
New Perspectives in Dried Blood Spot Biomarkers for Lysosomal Storage Diseases

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
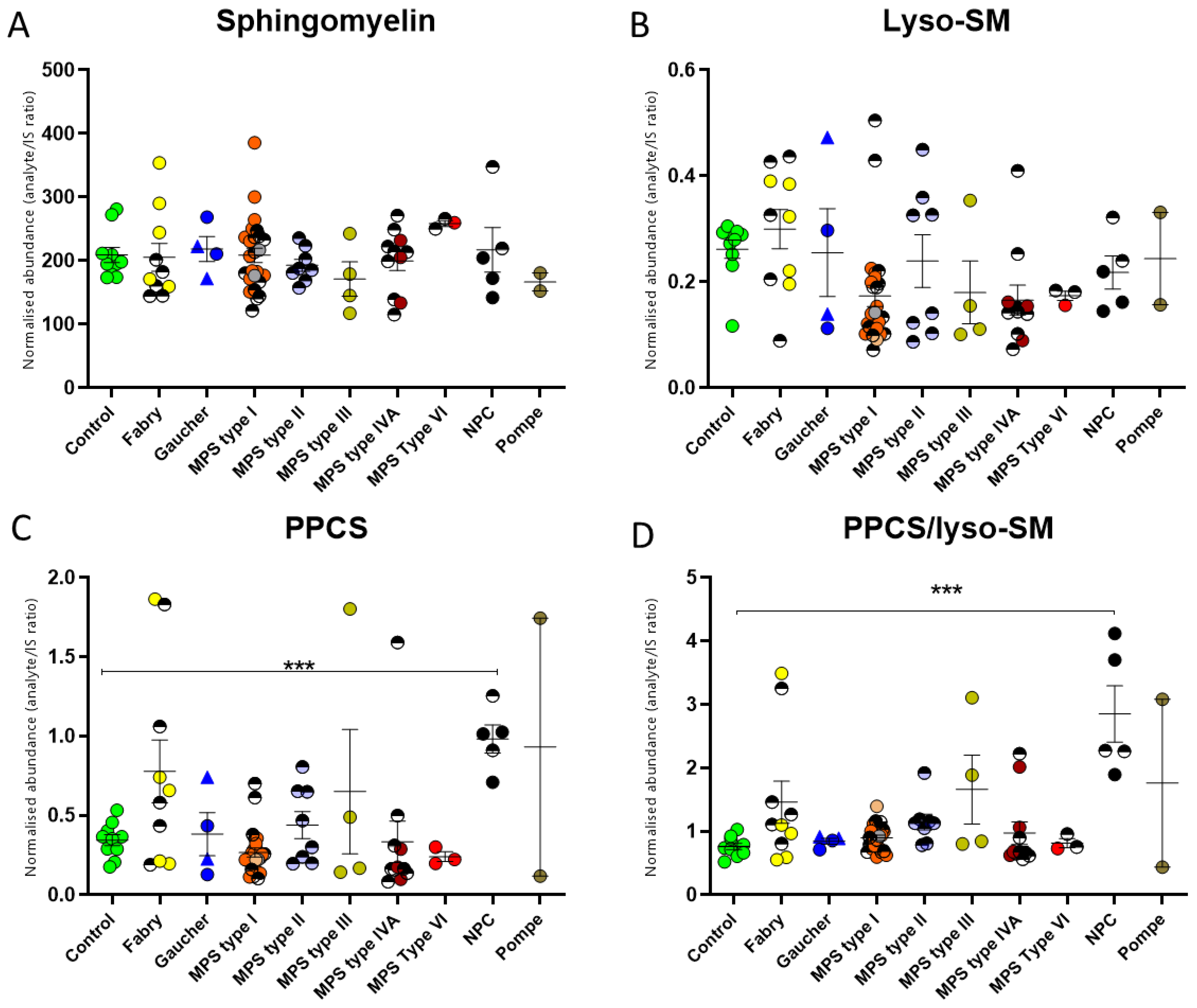
2.1. Dried Blood Spot Glycosphingolipids in Lysosomal Storage Diseases
2.2. Lyso-Glycosphingolipids in the Lysosomal Storage Diseases
2.3. Dried Blood Spot NPC Biomarkers in Lysosomal Storage Diseases
3. Discussion
4. Materials and Methods
4.1. Reagents and Standards
4.2. Samples
4.3. Dried Blood Spot Sample Preparation
4.4. UPLC–MS/MS Analysis
4.5. Data and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Papandreou, A.; Doykov, I.; Spiewak, J.; Komarov, N.; Habermann, S.; Kurian, M.A.; Mills, P.B.; Mills, K.; Gissen, P.; Heywood, W.E.; et al. Niemann-Pick type C disease as proof-of-concept for intelligent biomarker panel selection in neurometabolic disorders. Dev. Med. Child Neurol. 2022, 64, 1539–1546. [Google Scholar] [CrossRef] [PubMed]
- Dinur, T.; Bauer, P.; Beetz, C.; Kramp, G.; Cozma, C.; Iurascu, M.I.; Becker-Cohen, M.; Istaiti, M.; Rolfs, A.; Zimran, A.; et al. Gaucher Disease Diagnosis Using Lyso-Gb1 on Dry Blood Spot Samples: Time to Change the Paradigm? Int. J. Mol. Sci. 2022, 23, 1627. [Google Scholar] [CrossRef] [PubMed]
- Malvagia, S.; Ferri, L.; Della Bona, M.; Borsini, W.; Cirami, C.L.; Dervishi, E.; Feriozzi, S.; Gasperini, S.; Motta, S.; Mignani, R.; et al. Multicenter evaluation of use of dried blood spot compared to conventional plasma in measurements of globotriaosylsphingosine (LysoGb3) concentration in 104 Fabry patients. Clin. Chem. Lab. Med. 2021, 59, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Khaledi, H.; Gelb, M.H. Tandem Mass Spectrometry Enzyme Assays for Multiplex Detection of 10-Mucopolysaccharidoses in Dried Blood Spots and Fibroblasts. Anal. Chem. 2020, 92, 11721–11727. [Google Scholar] [CrossRef] [PubMed]
- Young, E.; Mills, K.; Morris, P.; Vellodi, A.; Lee, P.; Waldek, S.; Winchester, B. Is globotriaosylceramide a useful biomarker in Fabry disease? Acta Paed. 2005, 94, 51–54; discussion 37–38. [Google Scholar] [CrossRef]
- Tang, C.; Jia, X.; Tang, F.; Liu, S.; Jiang, X.; Zhao, X.; Sheng, H.; Peng, M.; Liu, L.; Huang, Y. Detection of glucosylsphingosine in dried blood spots for diagnosis of Gaucher disease by LC-MS/MS. Clin. Biochem. 2021, 87, 79–84. [Google Scholar] [CrossRef]
- Viana, G.M.; Priestman, D.A.; Platt, F.M.; Khan, S.; Tomatsu, S.; Pshezhetsky, A.V. Brain Pathology in Mucopolysaccharidoses (MPS) Patients with Neurological Forms. J. Clin. Med. 2020, 9, 396. [Google Scholar] [CrossRef] [Green Version]
