
Introducing and Implementing Genetic Assessment in Cardio-Obstetrics Clinical Practice: Clinical and Genetic Workup of Patients with Cardiomyopathy
 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
2.1. Overview of the Corniche Hospital Cardio-Obstetrics Clinic Referrals
2.2. Clinical and Genetic Workups of Five Patients
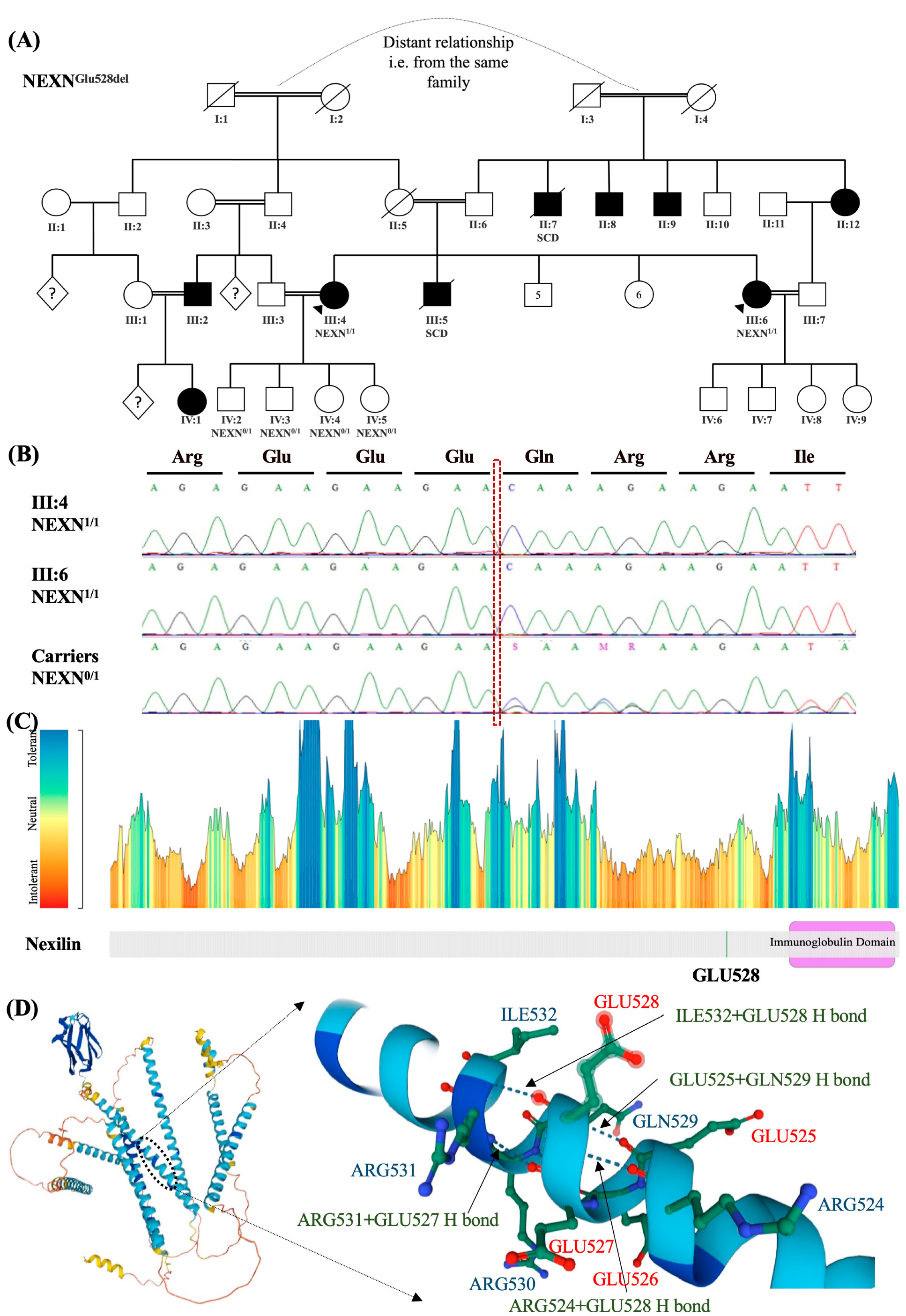
2.2.1. Patient-1
2.2.2. Patient-2
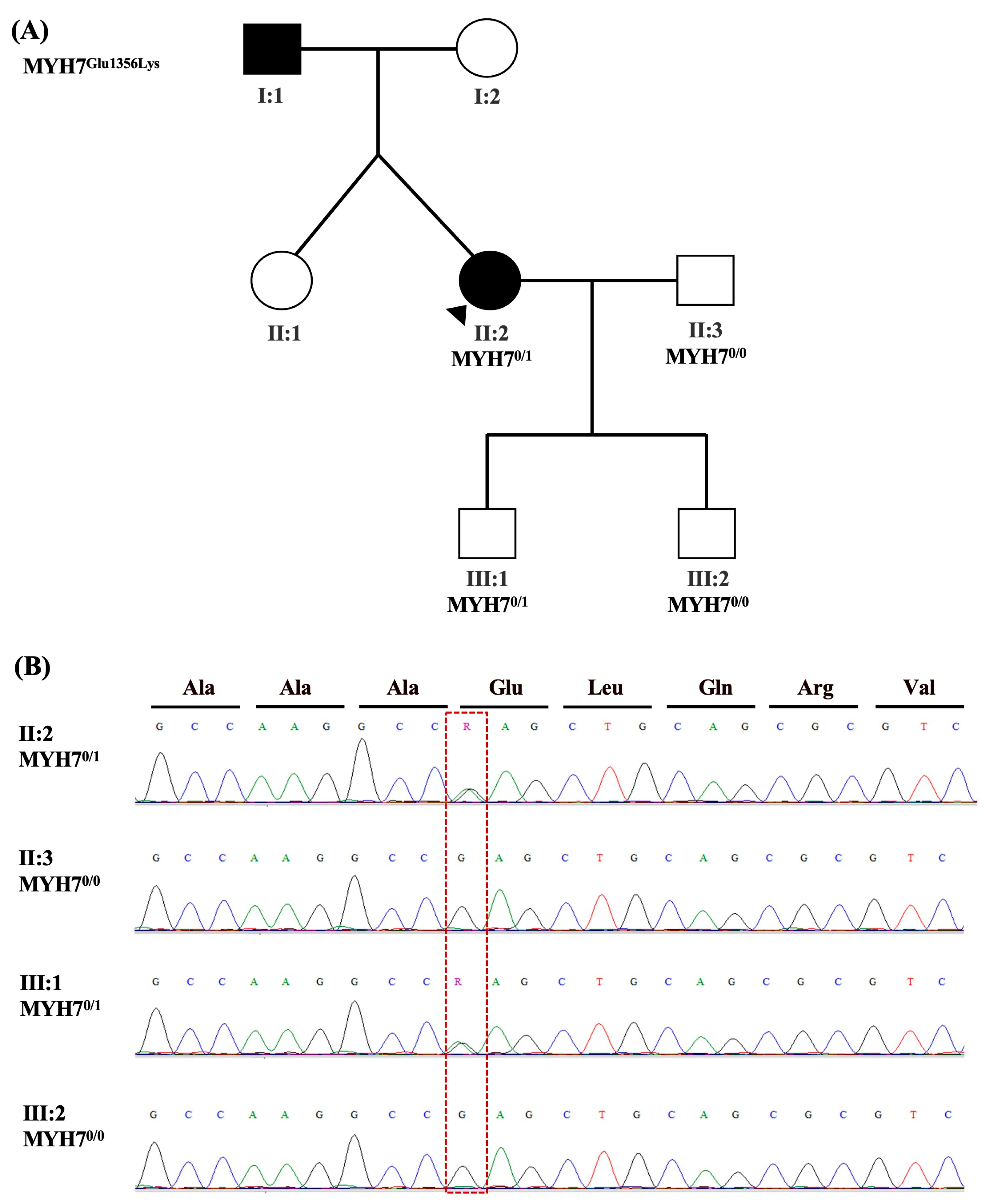
2.2.3. Patient-3
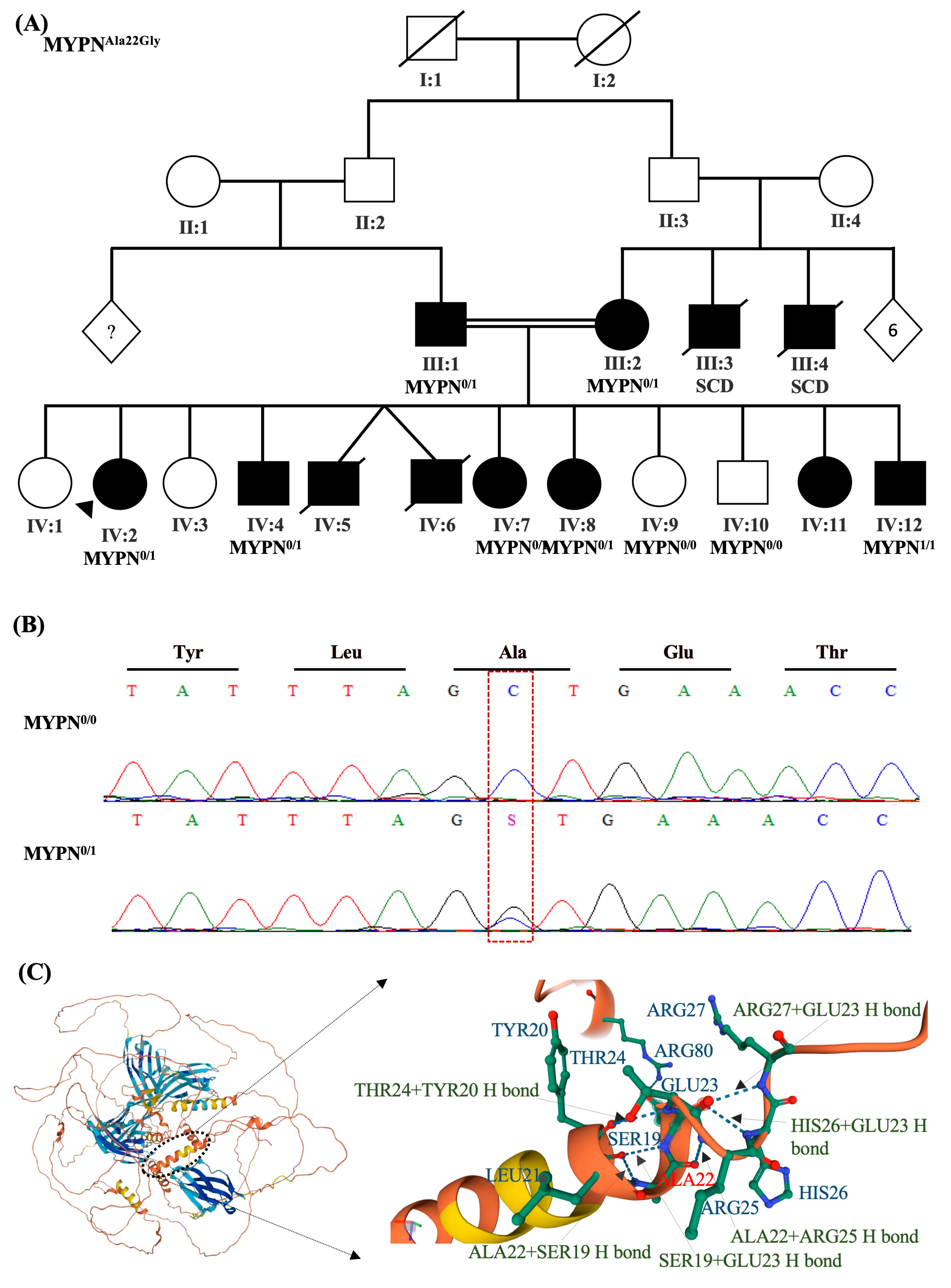
2.2.4. Patient-4
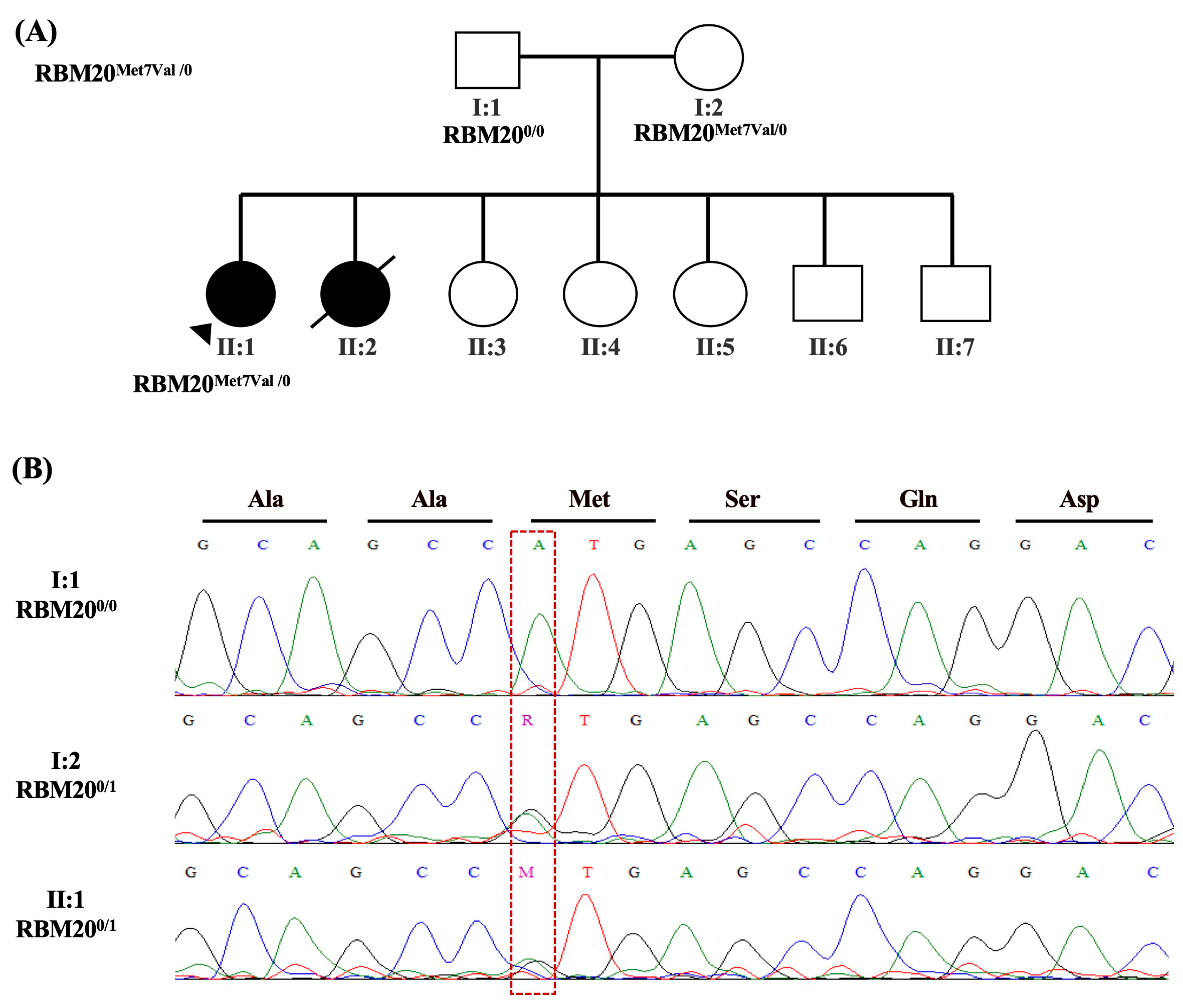
2.2.5. Patient-5
3. Discussion
3.1. Spectrum and Impact of Cardiovascular Disease Seen in Women from the United Arab Emirates
3.2. Clinical and Genetic Risk Assessment and Care during Pregnancy at the COB
3.3. Preconception Risk Assessment and Care
3.4. Family Clinical Screening and Genetic Risk Assessment
3.5. Strengths and Limitations of the Study
4. Materials and Methods
4.1. Material
4.2. DNA Isolation and Whole-Exome Sequencing
4.3. Variant Identification, Filtering, and Classification
4.4. Segregation Analysis
5. Conclusions
- Risk stratification of disease severity;
- Full understanding of the underlying etiology (inheritable, post-partum cardiomyopathy, valve-related, congenital heart disease);
- Risk of transmission to offspring;
- Projected effect of pregnancy on disease progression;
- Impact of medication/treatment on fetal growth during the breastfeeding period;
- Facilitating access to a multi-disciplinary team to prove cardio obstetric care.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soomro, S.; Kumar, R.; Shaikh, A.A. Twenty-Eight Pregnant Women with Cardiac Diseases: A Review of Maternal and Fetal Outcomes in Pakistan. Cureus 2019, 11, e6034. [Google Scholar] [CrossRef]
- Gad, M.M.; Elgendy, I.Y.; Mahmoud, A.N.; Saad, A.M.; Isogai, T.; Sande Mathias, I.; Misbah Rameez, R.; Chahine, J.; Jneid, H.; Kapadia, S.R. Disparities in Cardiovascular Disease Outcomes Among Pregnant and Post-Partum Women. J. Am. Heart Assoc. 2021, 10, e017832. [Google Scholar] [CrossRef] [PubMed]
- van Hagen, I.M.; Roos-Hesselink, J.W. Pregnancy in Congenital Heart Disease: Risk Prediction and Counselling. Heart 2020, 106, 1853–1861. [Google Scholar] [CrossRef]
- Hunter, S.; Robson, S.C. Adaptation of the Maternal Heart in Pregnancy. Heart 1992, 68, 540–543. [Google Scholar] [CrossRef]
- Poppas, A.; Shroff, S.G.; Korcarz, C.E.; Hibbard, J.U.; Berger, D.S.; Lindheimer, M.D.; Lang, R.M. Serial Assessment of the Cardiovascular System in Normal Pregnancy: Role of Arterial Compliance and Pulsatile Arterial Load. Circulation 1997, 95, 2407–2415. [Google Scholar] [CrossRef]
- Hendricks, C.H.; Quilligan, E.J. Cardiac Output during Labor. Am. J. Obstet. Gynecol. 1956, 71, 953–972. [Google Scholar] [CrossRef]
- Ueland, K.; Hansen, J.M. Maternal Cardiovascular Dynamics. Am. J. Obstet. Gynecol. 1969, 103, 1–7. [Google Scholar] [CrossRef]
