
Inhibition of p90RSK Ameliorates PDGF-BB-Mediated Phenotypic Change of Vascular Smooth Muscle Cell and Subsequent Hyperplasia of Neointima
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
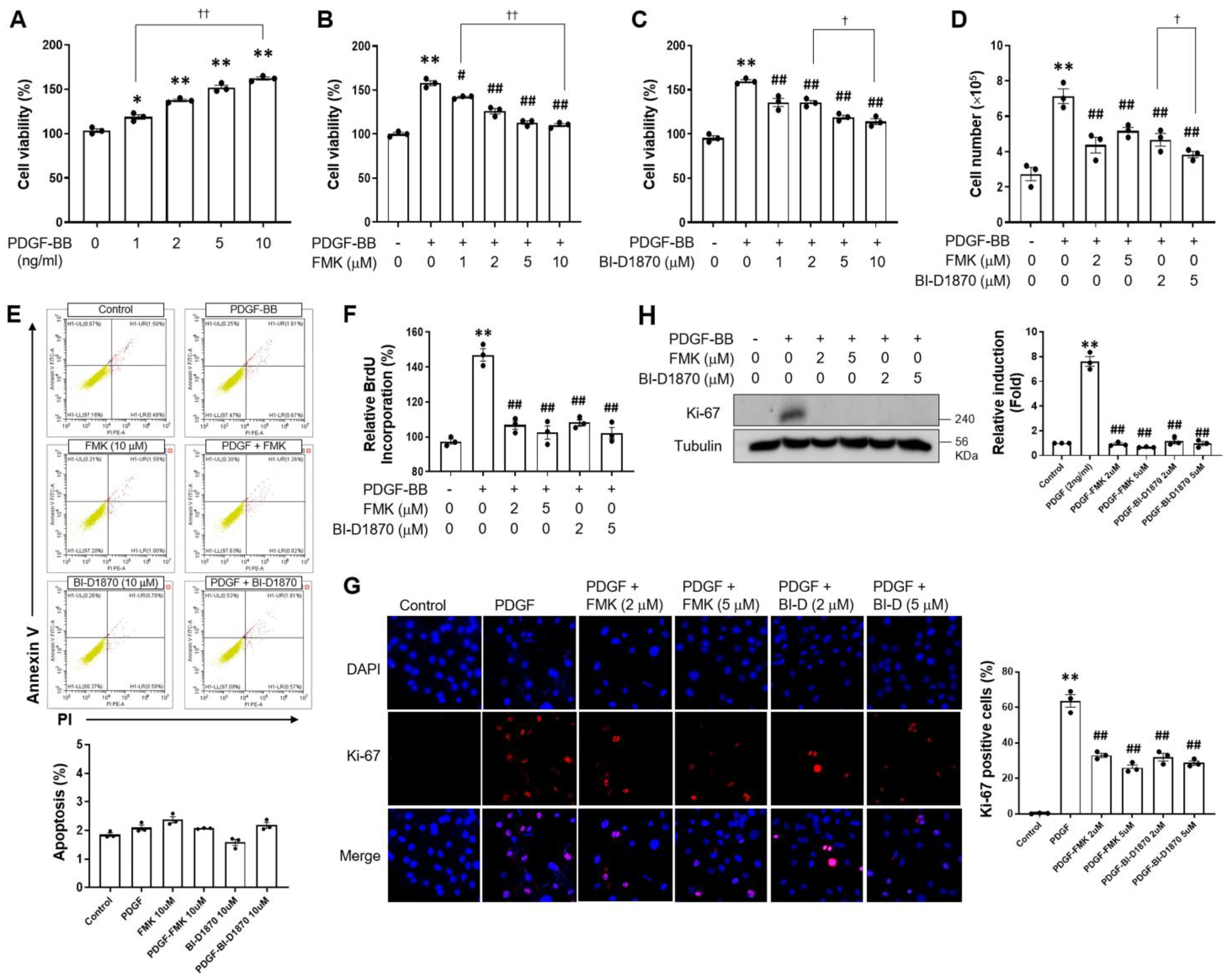
2.1. p90RSK Is Involved in PDGF-BB-Induced Cell Proliferation in VSMCs
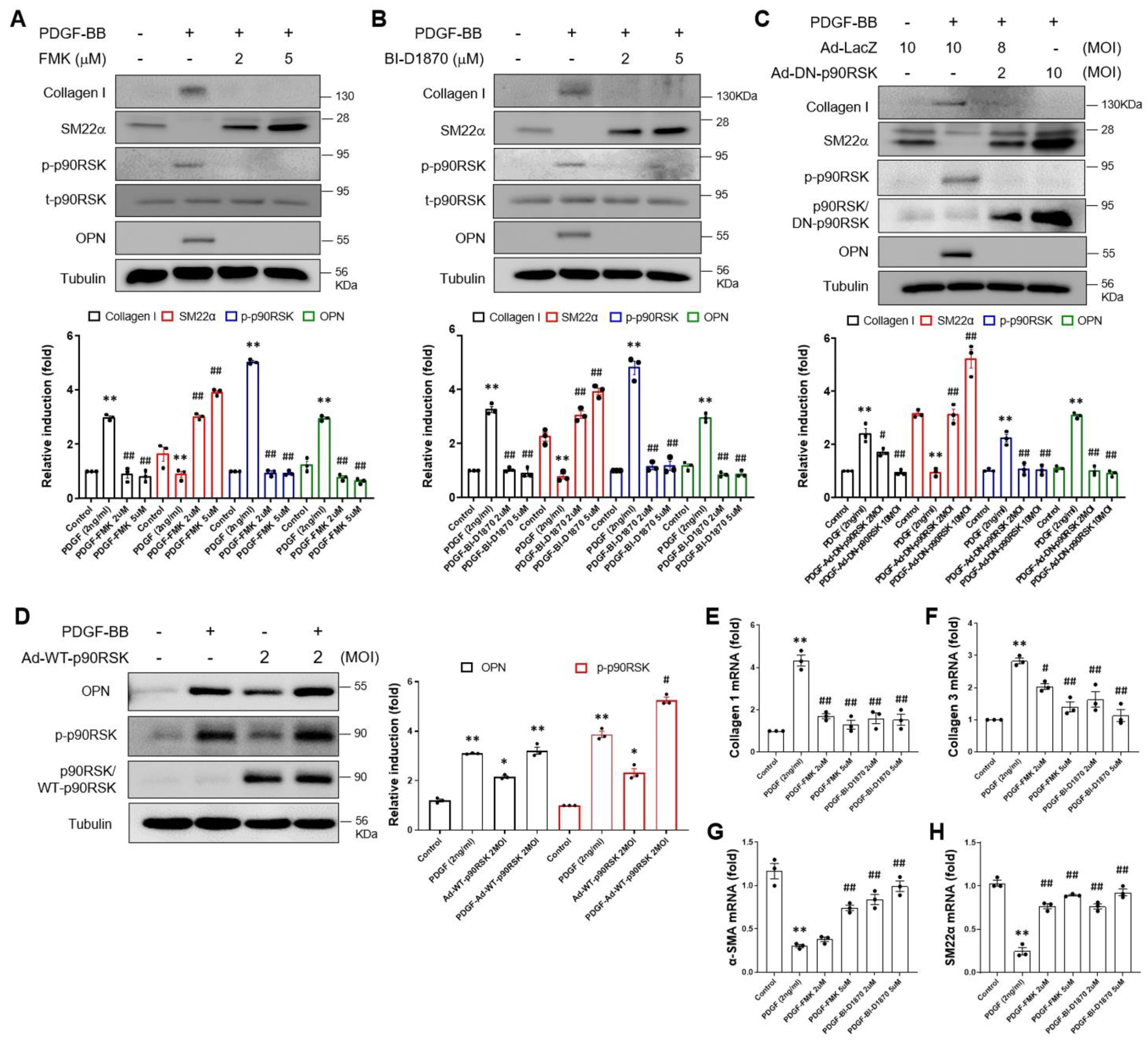
2.2. p90RSK Is Involved in PDGF-BB-Mediated Phenotypic Switching
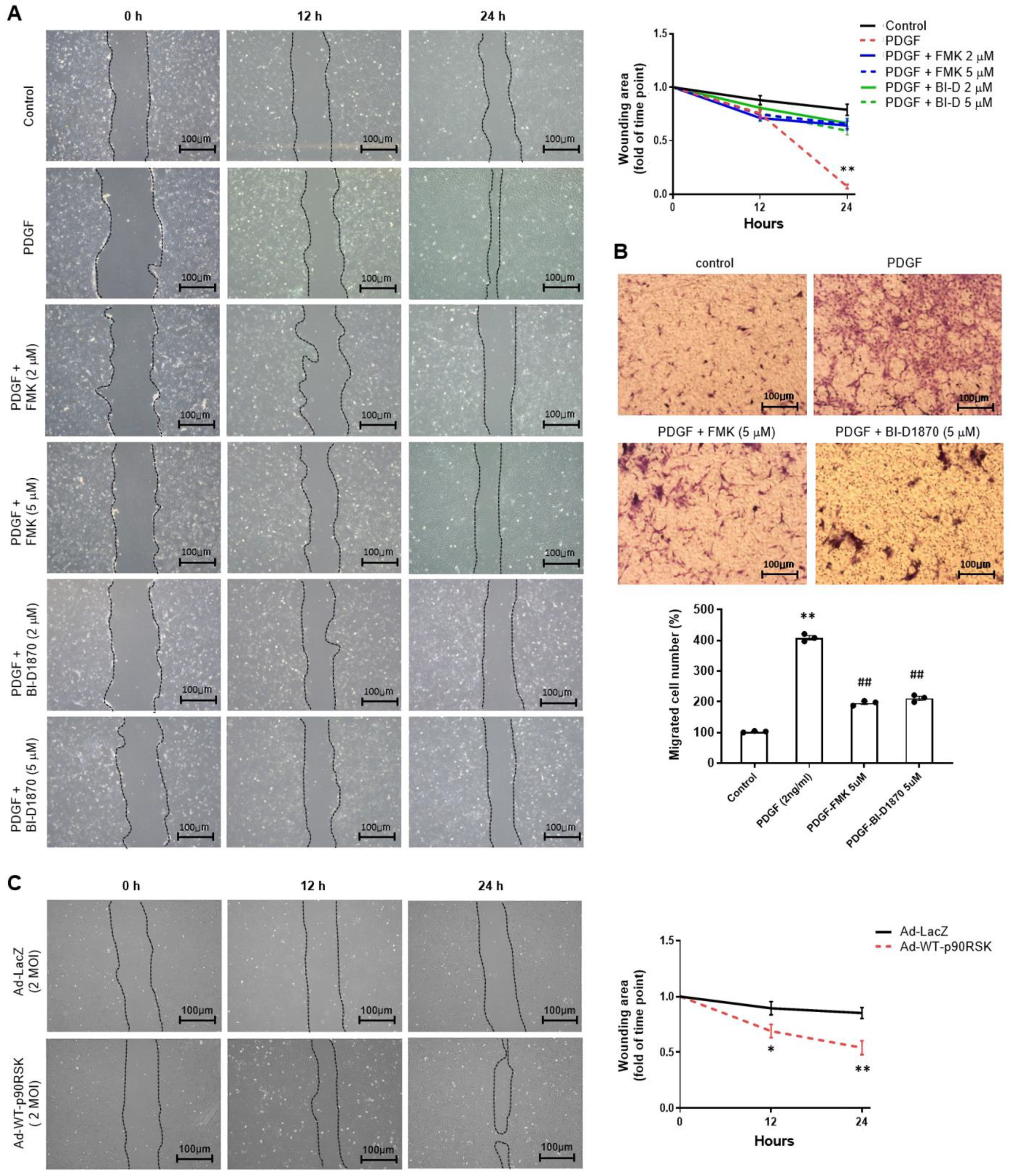
2.3. p90RSK Regulates PDGF-BB-Induced Migration of VSMCs
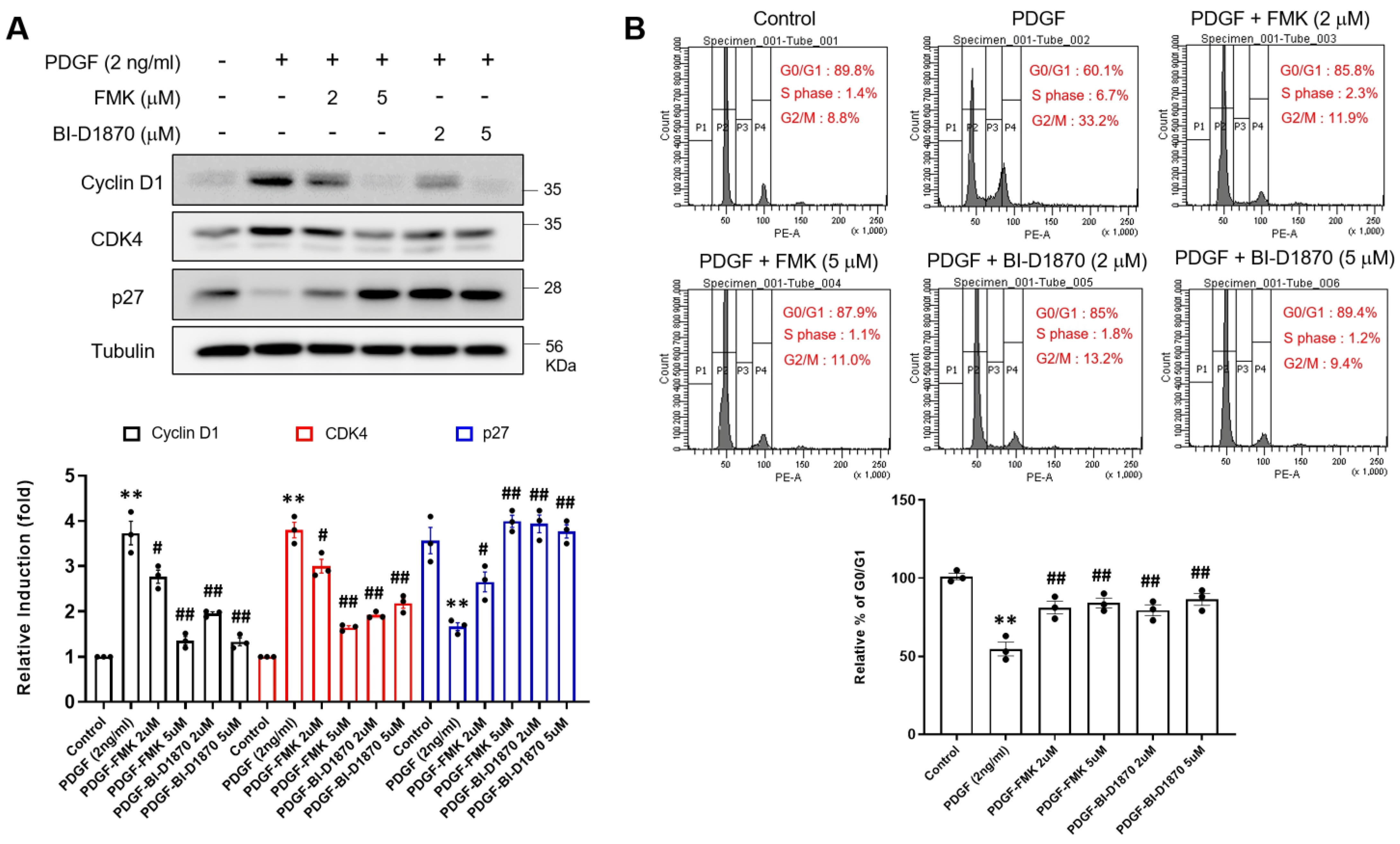
2.4. p90RSK Inhibition Prevents PDGF-BB-Induced Cell Cycle Progression of Rat Primary VSMCs
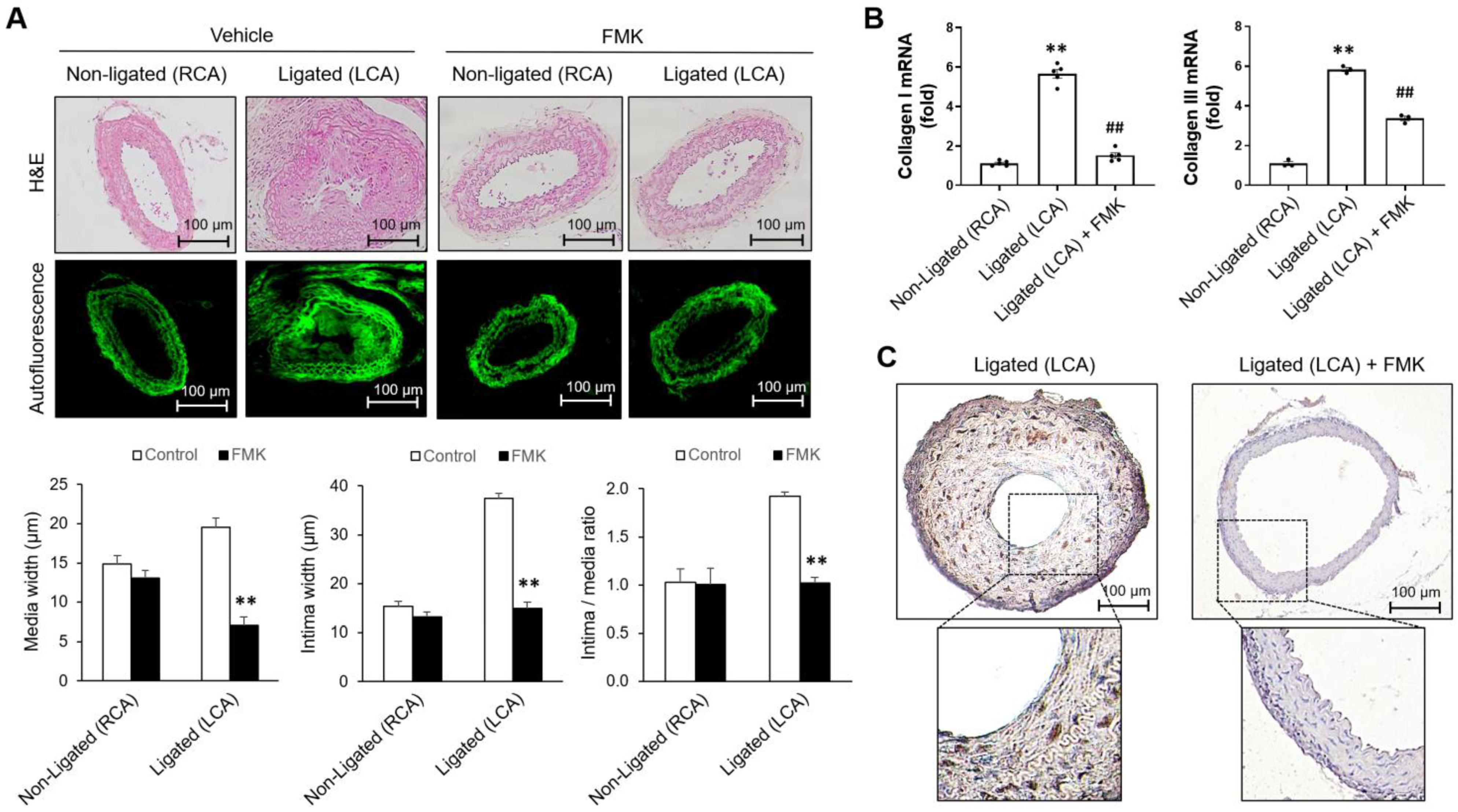
2.5. FMK Attenuates Neointimal Hyperplasia Induced by Carotid Artery Ligation
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Vascular Smooth Muscle Cell Culture
4.3. Animals
4.4. Western Blotting Assay
4.5. Quantitative Real-Time RT-PCR
4.6. MTT Assay
4.7. BrdU Incorporation Assay
4.8. Adenoviral Transduction
4.9. Ki-67 Immunofluorescent Staining
4.10. Flow Cytometric Assay
4.11. Annexin V-FITC Apoptosis
4.12. Cell Migration Assay
4.13. Carotid Artery Ligation Model
4.14. Histology
4.15. Immunohistochemistry
4.16. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, H.; Han, J.H.; Kim, S.; Kim, S.; Cho, D.H.; Woo, C.H. Anti-malarial Drugs Reduce Vascular Smooth Muscle Cell Proliferation via Activation of AMPK and Inhibition of Smad3 Signaling. J. Lipid Atheroscler. 2019, 8, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.-H.; Wang, N.; Zhao, D.-W.; Zheng, D.-L.; Zheng, L.; Xing, W.-J.; Ma, W.-J.; Bao, L.-Y.; Dong, J.; Zhang, T.-C. STAT3 Protein Regulates Vascular Smooth Muscle Cell Phenotypic Switch by Interaction with Myocardin. J. Biol. Chem. 2015, 290, 19641–19652. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Zhang, L.; Yao, L.