- Soldati, C.; Lopez-Fabuel, I.; Wanderlingh, L.G.; Garcia-Macia, M.; Monfregola, J.; Esposito, A.; Napolitano, G.; Guevara-Ferrer, M.; Scotto Rosato, A.; Krogsaeter, E.K.; et al. Repurposing of tamoxifen ameliorates CLN3 and CLN7 disease phenotype. EMBO Mol. Med. 2021, 13, e13742. [Google Scholar] [CrossRef]
- Sidhu, R.; Mondjinou, Y.; Qian, M.; Song, H.; Kumar, A.B.; Hong, X.; Hsu, F.F.; Dietzen, D.J.; Yanjanin, N.M.; Porter, F.D.; et al. N-acyl-O-phosphocholineserines: Structures of a novel class of lipids that are biomarkers for Niemann-Pick C1 disease. J. Lipid Res. 2019, 60, 1410–1424. [Google Scholar] [CrossRef]
- Kasapkara, Ç.S.; Ceylan, A.C.; Yılmaz, D.; Kıreker Köylü, O.; Yürek, B.; Civelek Ürey, B.; Gündüz, M. CLN3-Associated NCL Case with a Preliminary Diagnosis of Niemann Pick Type C. Mol. Syndromol. 2022, 14, 30–34. [Google Scholar] [CrossRef]
- Boutin, M.; Menkovic, I.; Martineau, T.; Vaillancourt-Lavigueur, V.; Toupin, A.; Auray-Blais, C. Separation and Analysis of Lactosylceramide, Galabiosylceramide, and Globotriaosylceramide by LC-MS/MS in Urine of Fabry Disease Patients. Anal. Chem. 2017, 89, 13382–13390. [Google Scholar] [CrossRef] [PubMed]
- Heywood, W.E.; Doykov, I.; Spiewak, J.; Hallqvist, J.; Mills, K.; Nowak, A. Global glycosphingolipid analysis in urine and plasma of female Fabry disease patients. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2726–2735. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.; Morris, P.; Lee, P.; Vellodi, A.; Waldek, S.; Young, E.; Winchester, B. Measurement of urinary CDH and CTH by tandem mass spectrometry in patients hemizygous and heterozygous for Fabry disease. J. Inherit. Metab. Dis. 2005, 28, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Carnicer-Caceres, C.; Arranz-Amo, J.A.; Cea-Arestin, C.; Camprodon-Gomez, M.; Moreno-Martinez, D.; Lucas-Del-Pozo, S.; Molto-Abad, M.; Tigri-Santina, A.; Agraz-Pamplona, I.; Rodriguez-Palomares, J.F.; et al. Biomarkers in Fabry Disease. Implications for Clinical Diagnosis and Follow-up. J. Clin. Med. 2021, 10, 1664. [Google Scholar] [CrossRef] [PubMed]
- Kusano, E.; Saito, O.; Akimoto, T.; Asano, Y. Fabry disease: Experience of screening dialysis patients for Fabry disease. Clin. Exp. Nephrol. 2014, 18, 269–273. [Google Scholar] [CrossRef]