- Adams, J.Q.; Alexander, A.M. Alterations in Cardiovascular Physiology during Labor. Obstet. Gynecol. 1958, 12, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Rokey, R.; Miller, J.; Cotton, D.B. Maternal Hemodynamic Effects of Uterine Contractions by M-Mode and Pulsed-Doppler Echocardiography. Am. J. Obstet. Gynecol. 1989, 161, 974–977. [Google Scholar] [CrossRef]
- Davis, M.B.; Arany, Z.; McNamara, D.M.; Goland, S.; Elkayam, U. Peripartum Cardiomyopathy. J. Am. Coll. Cardiol. 2020, 75, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Lindley, K.J.; Bairey Merz, C.N.; Asgar, A.W.; Bello, N.A.; Chandra, S.; Davis, M.B.; Gomberg-Maitland, M.; Gulati, M.; Hollier, L.M.; Krieger, E.V.; et al. Management of Women With Congenital or Inherited Cardiovascular Disease From Pre-Conception Through Pregnancy and Postpartum. J. Am. Coll. Cardiol. 2021, 77, 1778–1798. [Google Scholar] [CrossRef] [PubMed]
- Schaufelberger, M. Cardiomyopathy and Pregnancy. Heart 2019, 105, 1543–1551. [Google Scholar] [CrossRef]
- Goli, R.; Li, J.; Brandimarto, J.; Levine, L.D.; Riis, V.; McAfee, Q.; DePalma, S.; Haghighi, A.; Seidman, J.G.; Seidman, C.E.; et al. Genetic and Phenotypic Landscape of Peripartum Cardiomyopathy. Circulation 2021, 143, 1852–1862. [Google Scholar] [CrossRef]
- Saberi, S. Hypertrophic Cardiomyopathy in Pregnancy. Cardiol. Clin. 2021, 39, 143–150. [Google Scholar] [CrossRef]
- Millington, S.; Magarey, J.; Dekker, G.A.; Clark, R.A. Cardiac Conditions in Pregnancy and the Role of Midwives: A Discussion Paper. Nurs. Open 2019, 6, 722–732. [Google Scholar] [CrossRef]
- Van Tintelen, J.P.; Pieper, P.G.; Van Spaendonck-Zwarts, K.Y.; Van Den Berg, M.P. Pregnancy, Cardiomyopathies, and Genetics. Cardiovasc. Res. 2014, 101, 571–578. [Google Scholar] [CrossRef]
- Endorsed by the European Society of Gynecology (ESG), the Association for European Paediatric Cardiology (AEPC), and the German Society for Gender Medicine (DGesGM); Authors/Task Force Members; Regitz-Zagrosek, V.; Blomstrom Lundqvist, C.; Borghi, C.; Cifkova, R.; Ferreira, R.; Foidart, J.-M.; Gibbs, J.S.R.; Gohlke-Baerwolf, C.; et al. ESC Guidelines on the Management of Cardiovascular Diseases during Pregnancy: The Task Force on the Management of Cardiovascular Diseases during Pregnancy of the European Society of Cardiology (ESC). Eur. Heart J. 2011, 32, 3147–3197. [Google Scholar] [CrossRef] [PubMed]
- Wiel, L.; Baakman, C.; Gilissen, D.; Veltman, J.A.; Vriend, G.; Gilissen, C. MetaDome: Pathogenicity Analysis of Genetic Variants through Aggregation of Homologous Human Protein Domains. Hum. Mutat. 2019, 40, 1030–1038. [Google Scholar] [CrossRef]
- van Hagen, I.M.; Boersma, E.; Johnson, M.R.; Thorne, S.A.; Parsonage, W.A.; Escribano Subías, P.; Leśniak-Sobelga, A.; Irtyuga, O.; Sorour, K.A.; Taha, N.; et al. Global Cardiac Risk Assessment in the Registry Of Pregnancy And Cardiac Disease: Results of a Registry from the European Society of Cardiology: Cardiac Risk Assessment in the ROPAC. Eur. J. Heart Fail. 2016, 18, 523–533. [Google Scholar] [CrossRef]
