of 4 °C until the time of processing. The cells were treated with 100 µM boldine or 1% DMSO. Fluorescence in situ hybridization (FISH) was further performed. The cells were rehydrated with 1× PBS and pre-hybridized for 10 min at room temperature with the pre-hybridization buffer (40% formamide deionized, 2× SSC). Hybridization was performed in a dark chamber at 37 °C using the hybridization buffer (30% formamide, 2× SSC, 0.02% BSA, 1 mg/mL tRNA from yeast (Sigma), 2 mM sodium metavanadate, 1 μm/mL sperm DNA from denatured herring, 10% dextran sulfate, and 1 ng/µL Cy3-RNA probe 5′CAGCAGCAGCAGCAGCA3′-Cy3 (Sigma)). After hybridization, the cells were washed with the prehybridization buffer for 15 min at 45 °C (two washes) and once with 1× PBS at room temperature. The coverslips were mounted with the Vectashield solution with DAPI. The number of foci per cell nucleus were quantified in the Leica optical microscope using the 63× objective, quantifying the number of the foci present in three different fields, in each of which 25 cells were counted. The number of foci obtained was divided by the number of cells observed and analyzed using a two-tailed Student t-test and p = 0.05.
- Quantification of MBNL1 distribution via immunodetection: After the transdifferentiation process (see previous bullet), the compounds to be tested were added and left to act for 2 h. The cells were washed twice with 1× PBS and fixed for 15 min with 4% PFA at room temperature. Afterward, they were washed with 1× PBS for 5 min and stored in 1× PBS at 4 °C overnight. The cells were treated with 100 µM boldine or 1% DMSO. The whole process was carried out in a humid and dark chamber. A further immunohistochemistry approach involved the samples being washed with PBST (Triton × 0.3%) for 5 min (2 washes), after which 300 µL of blocking solution (1% donkey serum in PBST) was added for 30 min. After that time, the first antibody was added, i.e., 300 µL of anti-MBNL1 (Sigma) diluted in a ratio of 1:100 in the blocking solution. The antibody was left to incubate overnight at 4 °C. After three 5 min washes with PBST, the second antibody was added to each sample, which was 300 µL biotinylated anti-mouse (Fisher Scientific; Waltham, MA, USA) dissolved in the blocking solution in a ratio of 1:200, for 45 min at room temperature. Next, the samples were washed for 5 min with PBST (3 washes). For signal amplification, 300 µL was added to each sample of the Vectastain Elite ABC kit reagent AB (40 A:40 B:920 PBST ratio, prepared at least 30 min before use) and left to incubate for 45 min at room temperature. Subsequently, the samples were washed for 5 min with PBST (3 washes). Finally, 300 µL of Avidin Alexa Fluor 488 (Fisher Scientific) in a 1:200 PBST ratio was added to each sample and left to incubate for 45 min at room temperature. After 3 washes with PBS of a 5 min duration, the samples were mounted using Vestashield with DAPI. The images were taken by using an Olympus confocal microscope FluoView FV100 (SCIE), adjusting the blue channel conditions for DNA and blue channel green for MBNL1. The images were analyzed with the Image J program (NIH).
- Quantification of alternative splicing via semi-quantitative RT-PCR: The cells were seeded in 60 mm petri dishes at a density of 125,000 cells/plate, putting 3 mL of cells in each well. The fibroblasts were fixed for 24 h, and the fibroblast proliferation medium was changed by means of transdifferentiation into myoblasts. One day after the medium change, the compounds were added at the desired concentration and left to act for 24 h. Afterward, the cells were washed with 1× PBS and collected with a scraper. RNA was extracted from the cells collected in the previous section with the RNAGENTM Tissue Plus (Zygem; Solana Beach, CA, USA) kit, following the manufacturer’s instructions, using 8 µL of the RNA obtained as a template for reverse transcription. For the PCRs of cTNT, SERCA, and GAPDH, 1 µL of the cDNA obtained was used as template together with 10 µL Flexi Gotag buffer, 3 µL Mg2+, 1 µL dNTPs (10 mM), 1 µL forward primer (cTNT F/SERCA F/GADPH F) (10 mM), 1 µL of primer reverse (cTNT R/SERCA R/GADPH R) (10 mM), 0.25 µL GoTaq polymerase, and DNase-free H2O, to reach a final volume of 50 µL. The standard amplification conditions were used with an annealing temperature and a number of specific cycles for each gene (Supplementary Information). The PCR products were separated on a 2.5% agarose gel. The amplicon expected in all cases was two bands of different sizes: 244 bp and 201 bp in SERCA, and 132 bp and 110 bp in cTNT. The intensity of the bands was quantified with the Image J program (NIH) to obtain the percentages of inclusion of SERCA exon 22, and cTNR exon 5, normalized with the values of the GAPDH gene. The between-group differences were analyzed using a two-tailed Student t-test and p = 0.05. The designed primers and amplification temperatures for the reactions are detailed in Table S1.