- Giese, A.K.; Mascher, H.; Grittner, U.; Eichler, S.; Kramp, G.; Lukas, J.; te Vruchte, D.; Al Eisa, N.; Cortina-Borja, M.; Porter, F.D.; et al. A novel, highly sensitive and specific biomarker for Niemann-Pick type C1 disease. Orphanet J. Rare Dis. 2015, 10, 78. [Google Scholar] [CrossRef] [Green Version]
- Kuchar, L.; Sikora, J.; Gulinello, M.E.; Poupetova, H.; Lugowska, A.; Malinova, V.; Jahnova, H.; Asfaw, B.; Ledvinova, J. Quantitation of plasmatic lysosphingomyelin and lysosphingomyelin-509 for differential screening of Niemann-Pick A/B and C diseases. Anal. Biochem. 2017, 525, 73–77. [Google Scholar] [CrossRef]
- Sitarska, D.; Lugowska, A. Laboratory diagnosis of the Niemann-Pick type C disease: An inherited neurodegenerative disorder of cholesterol metabolism. Metab. Brain Dis. 2019, 34, 1253–1260. [Google Scholar] [CrossRef] [Green Version]
- Marques, A.R.; Mirzaian, M.; Akiyama, H.; Wisse, P.; Ferraz, M.J.; Gaspar, P.; Ghauharali-van der Vlugt, K.; Meijer, R.; Giraldo, P.; Alfonso, P.; et al. Glucosylated cholesterol in mammalian cells and tissues: Formation and degradation by multiple cellular beta-glucosidases. J. Lipid Res. 2016, 57, 451–463. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, S.; Haberkant, P.; Bhardwaj, M.; Tongue, P.; Ferraz, M.J.; Halter, D.; Sprong, H.; Schmid, R.; Aerts, J.; Sullo, N.; et al. Cytosolic glucosylceramide regulates endolysosomal function in Niemann-Pick type C disease. Neurobiol. Dis. 2019, 127, 242–252. [Google Scholar] [CrossRef]
- Hollak, C.E.; van Weely, S.; van Oers, M.H.; Aerts, J.M. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J. Clin. Investig. 1994, 93, 1288–1292. [Google Scholar] [CrossRef] [Green Version]
- Boot, R.G.; Renkema, G.H.; Verhoek, M.; Strijland, A.; Bliek, J.; de Meulemeester, T.M.; Mannens, M.M.; Aerts, J.M. The human chitotriosidase gene. Nature of inherited enzyme deficiency. J. Biol. Chem. 1998, 273, 25680–25685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murugesan, V.; Chuang, W.L.; Liu, J.; Lischuk, A.; Kacena, K.; Lin, H.; Pastores, G.M.; Yang, R.; Keutzer, J.; Zhang, K.; et al. Glucosylsphingosine is a key biomarker of Gaucher disease. Am. J. Hematol. 2016, 91, 1082–1089. [Google Scholar] [CrossRef] [Green Version]
- Dekker, N.; van Dussen, L.; Hollak, C.E.; Overkleeft, H.; Scheij, S.; Ghauharali, K.; van Breemen, M.J.; Ferraz, M.J.; Groener, J.E.; Maas, M.; et al. Elevated plasma glucosylsphingosine in Gaucher disease: Relation to phenotype, storage cell markers, and therapeutic response. Blood 2011, 118, e118–e127. [Google Scholar] [CrossRef] [Green Version]