- Roos-Hesselink, J.; Baris, L.; Johnson, M.; De Backer, J.; Otto, C.; Marelli, A.; Jondeau, G.; Budts, W.; Grewal, J.; Sliwa, K.; et al. Pregnancy Outcomes in Women with Cardiovascular Disease: Evolving Trends over 10 Years in the ESC Registry Of Pregnancy And Cardiac Disease (ROPAC). Eur. Heart J. 2019, 40, 3848–3855. [Google Scholar] [CrossRef] [PubMed]
- Bottega, N.; Malhamé, I.; Guo, L.; Ionescu-Ittu, R.; Therrien, J.; Marelli, A. Secular Trends in Pregnancy Rates, Delivery Outcomes, and Related Health Care Utilization among Women with Congenital Heart Disease. Congenit. Heart Dis. 2019, 14, 735–744. [Google Scholar] [CrossRef]
- Vrselja, A.; Pillow, J.J.; Black, M.J. Effect of Preterm Birth on Cardiac and Cardiomyocyte Growth and the Consequences of Antenatal and Postnatal Glucocorticoid Treatment. J. Clin. Med. 2021, 10, 3896. [Google Scholar] [CrossRef] [PubMed]
- Rivera, F.B.; Magalong, J.V.; Tantengco, O.A.; Mangubat, G.F.; Villafuerte, M.G.; Volgman, A.S. Maternal and Neonatal Outcomes among Pregnant Women with Cardiovascular Disease in the Philippines: A Retrospective Cross-Sectional Study from 2015–2019. J. Matern.-Fetal Neonatal Med. 2022, 35, 9922–9933. [Google Scholar] [CrossRef]
- Makino, Y.; Matsuda, Y.; Mitani, M.; Shinohara, T.; Matsui, H. Risk Factors Associated with Preterm Delivery in Women with Cardiac Disease. J. Cardiol. 2012, 59, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Miseljic, N.; Ibrahimovic, S. Health Implications of Increased Cesarean Section Rates. Mater. Socio-Med. 2020, 32, 123. [Google Scholar] [CrossRef] [PubMed]
- Tutarel, O. Mode of Delivery for Pregnant Women with Heart Disease. Heart 2015, 101, 504–505. [Google Scholar] [CrossRef]
- Eggleton, E.J.; McMurrugh, K.J.; Aiken, C.E. Maternal Pregnancy Outcomes in Women with Cardiomyopathy: A Systematic Review and Meta-Analysis. Am. J. Obstet. Gynecol. 2022, 227, 582–592. [Google Scholar] [CrossRef]
- Ware, J.S.; Li, J.; Mazaika, E.; Yasso, C.M.; DeSouza, T.; Cappola, T.P.; Tsai, E.J.; Hilfiker-Kleiner, D.; Kamiya, C.A.; Mazzarotto, F.; et al. Shared genetic predisposition in peripartum and dilated cardiomyopathies. N. Engl. J. Med. 2016, 374, 233–241. [Google Scholar] [CrossRef]
- Safavi, M.; Honarmand, A.; Kashefi, P.; Heidari, S.M.; Safavi, A.; Hekmat, R. Post-delivery cardiomyopathy in a patient admitted to critical care unit; a rare case report. J. Reprod. Infertil. 2011, 12, 37–41. [Google Scholar] [PubMed]
- Rosenbaum, A.N.; Agre, K.E.; Pereira, N.L. Genetics of Dilated Cardiomyopathy: Practical Implications for Heart Failure Management. Nat. Rev. Cardiol. 2020, 17, 286–297. [Google Scholar] [CrossRef]
- Gerull, B.; Klaassen, S.; Brodehl, A. The Genetic Landscape of Cardiomyopathies. In Genetic Causes of Cardiac Disease; Erdmann, J., Moretti, A., Eds.; Cardiac and Vascular Biology; Springer: Cham, Switzerland, 2019; Volume 7. [Google Scholar] [CrossRef]