- Quantification of MBNL1 and DMPK expression levels via qRT-PCR: After the compounds were administered and the total RNA extracted, the DMPK F/DMPK R/MBNL1 F/MBNL1 R primers (Supplementary Information) were mixed with the cDNA samples and the Master Mix of the Power SYBR Green PCR kit (Applied Biosystems; Waltham, MA, USA), following the manufacturer’s instructions. The amplification in real time and subsequent quantification were carried out using Step One Plus (Applied Biosystems), following the manufacturer’s protocol. The designed primers and amplification temperatures for the reactions are detailed in Table S1.
- Sodium ion channel electrophysiological study: Bovine chromaffin cells were isolated following a standard protocol [60], modified according to [61]. The cells were suspended in medium of Dulbecco MEM (DMEM), supplemented with 7.5% fetal bovine serum, 10 µM of arabinose cytosine, 10 µM fluorodeoxyuridine, 50 IU/mL penicillin, and 50 mg/mL streptomycin. For the ion current measurements, the cells were seeded in 24-well plates on 1 cm diameter circular coverslips, with a density of 105 cells per well. The inward currents into the cell through sodium channels (Ina) dependent on voltage were recorded using (whole cell) the patch clamp technique [62]. During the preparation of the cell membrane sealing process, the chamber contained a control Tyrode solution composed of 137 mM NaCl, 5.3 mM KCl, CaCl2, 2 mM, 1 mM MgCl2, and 10 mM HEPES pH 7.4. Once the membrane was broken, with the whole cell configuration of the patch clamp technique well established, the cell was rapidly perfused with an extracellular solution of a composition similar to the chamber solution, modified according to the current recorded: 0 mM Ca2+ for INa measurement. For recording the input ion currents, the cells were dialyzed with an intracellular solution formed of 100 mM CsCl, 14 mM EGTA, 20 mM TEA.CL, 10 NaCl mM, 5 mM Mg-ATP, 0.3 mM Na-GTP, and 20 mM HEPES/CsOH pH = 7.3. The current was recorded using an EP-10 amplifier (HEKA Electronic; Stuttgart, Germany) with 2–5 MΩ resistance electrodes. The data were acquired at a frequency between 5 and 10 kHz, and were later filtered to a frequency of 1 or 2 kHzData on weak currents (>25 pA), or resistances in a series > 20 Ω were recorded. Data analysis was carried out with HIEKA Elektronik and Igor Pro (Wavemetrics; Portland, OR, USA) PULSO programs.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thornton, C.A. Myotonic dystrophy. Neurol. Clin. 2014, 32, 705–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Q.; Zhang, Y.; He, J.; Huang, K. Global prevalence of myotonic dystrophy: An updated systematic review and meta-analysis. Neuroepidemiology 2022, 56, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.E.; Butterfield, R.J.; Mayne, K.; Newcomb, T.; Imburgia, C.; Dunn, D.; Duval, B.; Feldkamp, M.L.; Weiss, R.B. Population-Based Prevalence of Myotonic Dystrophy Type 1 Using Genetic Analysis of Statewide Blood Screening Program. Neurology 2021, 96, e1045–e1053. [Google Scholar] [CrossRef] [PubMed]
- Harper, P.S. Myotonic Dystrophy, 3rd ed.; Saunders, W.B., Ed.; OUP Oxford: Oxford, UK, 2001. [Google Scholar]
- Xia, G.; Ashizawa, T. Dynamic changes of nuclear RNA foci in proliferating DM1 cells. Histochem. Cell Biol. 2015, 143, 557–564. [Google Scholar] [CrossRef] [Green Version]