-L.; Zheng, F.; Wang, L.; Yang, J.-Y.; Guo, L.-Y.; Li, X.-Y.; Yan, Y.-W.; Pan, Y.-M.; et al. S100B promotes injury-induced vascular remodeling through modulating smooth muscle phenotype. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2772–2782. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.-H.; Huang, L.; Zhang, X.; Zhang, P.; Zhang, S.-M.; Guan, H.; Zhang, Y.; Zhu, X.-Y.; Tian, S.; Deng, K.; et al. Mindin regulates vascular smooth muscle cell phenotype and prevents neointima formation. Clin. Sci. 2015, 129, 129–145. [Google Scholar] [CrossRef]
- Cohen, M.S.; Zhang, C.; Shokat, K.M.; Taunton, J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science 2005, 308, 1318–1321. [Google Scholar] [CrossRef]
- Hall, C.; Nelson, D.M.; Ye, X.; Baker, K.; DeCaprio, J.A.; Seeholzer, S.; Lipinski, M.; Adams, P.D. HIRA, the human homologue of yeast Hir1p and Hir2p, is a novel cyclin-cdk2 substrate whose expression blocks S-phase progression. Mol. Cell. Biol. 2001, 21, 1854–1865. [Google Scholar] [CrossRef]
- Adams, P.D. Regulation of the retinoblastoma tumor suppressor protein by cyclin/cdks. Biochim. Biophys. Acta 2001, 1471, M123–M133. [Google Scholar] [CrossRef]
- Bornfeldt, K.E.; Raines, E.W.; Nakano, T.; Graves, L.M.; Krebs, E.G.; Ross, R. Insulin-like growth factor-I and platelet-derived growth factor-BB induce directed migration of human arterial smooth muscle cells via signaling pathways that are distinct from those of proliferation. J. Clin. Invest. 1994, 93, 1266–1274. [Google Scholar] [CrossRef]
- Heldin, C.H.; Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 1999, 79, 1283–1316. [Google Scholar] [CrossRef]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis--An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G.; Libby, P.; Gersh, B.; Yellon, D.; Böhm, M.; Lopaschuk, G.; Opie, L. Cardiovascular remodelling in coronary artery disease and heart failure. Lancet 2014, 383, 1933–1943. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Owens, G.K. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc. Res. 2012, 95, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.B.; Benditt, E.P. sis (platelet-derived growth factor B chain) gene transcript levels are elevated in human atherosclerotic lesions compared to normal artery. Proc. Natl. Acad. Sci. USA 1987, 84, 1099–1103. [Google Scholar] [CrossRef]
- Rubin, K.; Tingström, A.; Hansson, G.K.; Larsson, E.; Rönnstrand, L.; Klareskog, L.; Claesson-Welsh, L.; Heldin, C.H.; Fellström, B.; Terracio, L. Induction of B-type receptors for platelet-derived growth factor in vascular inflammation: Possible implications for development of vascular proliferative lesions. Lancet 1988, 1, 1353–1356. [Google Scholar] [CrossRef]
- Tanizawa, S.; Ueda, M.; van der Loos, C.M.; van der Wal, A.C.; Becker, A.E. Expression of platelet derived growth factor B chain and beta receptor in human coronary arteries after percutaneous transluminal coronary angioplasty: An immunohistochemical study. Heart 1996, 75, 549–556. [Google Scholar] [CrossRef]
- Ueda, M.; Becker, A.E.; Kasayuki, N.; Kojima, A.; Morita, Y.; Tanaka, S. In situ detection of platelet-derived growth factor-A and -B chain mRNA in human coronary arteries after percutaneous transluminal coronary angioplasty. Am. J. Pathol. 1996, 149, 831–843. [Google Scholar]
- Ha, J.M.; Yun, S.J.; Kim, Y.W.; Jin, S.Y.; Lee, H.S.; Song, S.H.; Shin, H.K.; Bae, S.S. Platelet-derived growth factor regulates vascular smooth muscle phenotype via mammalian target of rapamycin complex 1. Biochem. Biophys. Res. Commun. 2015, 464, 57–62. [Google Scholar] [CrossRef]