- Meyer, T.; Ruppert, V.; Ackermann, S.; Richter, A.; Perrot, A.; Sperling, S.R.; Posch, M.G.; Maisch, B.; Pankuweit, S.; German Competence Network Heart Failure. Novel Mutations in the Sarcomeric Protein Myopalladin in Patients with Dilated Cardiomyopathy. Eur. J. Hum. Genet. 2013, 21, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Van Driest, S.L.; Jaeger, M.A.; Ommen, S.R.; Will, M.L.; Gersh, B.J.; Tajik, A.J.; Ackerman, M.J. Comprehensive Analysis of the Beta-Myosin Heavy Chain Gene in 389 Unrelated Patients With Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 602–610. [Google Scholar] [CrossRef]
- Lu, C.; Wu, W.; Liu, F.; Yang, K.; Li, J.; Liu, Y.; Wang, R.; Si, N.; Gao, P.; Liu, Y.; et al. Molecular analysis of inherited cardiomyopathy using next generation semiconductor sequencing technologies. J. Transl. Med. 2018, 16, 241. [Google Scholar] [CrossRef]
- Wolny, M.; Colegrave, M.; Colman, L.; White, E.; Knight, P.J.; Peckham, M. Cardiomyopathy mutations in the tail of β-cardiac myosin modify the coiled-coil structure and affect integration into thick filaments in muscle sarcomeres in adult cardiomyocytes. J. Biol. Chem. 2013, 288, 31952–31962, Erratum in J. Transl. Med. 2018, 16, 241. [Google Scholar] [CrossRef] [PubMed]
- Johansson, J.; Frykholm, C.; Ericson, K.; Kazamia, K.; Lindberg, A.; Mulaiese, N.; Falck, G.; Gustafsson, P.; Lidéus, S.; Gudmundsson, S.; et al. Loss of Nexilin Function Leads to a Recessive Lethal Fetal Cardiomyopathy Characterized by Cardiomegaly and Endocardial Fibroelastosis. Am. J. Med. Genet. Part A 2022, 188, 1676–1687. [Google Scholar] [CrossRef] [PubMed]
- Al-Hassnan, Z.N.; Almesned, A.; Tulbah, S.; Al-Manea, W.; Al-Fayyadh, M. Identification of a Novel Homozygous NEXN Gene Mutation in Recessively Inherited Dilated Cardiomyopathy. J. Saudi Heart Assoc. 2013, 25, 171–172. [Google Scholar] [CrossRef]
- Bruyndonckx, L.; Vogelzang, J.L.; Bugiani, M.; Straver, B.; Kuipers, I.M.; Onland, W.; Nannenberg, E.A.; Clur, S.; Crabben, S.N. Childhood Onset Nexilin Dilated Cardiomyopathy: A Heterozygous and a Homozygous Case. Am. J. Med. Genet. Part A 2021, 185, 2464–2470. [Google Scholar] [CrossRef]
- Kühnisch, J.; Herbst, C.; Al-Wakeel-Marquard, N.; Dartsch, J.; Holtgrewe, M.; Baban, A.; Mearini, G.; Hardt, J.; Kolokotronis, K.; Gerull, B.; et al. Targeted Panel Sequencing in Pediatric Primary Cardiomyopathy Supports a Critical Role of TNNI3. Clin. Genet. 2019, 96, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Ringpfeil, F.; McGuigan, K.; Fuchsel, L.; Kozic, H.; Larralde, M.; Lebwohl, M.; Uitto, J. Pseudoxanthoma elasticum is a recessive disease characterized by compound heterozygosity. J. Investig. Dermatol. 2006, 126, 782–786. [Google Scholar] [CrossRef]
- Dibi, A.; El Fahime, E.L.; Mouane, N.; Dafiri, R.; Bentahila, A. Pseudo-xanthome élastique: Une cause rare d’hémorragies digestives chez l’enfant [Pseudoxanthoma elasticum: A rare cause of gastrointestinal bleeding in children]. Arch. Pediatr. 2016, 23, 591–594. [Google Scholar] [CrossRef]