- Chau, A.; Kalsotra, A. Developmental insights into the pathology of and therapeutic strategies for DM1: Back to the basics. Dev. Dyn. 2015, 244, 377–390. [Google Scholar] [CrossRef]
- Cerro-Herreros, E.; Sabater-Arcis, M.; Fernandez-Costa, J.M.; Moreno, N.; Perez-Alonso, M.; Llamusi, B.; Artero, R. miR-23b and miR-218 silencing increase Muscleblind-like expression and alleviate myotonic dystrophy phenotypes in mammalian models. Nat. Commun. 2018, 9, 2482. [Google Scholar] [CrossRef]
- Pascual-Gilabert, M.; Artero, R.; López-Castel, A. The myotonic dystrophy type 1 drug development pipeline: 2022 edition. Drug Discov. Today 2023, 28, 103489. [Google Scholar] [CrossRef]
- Heatwole, C.; Luebbe, E.; Rosero, S.; Eichinger, K.; Martens, W.; Hilbert, J.; Dekdebrun, J.; Dilek, N.; Zizzi, C.; Johnson, N.; et al. Mexiletine in Myotonic Dystrophy Type 1. A Randomized, Double-Blind, Placebo-Controlled Trial. Neurology 2021, 96, e228–e240. [Google Scholar] [CrossRef]
- Bassez, G.; Audureau, E.; Hogrel, J.-Y.; Arrouasse, R.; Baghdoyan, S.; Bhugaloo, H.; Gourlay-Chu, M.-L.; Corvoisier, P.L.; Peschanski, M. Improved mobility with metformin in patients with myotonic dystrophy type 1: A randomized controlled trial. Brain 2018, 141, 2855–2865. [Google Scholar] [CrossRef] [Green Version]
- Nakamori, M.; Taylor, K.; Mochizuki, H.; Sobczak, K.; Takahashi, M.P. Oral administration of erythromycin decreases RNA toxicity in myotonic dystrophy. Ann. Clin. Transl. Neurol. 2016, 3, 42–54. [Google Scholar] [CrossRef]
- Horrigan, J.; Gomes, T.B.; Snape, M.; Nikolenko, N.; McMorn, A.; Evans, S.; Yaroshinsky, A.; Della Pasqua, O.; Oosterholt, S.; Lochmüller, H. A Phase 2 Study of AMO-02 (Tideglusib) in Congenital and Childhood-Onset Myotonic Dystrophy Type 1 (DM1). Pediatr. Neurol. 2020, 112, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Gilabert, M. Nutritional Management of Myotonic Dystrophy Type 1. Ph.D. Thesis, Universitat de Barcelona, Barcelona, Spain, 4 October 2022. [Google Scholar]
- Myogem Health Company. CORDIS EU Research Results. Available online: https://cordis.europa.eu/project/id/875615/reporting (accessed on 26 April 2023).
- O’Brien, P.; Carrasco-Pozo, C.; Speisky, H. Boldine and its antioxidant or health-promoting properties. Chem. Biol. Interact. 2006, 159, 1–17. [Google Scholar] [CrossRef]
- Speisky, H.; Cassels, B.K. Boldo and boldine: An emerging case of natural drug development. Pharmacol. Res. 1994, 29, 1–12. [Google Scholar] [CrossRef]
- Hernández-Salinas, R.; Vielma, A.Z.; Arismendi, M.N.; Boric, M.P.; Sáez, J.C.; Velarde, V. Boldine prevents renal alterations in diabetic rats. J. Diabetes Res. 2013, 2013, 593672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivorra, M.D.; Martinez, F.; Serrano, A.; D’Ocon, P. Different mechanism of relaxation induced by aporphine alkaloids in rat uterus. J. Pharm. Pharmacol. 1993, 45, 439–443. [Google Scholar] [CrossRef] [PubMed]
- García-Alcover, I.; Colonques-Bellmunt, J.; Garijo, R.; Tormo, J.R.; Artero, R.; Álvarez-Abril, M.C.; López Castel, A.; Pérez-Alonso, M. Development of a Drosophila melanogaster spliceosensor system for in vivo high-throughput screening in myotonic dystrophy type 1. Dis. Model. Mech. 2014, 7, 1297–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drug Bank. Available online: https://go.drugbank.com/categories/DBCAT000691 (accessed on 22 April 2023).