- Shawky, N.M.; Segar, L. Sulforaphane inhibits platelet-derived growth factor-induced vascular smooth muscle cell proliferation by targeting mTOR/p70S6kinase signaling independent of Nrf2 activation. Pharmacol. Res. 2017, 119, 251–264. [Google Scholar] [CrossRef]
- Frödin, M.; Gammeltoft, S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol. Cell. Endocrinol. 1999, 151, 65–77. [Google Scholar] [CrossRef]
- Anjum, R.; Blenis, J. The RSK family of kinases: Emerging roles in cellular signalling. Nat. Rev. Mol. Cell Biol. 2008, 9, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Abe, J.; Okuda, M.; Huang, Q.; Yoshizumi, M.; Berk, B.C. Reactive oxygen species activate p90 ribosomal S6 kinase via Fyn and Ras. J. Biol. Chem. 2000, 275, 1739–1748. [Google Scholar] [CrossRef]
- da Costa-Pessoa, J.M.; Damasceno, R.S.; Machado, U.F.; Beloto-Silva, O.; Oliveira-Souza, M. High glucose concentration stimulates NHE-1 activity in distal nephron cells: The role of the Mek/Erk1/2/p90RSK and p38MAPK signaling pathways. Cell. Physiol. Biochem. 2014, 33, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Le, N.T.; Takei, Y.; Shishido, T.; Woo, C.H.; Chang, E.; Heo, K.S.; Lee, H.; Lu, Y.; Morrell, C.; Oikawa, M.; et al. p90RSK targets the ERK5-CHIP ubiquitin E3 ligase activity in diabetic hearts and promotes cardiac apoptosis and dysfunction. Circ. Res. 2012, 110, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Le, N.T.; Heo, K.S.; Takei, Y.; Lee, H.; Woo, C.H.; Chang, E.; McClain, C.; Hurley, C.; Wang, X.; Li, F.; et al. A crucial role for p90RSK-mediated reduction of ERK5 transcriptional activity in endothelial dysfunction and atherosclerosis. Circulation 2013, 127, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Morales-Ibanez, O.; Affò, S.; Rodrigo-Torres, D.; Blaya, D.; Millán, C.; Coll, M.; Perea, L.; Odena, G.; Knorpp, T.; Templin, M.F.; et al. Kinase analysis in alcoholic hepatitis identifies p90RSK as a potential mediator of