- Clapp, M.A.; Bernstein, S.N. Preconception Counseling for Women With Cardiac Disease. Curr. Treat. Options Cardiovasc. Med. 2017, 19, 67. [Google Scholar] [CrossRef] [PubMed]
- Regitz-Zagrosek, V.; Roos-Hesselink, J.W.; Bauersachs, J.; Blomström-Lundqvist, C.; Cífková, R.; De Bonis, M.; Iung, B.; Johnson, M.R.; Kintscher, U.; Kranke, P.; et al. 2018 ESC Guidelines for the Management of Cardiovascular Diseases during Pregnancy. Eur. Heart J. 2018, 39, 3165–3241. [Google Scholar] [CrossRef]
- Kasargod Prabhakar, C.R.; Pamment, D.; Thompson, P.J.; Chong, H.; Thorne, S.A.; Fox, C.; Morris, K.; Hudsmith, L.H. Pre-Conceptual Counselling in Cardiology Patients: Still Work to Do and Still Missed Opportunities. A Comparison between 2015 and 2019 in Women with Cardiac Disease Attending Combined Obstetric Cardiology Clinics. Should the European Guidelines Change Anything? Cardiol. Young 2022, 32, 64–70. [Google Scholar] [CrossRef]
- Meah, V.L.; Backx, K.; Davenport, M.H. International Working Group on Maternal Hemodynamics Functional Hemodynamic Testing in Pregnancy: Recommendations of the International Working Group on Maternal Hemodynamics. Ultrasound Obstet. Gynecol. 2018, 51, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Kuliev, A.; Rechitsky, S. Preimplantation Genetic Testing (PGT) for Cardiac Disease. J. Invasive Non-Invasive Cardiol. 2018, 2, 1–4. [Google Scholar] [CrossRef]
- Musunuru, K.; Hershberger, R.E.; Day, S.M.; Klinedinst, N.J.; Landstrom, A.P.; Parikh, V.N.; Prakash, S.; Semsarian, C.; Sturm, A.C.; American Heart Association Council on Genomic and Precision Medicine; et al. Genetic Testing for Inherited Cardiovascular Diseases: A Scientific Statement From the American Heart Association. Circ. Genom. Precis. Med. 2020, 13, e000067. [Google Scholar] [CrossRef]
- Morales, A.; Painter, T.; Li, R.; Siegfried, J.D.; Li, D.; Norton, N.; Hershberger, R.E. Rare Variant Mutations in Pregnancy-Associated or Peripartum Cardiomyopathy. Circulation 2010, 121, 2176–2182. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Cardiac Phenotype | Genetic Mutation | ACMG Classification Criteria | ACMG Classification Criteria Description | ACMG Classification-on | * MAF | § Homo count | dbSNP rsID | Segregation Analysis Results | Gene OMIM ID: Associated Cardiovascular-Related Disease |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Dilated cardiomyopathy | NEXN (NM_144573.4): c.1582_1584del (p. Glu528del) | PP3, BP3 | NEXN: Gerp++ predicts conserved at this location, PhyloP predicts conserved at this location, NEXN: Transcript has inframe_deletion ontology, and is repeated 3 times | VUS/ Conflicting | 1.5 × 104 | 0 | rs764505909 | Homozygous in patient, and her sister. | 613121: Cardiomyopathy |
| ABCC6 (NM_001171.6): c.3491G>A (p. Arg1164Gln) | PM2, PS1 | ABCC6: Maximum allele frequency 0.009799% is below rare threshold of 0.01% for max AF source Annotate-gnomAD Exomes Variant Frequencies 2.1.1, BROAD, ABCC6: The variant causes the same pathogenic mutation as a pathogenic variant in the same amino acid for the following sources: ClinVar. | Likely pathogenic | 2.39 × 105 | 0 | rs63750457 | Heterozygous in patient, and her sister | 603234: Atrial calcification and premature atherosclerosis; Pseudoxanthoma elasticum | ||