- Fuentes-Barros, G.; Castro-Saavedra, S.; Liberona, L.; Acevedo-Fuentes, W.; Tirapegui, C.; Mattar, C.; Cassels, B.K. Variation of the alkaloid content of Peumus boldus (boldo). Fitoterapia 2018, 127, 179–185. [Google Scholar] [CrossRef]
- Arandel, L.; Polay Espinoza, M.; Matloka, M.; Bazinet, A.; De Dea Diniz, D.; Naouar, N.; Rau, F.; Jollet, A.; Edom-Vovard, F.; Mamchaoui, K.; et al. Immortalized human myotonic dystrophy muscle cell lines to assess therapeutic compounds. Dis. Model. Mech. 2017, 10, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Nakamori, M.; Lueck, J.D.; Pouliquin, P.; Aoike, F.; Fujimura, H.; Dirksen, R.T.; Takahashi, M.P.; Dulhunty, A.F.; Sakoda, S. Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase in myotonic dystrophy type 1. Hum. Mol. Genet. 2005, 14, 2189–2200. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Miller, J.W.; Mankodi, A.; Kanadia, R.N.; Yuan, Y.; Moxley, R.T.; Swanson, M.S.; Thornton, C.A. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 2006, 15, 2087–2089. [Google Scholar] [CrossRef]
- Álvarez-Abril, M.C.; (Valentia BioPharma, Paterna, Spain); García-Alcover, I.; (Valentia BioPharma, Paterna, Spain); Colonques-Bellmunt, J.; (Valentia BioPharma, Paterna, Spain); Garijo, R.; (Valentia BioPharma, Paterna, Spain); Pérez-Alonso, M.; (University of Valencia, Burjasot, Spain); Artero, R.; (University of Valencia, Burjasot, Spain); López-Castel, A.; (University of Valencia, Burjasot, Spain). AS modulation in patient-derived cells. Personal communication, 2015. [Google Scholar]
- Mankodi, A.; Logigian, E.; Callahan, L.; McClain, C.; White, R.; Henderson, D.; Krym, M.; Thornton, C.A. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 2000, 289, 1769–1773. [Google Scholar] [CrossRef] [Green Version]
- Lueck, J.D.; Mankodi, A.; Swanson, M.S.; Thornton, C.A.; Dirksen, R.T. Muscle chloride channel dysfunction in two mouse models of myotonic dystrophy. J. Gen. Physiol. 2007, 129, 79–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez-Abril, M.C.; (Valentia BioPharma, Paterna, Spain); García-Alcover, I.; (Valentia BioPharma, Paterna, Spain); Colonques-Bellmunt, J.; (Valentia BioPharma, Paterna, Spain); Garijo, R.; (Valentia BioPharma, Paterna, Spain); Pérez-Alonso, M.; (University of Valencia, Burjasot, Spain); Artero, R.; (University of Valencia, Burjasot, Spain); López-Castel, A.; (University of Valencia, Burjasot, Spain). AS modulation in HSALR mouse model. Personal communication, 2015. [Google Scholar]
- Álvarez-Abril, M.C.; (Valentia BioPharma, Paterna, Spain); García-Alcover, I.; (Valentia BioPharma, Paterna, Spain); Colonques-Bellmunt, J.; (Valentia BioPharma, Paterna, Spain); Garijo, R.; (Valentia BioPharma, Paterna, Spain); Pérez-Alonso, M.; (University of Valencia, Burjasot, Spain); Artero, R.; (University of Valencia, Burjasot, Spain); López-Castel, A.; (University of Valencia, Burjasot, Spain). Myotonia quantification in HSALR mouse model after 5 days of treatment. Personal communication, 2015. [Google Scholar]