liver fibrogenesis. Gut 2016, 65, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.; Ding, B.; Shishido, T.; Lerner-Marmarosh, N.; Wang, N.; Maekawa, N.; Berk, B.C.; Takeishi, Y.; Yan, C.; Blaxall, B.C.; et al. Role of p90 ribosomal S6 kinase-mediated prorenin-converting enzyme in ischemic and diabetic myocardium. Circulation 2006, 113, 1787–1798. [Google Scholar] [CrossRef]
- Lin, L.; White, S.A.; Hu, K. Role of p90RSK in Kidney and Other Diseases. Int. J. Mol. Sci. 2019, 20, 972. [Google Scholar] [CrossRef]
- Smith, J.A.; Poteet-Smith, C.E.; Xu, Y.; Errington, T.M.; Hecht, S.M.; Lannigan, D.A. Identification of the first specific inhibitor of p90 ribosomal S6 kinase (RSK) reveals an unexpected role for RSK in cancer cell proliferation. Cancer Res. 2005, 65, 1027–1034. [Google Scholar] [CrossRef]
- Sapkota, G.P.; Cummings, L.; Newell, F.S.; Armstrong, C.; Bain, J.; Frodin, M.; Grauert, M.; Hoffmann, M.; Schnapp, G.; Steegmaier, M.; et al. BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem. J. 2007, 401, 29–38. [Google Scholar] [CrossRef]
- Louis, S.F.; Zahradka, P. Vascular smooth muscle cell motility: From migration to invasion. Exp. Clin. Cardiol. 2010, 15, e75–e85. [Google Scholar] [PubMed]
- Rudijanto, A. The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis. Acta Med. Indones. 2007, 39, 86–93. [Google Scholar] [PubMed]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Vascular smooth muscle cell in atherosclerosis. Acta Physiol. 2015, 214, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Kim, S.; Kim, S.; Lee, H.; Park, S.Y.; Woo, C.H. FMK, an Inhibitor of p90RSK, Inhibits High Glucose-Induced TXNIP Expression via Regulation of ChREBP in Pancreatic β Cells. Int. J. Mol. Sci. 2019, 20, 4424. [Google Scholar] [CrossRef]