| MYH11 (NM_001040113.2): c.2383G>A (p. Asp795Asn) | PM2, PP2, PP3 | MYH11: Maximum allele frequency 0.000879% is below rare threshold of 0.01% for max AF source Annotate-gnomAD Exomes Variant Frequencies 2.1.1, BROAD, MYH11: Missense variant in gene with disease commonly caused by missense variants, MYH11: Gerp++ predicts conserved at this location, PhyloP predicts conserved at this location, sift predicts this variant is: ’Damaging’, Polyphon predicts that this variant is ’Damaging’ | VUS/Weak pathogenic | 3.98 × 106 | 0 | rs768858261 | Heterozygous in patient, and her sister | 160745: Familial Thoracic Aortic Aneurysm and Aortic Dissection | ||
| TCAP (NM_003673.4): c.113G>T (p. Cys38Phe) | PM2, BP1 | TCAP: gnomAD genomes homozygous allele count = 0 is less than 2 for AD/AR gene TCAP, good gnomAD genomes coverage = 31.0. gnomAD exomes homozygous allele count = 0 is less than 2 for AD/AR gene TCAP, good gnomAD exomes coverage = 41.0, TCAP: 8 out of 22 non-VUS missense variants in gene TCAP are benign = 36.4% which is more than threshold of 33.1% | VUS/ Conflicting | 1.08 × 104 | 0 | rs375310569 | Heterozygous in patient, and her sister | 604488: Cardiomyopathy | ||
| KCNJ11 (NM_000525.4): c.112A>G (p. Lys38Glu) | PM2, PP2, PM1, PP3 | KCNJ11:Variant is missing from all sub population sources.,KCNJ11:Missense variant in gene with disease commonly caused by missense variants, KCNJ11:There are no benign variants within 3 amino acids of this variant, and 2 pathogenic/0 benign variants within 6 amino acids of this variant.,KCNJ11:Gerp++ predicts conserved at this location, PhyloP predicts conserved at this location, Sift predicts this variant is: ’Damaging’, Polyphon predicts that this variant is ’Damaging’ | Likely pathogenic | 0 | 0 | Heterozygous in patient, and her sister | 600937: Sudden Cardiac Death | |||
| 2 | Dilated cardiomyopathy | NEXN (NM_144573.4): c.1582_1584del (p. Glu528del) | PP3, BP3 | NEXN: Gerp++ predicts conserved at this location, PhyloP predicts conserved at this location, NEXN: Transcript has inframe_deletion ontology, and is repeated 3 times | VUS/ Conflicting | 1.53 × 104 | 0 | rs764505909 | Homozygous in patient, and her sister. Heterozygous in all patient’s children | 613121: Cardiomyopathy |
| ABCC6 (NM_001171.6): c.3491G>A (p.Arg1164Gln) | PM2, PS1 | ABCC6: Maximum allele frequency 0.009799% is below rare threshold of 0.01% for max AF source Annotate-gnomAD Exomes Variant Frequencies 2.1.1, BROAD, ABCC6: The variant causes the same pathogenic mutation as a pathogenic variant in the same amino acid for the following sources: ClinVar. | Likely pathogenic | 2.39 × 105 | 0 | rs63750457 | Heterozygous in patient, her sister and her 4 children | 603234: Atrial calcification and premature atherosclerosis; Pseudoxanthoma elasticum | ||