- Álvarez-Abril, M.C.; (Valentia BioPharma, Paterna, Spain); García-Alcover, I.; (Valentia BioPharma, Paterna, Spain); Colonques-Bellmunt, J.; (Valentia BioPharma, Paterna, Spain); Garijo, R.; (Valentia BioPharma, Paterna, Spain); Pérez-Alonso, M.; (University of Valencia, Burjasot, Spain); Artero, R.; (University of Valencia, Burjasot, Spain); López-Castel, A.; (University of Valencia, Burjasot, Spain). Myotonia quantification in HSALR mouse model after 21 days of treatment. Personal communication, 2015. [Google Scholar]
- Trip, J.; Drost, G.; van Engelen, B.G.; Faber, C.G. Drug treatment for myotonia. Cochrane Database Syst. Rev. 2006, 2006, CD004762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estévez, R.; Schroeder, B.C.; Accardi, A.; Jentsch, T.J.; Pusch, M. Conservation of chloride channel structure revealed by an inhibitor binding site in ClC-1. Neuron 2003, 38, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desaphy, J.F.; Costanza, T.; Carbonara, R.; Conte-Camerino, D. In vivo evaluation of anti-myotonic efficacy of β-adrenergic drugs in a rat model of myotonia. Neuropharmacology 2013, 65, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Warf, M.B.; Nakamori, M.; Matthys, C.M.; Thornton, C.A.; Berglund, J.A. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 18551–18856. [Google Scholar] [CrossRef] [Green Version]
- Mao, C.; Flavin, K.G.; Wang, S.; Dodson, R.; Ross, J.; Shapiro, D.J. Analysis of RNA-protein interactions by a microplate-based fluorescence anisotropy assay. Anal. Biochem. 2006, 350, 222–232. [Google Scholar] [CrossRef]
- Garcia-Lopez, A.; Monferrer, L.; Garcia-Alcover, I.; Vicente-Crespo, M.; Alvarez-Abril, M.C.; Artero, R.D. Genetic and chemical modifiers of a CUG toxicity model in Drosophila. PLoS ONE 2008, 3, e1595. [Google Scholar] [CrossRef] [Green Version]
- Neault, N.; O’Reilly, S.; Baig, A.T.; Plaza-Diaz, J.; Azimi, M.; Farooq, F.; Baird, S.D.; MacKenzie, A. High-throughput kinome-RNAi screen identifies protein kinase R activator (PACT) as a novel genetic modifier of CUG foci integrity in myotonic dys-trophy type 1 (DM1). PLoS ONE 2021, 16, e0256276. [Google Scholar] [CrossRef]
- Nguyen, Q.; Yokota, T. Degradation of Toxic RNA in Myotonic Dystrophy Using Gapmer Antisense Oligonucleotides. Methods Mol. Biol. 2020, 2176, 99–109. [Google Scholar] [CrossRef]
- García, M.T.; Carreño, D.; Tirado-Vélez, J.M.; Ferrándiz, M.J.; Rodrigues, L.; Gracia, B.; Amblar, M.; Ainsa, J.A.; de la Campa, A.G. Boldine-Derived Alkaloids Inhibit the Activity of DNA Topoisomerase I and Growth of Mycobacterium tuberculosis. Front. Microbiol. 2018, 9, 1659. [Google Scholar] [CrossRef] [PubMed]
- Noureini, S.K.; Wink, M. Dose-dependent cytotoxic effects of boldine in HepG-2 cells-telomerase inhibition and apoptosis induction. Molecules 2015, 20, 3730–3743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paydar, M.; Kamalidehghan, B.; Wong, Y.L.; Wong, W.F.; Looi, C.Y.; Mustafa, M.R. Evaluation of cytotoxic and chemotherapeutic properties of boldine in breast cancer using in vitro and in vivo models. Drug Des. Dev. Ther. 2014, 8, 719–733. [Google Scholar] [CrossRef] [Green Version]