- Mao, X.; Debenedittis, P.; Sun, Y.; Chen, J.; Yuan, K.; Jiao, K.; Chen, Y. Vascular smooth muscle cell Smad4 gene is important for mouse vascular development. Arter. Thromb. Vasc. Biol. 2012, 32, 2171–2177. [Google Scholar] [CrossRef]
- Rzucidlo, E.M.; Martin, K.A.; Powell, R.J. Regulation of vascular smooth muscle cell differentiation. J. Vasc. Surg. 2007, 45, A25–A32. [Google Scholar] [CrossRef]
- Dalby, K.N.; Morrice, N.; Caudwell, F.B.; Avruch, J.; Cohen, P. Identification of regulatory phosphorylation sites in mitogen-activated protein kinase (MAPK)-activated protein kinase-1a/p90rsk that are inducible by MAPK. J. Biol. Chem. 1998, 273, 1496–1505. [Google Scholar] [CrossRef]
- Clark, D.E.; Errington, T.M.; Smith, J.A.; Frierson, H.F., Jr.; Weber, M.J.; Lannigan, D.A. The serine/threonine protein kinase, p90 ribosomal S6 kinase, is an important regulator of prostate cancer cell proliferation. Cancer Res. 2005, 65, 3108–3116. [Google Scholar] [CrossRef]
- Gerthoffer, W.T. Mechanisms of vascular smooth muscle cell migration. Circ. Res. 2007, 100, 607–621. [Google Scholar] [CrossRef]
- Hu, W.; Huang, Y. Targeting the platelet-derived growth factor signalling in cardiovascular disease. Clin. Exp. Pharmacol. Physiol. 2015, 42, 1221–1224. [Google Scholar] [CrossRef]
- Hantschel, O. Unexpected off-targets and paradoxical pathway activation by kinase inhibitors. ACS Chem. Biol. 2015, 10, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Stathopoulou, K.; Schobesberger, S.; Bork, N.I.; Sprenger, J.U.; Perera, R.K.; Sotoud, H.; Geertz, B.; David, J.P.; Christ, T.; Nikolaev, V.O.; et al. Divergent off-target effects of RSK N-terminal and C-terminal kinase inhibitors in cardiac myocytes. Cell. Signal. 2019, 63, 109362. [Google Scholar] [CrossRef] [PubMed]