| MYH11 (NM_001040113.2): c.2383G>A (p. Asp795Asn) | PM2, PP2, PP3 | MYH11: Maximum allele frequency 0.000879% is below rare threshold of 0.01% for max AF source Annotate-gnomAD Exomes Variant Frequencies 2.1.1, BROAD, MYH11: Missense variant in gene with disease commonly caused by missense variants, MYH11: Gerp++ predicts conserved at this location, PhyloP predicts conserved at this location, sift predicts this variant is: ’Damaging’, Pollyphen predicts that this variant is ’Damaging’ | VUS/Weak pathogenic | 3.98 × 106 | 0 | rs768858261 | Heterozygous in patient, her sister and her 4 children | 160745: Familial Thoracic Aortic Aneurysm and Aortic Dissection | ||
| TCAP (NM_003673.4): c.113G>T (p. Cys38Phe) | PM2, BP1 | TCAP: gnomAD genomes homozygous allele count = 0 is less than 2 for AD/AR gene TCAP, good gnomAD genomes coverage = 31.0. gnomAD exomes homozygous allele count = 0 is less than 2 for AD/AR gene TCAP, good gnomAD exomes coverage = 41.0, TCAP: 8 out of 22 non-VUS missense variants in gene TCAP are benign = 36.4% which is more than threshold of 33.1% | VUS/ Conflicting | 1.08 × 104 | 0 | rs375310569 | Heterozygous in patient, her sister and her 3 children | 604488: Cardiomyopathy | ||
| KCNJ11 (NM_000525.4): c.112A>G (p. Lys38Glu) | PM2, PP2, PM1, PP3 | KCNJ11:Variant is missing from all sub population sources.,KCNJ11:Missense variant in gene with disease commonly caused by missense variants,KCNJ11:There are no benign variants within 3 amino acids of this variant, and 2 pathogenic/0 benign variants within 6 amino acids of this variant.,KCNJ11:Gerp++ predicts conserved at this location,PhyloP predicts conserved at this location, Sift predicts this variant is: ’Damaging’,Pollyphen predicts that this variant is ’Damaging’ | Likely pathogenic | 0 | 0 | Heterozygous in patient, her sister and her 2 children | 600937: Sudden Cardiac Death | |||
| 3 | Hypertrophic obstructive cardiomyopathy | MYH7 (NM_000257.4): c.4066G>A (p. Glu1356Lys) | PM2, PP2, PM1, PS1, PP3 | MYH7:Variant is missing from all sub population sources., MYH7:Missense variant in gene with disease commonly caused by missense variants,MYH7:There are no benign variants within 3 amino acids of this variant, and 2 pathogenic/0 benign variants within 6 amino acids of this variant.,MYH7:The variant causes the same pathogenic mutation as a pathogenic variant in the same amino acid for the following sources: ClinVar.,MYH7:Gerp++ predicts conserved at this location,PhyloP predicts conserved at this location, Sift predicts this variant is: ’Damaging’,Pollyphen predicts that this variant is ’Damaging. | Pathogenic | 0 | 0 | rs727503246| | Heterozygous in patient and her son. | 160760: Cardiomyopathy |