- Gerhardt, D.; Bertola, G.; Dietrich, F.; Figueiró, F.; Zanotto-Filho, A.; Moreira Fonseca, J.C.; Morrone, F.B.; Barrios, C.H.; Battastini, A.M.; Salbego, C.G. Boldine induces cell cycle arrest and apoptosis in T24 human bladder cancer cell line via regulation of ERK, AKT, and GSK-3β. Urol. Oncol. 2014, 32, 36.e1–36.e9. [Google Scholar] [CrossRef]
- D’Ambrosio, E.S.; Chuang, K.; David, W.S.; Amato, A.A.; Gonzalez-Perez, P. Frequency and type of cancers in myotonic dystrophy: A retrospective cross-sectional study. Muscle Nerve 2023. [Google Scholar] [CrossRef]
- García-Puga, M.; Saenz-Antoñanzas, A.; Gerenu, G.; Arrieta-Legorburu, A.; Fernández-Torrón, R.; Zulaica, M.; Saenz, A.; Elizazu, J.; Nogales-Gadea, G.; Gadalla, S.M.; et al. Senescence plays a role in myotonic dystrophy type 1. JCI Insight 2022, 7, e159357. [Google Scholar] [CrossRef]
- Mateos-Aierdi, A.J.; Goicoechea, M.; Aiastui, A.; Fernández-Torrón, R.; Garcia-Puga, M.; Matheu, A.; López de Munain, A. Muscle wasting in myotonic dystrophies: A model of premature aging. Front. Aging Neurosci. 2015, 7, 125. [Google Scholar] [CrossRef]
- Toro, C.A.; Johnson, K.; Hansen, J.; Siddiq, M.M.; Vásquez, W.; Zhao, W.; Graham, Z.A.; Sáez, J.C.; Iyengar, R.; Cardozo, C.P. Boldine modulates glial transcription and functional recovery in a murine model of contusion spinal cord injury. bioRxiv 2023. [Google Scholar] [CrossRef]
- Ozimski, L.L.; Sabater-Arcis, M.; Bargiela, A.; Artero, R. The hallmarks of myotonic dystrophy type 1 muscle dysfunction. Biol. Rev. Camb. Philos. Soc. 2021, 96, 716–730. [Google Scholar] [CrossRef]
- Nakamori, M.; Hamanaka, K.; Thomas, J.D.; Wang, E.T.; Hayashi, Y.K.; Takahashi, M.P.; Swanson, M.S.; Nishino, I.; Mochizuki, H. Aberrant Myokine Signaling in Congenital Myotonic Dystrophy. Cell Rep. 2017, 21, 1240–1252. [Google Scholar] [CrossRef] [Green Version]
- Milián, L.; Estellés, R.; Abarca, B.; Ballesteros, R.; Sanz, M.J.; Blázquez, M.A. Reactive oxygen species (ROS) generation inhibited by aporphine and phenanthrene alkaloids semi-synthesized from natural boldine. Chem. Pharm. Bull. 2004, 52, 696–699. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Kumar, V.; Singh, S.K.; Muthuswamy, S.; Agarwal, S. Imbalanced oxidant and antioxidant ratio in myotonic dystrophy type 1. Free Radic. Res. 2014, 48, 503–510. [Google Scholar] [CrossRef]
- Imbrici, P.; Altamura, C.; Pessia, M.; Mantegazza, R.; Desaphy, J.F.; Camerino, D.C. ClC-1 chloride channels: State-of-the-art research and future challenges. Front. Cell Neurosci. 2015, 9, 156. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, T.H.; Riisager, A.; de Paoli, F.V.; Chen, T.Y.; Nielsen, O.B. Role of physiological ClC-1 Cl- ion channel regulation for the excitability and function of working skeletal muscle. J. Gen. Physiol. 2016, 147, 291–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikonova, E.; Kao, S.Y.; Ravichandran, K.; Wittner, A.; Spletter, M.L. Conserved functions of RNA-binding proteins in muscle. Int. J. Biochem. Cell Biol. 2019, 110, 29–49. [Google Scholar] [CrossRef]