- Rolfs, A.; Giese, A.K.; Grittner, U.; Mascher, D.; Elstein, D.; Zimran, A.; Bottcher, T.; Lukas, J.; Hubner, R.; Golnitz, U.; et al. Glucosylsphingosine is a highly sensitive and specific biomarker for primary diagnostic and follow-up monitoring in Gaucher disease in a non-Jewish, Caucasian cohort of Gaucher disease patients. PLoS ONE 2013, 8, e79732. [Google Scholar] [CrossRef] [PubMed]
- Baydakova, G.; Ilyushkina, A.; Gaffke, L.; Pierzynowska, K.; Bychkov, I.; Lugowska, A.; Wegrzyn, G.; Tylki-Szymanska, A.; Zakharova, E. Elevated LysoGb3 Concentration in the Neuronopathic Forms of Mucopolysaccharidoses. Diagnostics 2020, 10, 155. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.; Mascher, H.; Mascher, D.; Legnini, E.; Hung, C.Y.; Dajnoki, A.; Chien, Y.H.; Marodi, L.; Hwu, W.L.; Bodamer, O.A. Analysis of lyso-globotriaosylsphingosine in dried blood spots. Ann. Lab. Med. 2013, 33, 274–278. [Google Scholar] [CrossRef] [Green Version]
- Lukas, J.; Scalia, S.; Eichler, S.; Pockrandt, A.M.; Dehn, N.; Cozma, C.; Giese, A.K.; Rolfs, A. Functional and Clinical Consequences of Novel alpha-Galactosidase A Mutations in Fabry Disease. Hum. Mutat. 2016, 37, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Cozma, C.; Yang, F.; Kramp, G.; Meyer, A.; Nesslauer, A.M.; Eichler, S.; Bottcher, T.; Witt, M.; Brauer, A.U.; et al. Glucosylsphingosine Causes Hematological and Visceral Changes in Mice-Evidence for a Pathophysiological Role in Gaucher Disease. Int. J. Mol. Sci. 2017, 18, 2192. [Google Scholar] [CrossRef] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spiewak, J.; Doykov, I.; Papandreou, A.; Hällqvist, J.; Mills, P.; Clayton, P.T.; Gissen, P.; Mills, K.; Heywood, W.E. New Perspectives in Dried Blood Spot Biomarkers for Lysosomal Storage Diseases. Int. J. Mol. Sci. 2023, 24, 10177. https://doi.org/10.3390/ijms241210177
Spiewak J, Doykov I, Papandreou A, Hällqvist J, Mills P, Clayton PT, Gissen P, Mills K, Heywood WE. New Perspectives in Dried Blood Spot Biomarkers for Lysosomal Storage Diseases. International Journal of Molecular Sciences. 2023; 24(12):10177. https://doi.org/10.3390/ijms241210177
Chicago/Turabian StyleSpiewak, Justyna, Ivan Doykov, Apostolos Papandreou, Jenny Hällqvist, Philippa Mills, Peter T. Clayton, Paul Gissen, Kevin Mills, and Wendy E. Heywood. 2023. "New Perspectives in Dried Blood Spot Biomarkers for Lysosomal Storage Diseases" International Journal of Molecular Sciences 24, no. 12: 10177. https://doi.org/10.3390/ijms241210177