| 4 | Dilated cardiomyopathy | MYPN (NM_001256267.1): c.65C>G (p. Ala22Gly) | BP4, BP1, PM2 | MYPN: MetaRNN = 0.034 is between 0.00692 and 0.108 ⇒ strong benign. MYPN:92 out of 122 non-VUS missense variants in gene MYPN are benign = 75.4% which is more than threshold of 33.1%. MYPN: gnomAD genomes homozygous allele count = 0 is less than 2 for AD/AR gene MYPN, good gnomAD genomes coverage = 30.9. gnomAD exomes homozygous allele count = 0 is less than 2 for AD/AR gene MYPN, good gnomAD exomes coverage = 72.5. | Likely benign | 1.67 × 104 | 0 | rs145142157 | Heterozygous in patient, her parents, and several siblings | 608517: Cardiomyopathy |
| 5 | Dilated cardiomyopathy | RBM20 (NM_001134363.3): c.19A>G (p. Met7Val) | PM2, PP2 | RBM20: Variant is missing from all sub population sources., RBM20: Missense variant in gene with disease commonly caused by missense variants | VUS/Likely Benign | 0 | 0 | Heterozygous in patient and her mother | 613171: Cardiomyopathy | |
| RBM20 (NM_001134363.3): c.1881-3C>T | BA1, BS2, PP3, BP7, BP6 | RBM20: Allele frequency is above 0.005 dominant threshold for source Annotate-gnomAD Exomes Variant Frequencies 2.1.1, BROAD., RBM20: Allele counts greater than 0 for dominant gene., RBM20: Gerp++ predicts conserved at this location, PhyloP predicts conserved at this location, RBM20:0 of 4 algorithms predict a disrupted splice site, RBM20: ClinVar Annotation has reputable ’Likely Benign’ classification | Likely benign | 9.98 × 104 | 0 | rs138436392 | Heterozygous in patient and her father | |||
| FLNC (NM_001458.5): c.3004C>T(p. Arg1002Trp) | PP2, PP3 | FLNC: Missense variant in gene with disease commonly caused by missense variants, FLNC: Gerp++ predicts conserved at this location, PhyloP predicts conserved at this location, sift predicts this variant is: ’Damaging’, Pollyphen predicts that this variant is ’Damaging’ | VUS/Weak Pathogenic | 2.64 × 105 | 0 | rs555764780 | Heterozygous in patient and her mother | 102565: Cardiomyopathy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mansoori, G.A.; Mahmeed, W.A.; Wani, S.; Salih, B.T.; Ansari, T.E.; Farook, F.; Farooq, Z.; Khair, H.; Zaręba, K.; Dhahouri, N.A.; et al. Introducing and Implementing Genetic Assessment in Cardio-Obstetrics Clinical Practice: Clinical and Genetic Workup of Patients with Cardiomyopathy. Int. J. Mol. Sci. 2023, 24, 9119. https://doi.org/10.3390/ijms24119119
Mansoori GA, Mahmeed WA, Wani S, Salih BT, Ansari TE, Farook F, Farooq Z, Khair H, Zaręba K, Dhahouri NA, et al. Introducing and Implementing Genetic Assessment in Cardio-Obstetrics Clinical Practice: Clinical and Genetic Workup of Patients with Cardiomyopathy. International Journal of Molecular Sciences. 2023; 24(11):9119. https://doi.org/10.3390/ijms24119119
Chicago/Turabian StyleMansoori, Ghadeera Al, Wael Al Mahmeed, Saleema Wani, Bashir Taha Salih, Tarek El Ansari, Fathima Farook, Zenab Farooq, Howaida Khair, Kornelia Zaręba, Nahid Al Dhahouri, and et al. 2023. "Introducing and Implementing Genetic Assessment in Cardio-Obstetrics Clinical Practice: Clinical and Genetic Workup of Patients with Cardiomyopathy" International Journal of Molecular Sciences 24, no. 11: 9119. https://doi.org/10.3390/ijms24119119