- Lau, Y.S.; Tian, X.Y.; Huang, Y.; Murugan, D.; Achike, F.I.; Mustafa, M.R. Boldine protects endothelial function in hyperglycemia-induced oxidative stress through an antioxidant mechanism. Biochem. Pharmacol. 2013, 85, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C.; Tasca, E. Fatigue in muscular dystrophies. Neuromuscul. Disord. 2012, 22 (Suppl. S3), S214–S220. [Google Scholar] [CrossRef] [Green Version]
- Liguori, S.; Moretti, A.; Toro, G.; Paoletta, M.; Palomba, A.; Barra, G.; Gimigliano, F.; Iolascon, G. Pain and Motor Function in Myotonic Dystrophy Type 1: A Cross-Sectional Study. Int. J. Environ. Res. Public Health 2023, 20, 5244. [Google Scholar] [CrossRef] [PubMed]
- Cea, L.A.; Fernández, G.; Arias-Bravo, G.; Castillo-Ruiz, M.; Escamilla, R.; Brañes, M.C.; Sáez, J.C. Blockade of Hemichannels Normalizes the Differentiation Fate of Myoblasts and Features of Skeletal Muscles from Dysferlin-Deficient Mice. Int. J. Mol. Sci. 2020, 21, 6025. [Google Scholar] [CrossRef]
- Yi, C.; Ezan, P.; Fernández, P.; Schmitt, J.; Sáez, J.C.; Giaume, C.; Koulakoff, A. Inhibition of glial hemichannels by boldine treatment reduces neuronal suffering in a murine model of Alzheimer’s disease. Glia 2017, 65, 1607–1625. [Google Scholar] [CrossRef]
- Nakamori, M.; Shimizu, H.; Ogawa, K.; Hasuike, Y.; Nakajima, T.; Sakurai, H.; Araki, T.; Okada, Y.; Kakita, A.; Mochizuki, H. Cell type-specific abnormalities of central nervous system in myotonic dystrophy type 1. Brain Commun. 2022, 4, fcac154. [Google Scholar] [CrossRef]
- Livett, B.G. Adrenal medullary chromaffin cells in vitro. Physiol. Rev. 1984, 64, 1103–1161. [Google Scholar] [CrossRef] [PubMed]
- Moro, M.A.; López, M.G.; Gandía, L.; Michelena, P.; García, A.G. Separation and culture of living adrena-line- and noradrenaline-containing cells from bovine adrenal medullae. Anal. Biochem. 1990, 185, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Hamill, O.P.; Marty, A.; Neher, E.; Sakmann, B.; Sigworth, F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell free membrane patches. Pfluguers Arch. Eur. J. Physiol. 1981, 391, 85–100. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Álvarez-Abril, M.C.; García-Alcover, I.; Colonques-Bellmunt, J.; Garijo, R.; Pérez-Alonso, M.; Artero, R.; López-Castel, A. Natural Compound Boldine Lessens Myotonic Dystrophy Type 1 Phenotypes in DM1 Drosophila Models, Patient-Derived Cell Lines, and HSALR Mice. Int. J. Mol. Sci. 2023, 24, 9820. https://doi.org/10.3390/ijms24129820
Álvarez-Abril MC, García-Alcover I, Colonques-Bellmunt J, Garijo R, Pérez-Alonso M, Artero R, López-Castel A. Natural Compound Boldine Lessens Myotonic Dystrophy Type 1 Phenotypes in DM1 Drosophila Models, Patient-Derived Cell Lines, and HSALR Mice. International Journal of Molecular Sciences. 2023; 24(12):9820. https://doi.org/10.3390/ijms24129820
Chicago/Turabian StyleÁlvarez-Abril, Mari Carmen, Irma García-Alcover, Jordi Colonques-Bellmunt, Raquel Garijo, Manuel Pérez-Alonso, Rubén Artero, and Arturo López-Castel. 2023. "Natural Compound Boldine Lessens Myotonic Dystrophy Type 1 Phenotypes in DM1 Drosophila Models, Patient-Derived Cell Lines, and HSALR Mice" International Journal of Molecular Sciences 24, no. 12: 9820. https://doi.org/10.3390/ijms24129820