- Neise, D.; Sohn, D.; Stefanski, A.; Goto, H.; Inagaki, M.; Wesselborg, S.; Budach, W.; Stühler, K.; Jänicke, R.U. The p90 ribosomal S6 kinase (RSK) inhibitor BI-D1870 prevents gamma irradiation-induced apoptosis and mediates senescence via RSK- and p53-independent accumulation of p21WAF1/CIP1. Cell Death Dis. 2013, 4, e859. [Google Scholar] [CrossRef] [PubMed]
- Pacurari, M.; Kafoury, R.; Tchounwou, P.B.; Ndebele, K. The Renin-Angiotensin-aldosterone system in vascular inflammation and remodeling. Int. J. Inflam. 2014, 2014, 689360. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Guo, Y.; Zhu, T.; Zhang, J.; Ma, P.X.; Chen, Y.E. Yap1 protein regulates vascular smooth muscle cell phenotypic switch by interaction with myocardin. J. Biol. Chem. 2012, 287, 14598–14605. [Google Scholar] [CrossRef]
- Han, J.H.; Lee, S.G.; Jung, S.H.; Lee, J.J.; Park, H.S.; Kim, Y.H.; Myung, C.S. Sesamin Inhibits PDGF-Mediated Proliferation of Vascular Smooth Muscle Cells by Upregulating p21 and p27. J. Agric. Food Chem. 2015, 63, 7317–7325. [Google Scholar] [CrossRef]
- Hwang, A.R.; Nam, J.O.; Kang, Y.J. Fluvastatin inhibits advanced glycation end products-induced proliferation, migration, and extracellular matrix accumulation in vascular smooth muscle cells by targeting connective tissue growth factor. Korean J. Physiol. Pharmacol. 2018, 22, 193–201. [Google Scholar] [CrossRef]
- Jaminon, A.; Reesink, K.; Kroon, A.; Schurgers, L. The Role of Vascular Smooth Muscle Cells in Arterial Remodeling: Focus on Calcification-Related Processes. Int. J. Mol. Sci. 2019, 20, 5694. [Google Scholar] [CrossRef]
- Lacolley, P.; Regnault, V.; Segers, P.; Laurent, S. Vascular Smooth Muscle Cells and Arterial Stiffening: Relevance in Development, Aging, and Disease. Physiol. Rev. 2017, 97, 1555–1617. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Herman, I.M.; Castellot, J.J., Jr. Regulation of vascular smooth muscle cell growth by endothelial-synthesized extracellular matrices. Arteriosclerosis 1987, 7, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Castellot, J.J., Jr.; Pukac, L.A.; Caleb, B.L.; Wright, T.C., Jr.; Karnovsky, M.J. Heparin selectively inhibits a protein kinase C-dependent mechanism of cell cycle progression in calf aortic smooth muscle cells. J. Cell Biol. 1989, 109 Pt 1, 3147–3155. [Google Scholar] [CrossRef]
- Nigro, P.; Abe, J.; Woo, C.H.; Satoh, K.; McClain, C.; O’Dell, M.R.; Lee, H.; Lim, J.H.; Li, J.D.; Heo, K.S.; et al. PKCzeta decreases eNOS protein stability via inhibitory phosphorylation of ERK5. Blood 2010, 116, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Nam, D.; Ni, C.W.; Rezvan, A.; Suo, J.; Budzyn, K.; Llanos, A.; Harrison, D.G.; Giddens, D.P.; Jo, H. A model of disturbed flow-induced atherosclerosis in mouse carotid artery by partial ligation and a simple method of RNA isolation from carotid endothelium. J. Vis. Exp. 2010, 40, 1861. [Google Scholar]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, A.-R.; Lee, H.-J.; Kim, S.; Park, S.-H.; Woo, C.-H. Inhibition of p90RSK Ameliorates PDGF-BB-Mediated Phenotypic Change of Vascular Smooth Muscle Cell and Subsequent Hyperplasia of Neointima. Int. J. Mol. Sci. 2023, 24, 8094. https://doi.org/10.3390/ijms24098094
Hwang A-R, Lee H-J, Kim S, Park S-H, Woo C-H. Inhibition of p90RSK Ameliorates PDGF-BB-Mediated Phenotypic Change of Vascular Smooth Muscle Cell and Subsequent Hyperplasia of Neointima. International Journal of Molecular Sciences. 2023; 24(9):8094. https://doi.org/10.3390/ijms24098094
Chicago/Turabian StyleHwang, Ae-Rang, Hee-Jung Lee, Suji Kim, Seong-Hee Park, and Chang-Hoon Woo. 2023. "Inhibition of p90RSK Ameliorates PDGF-BB-Mediated Phenotypic Change of Vascular Smooth Muscle Cell and Subsequent Hyperplasia of Neointima" International Journal of Molecular Sciences 24, no. 9: 8094. https://doi.org/10.3390/ijms24098094