
Identification of Potential Biomarkers for Group I Pulmonary Hypertension Based on Machine Learning and Bioinformatics Analysis


Abstract
:1. Introduction
2. Results
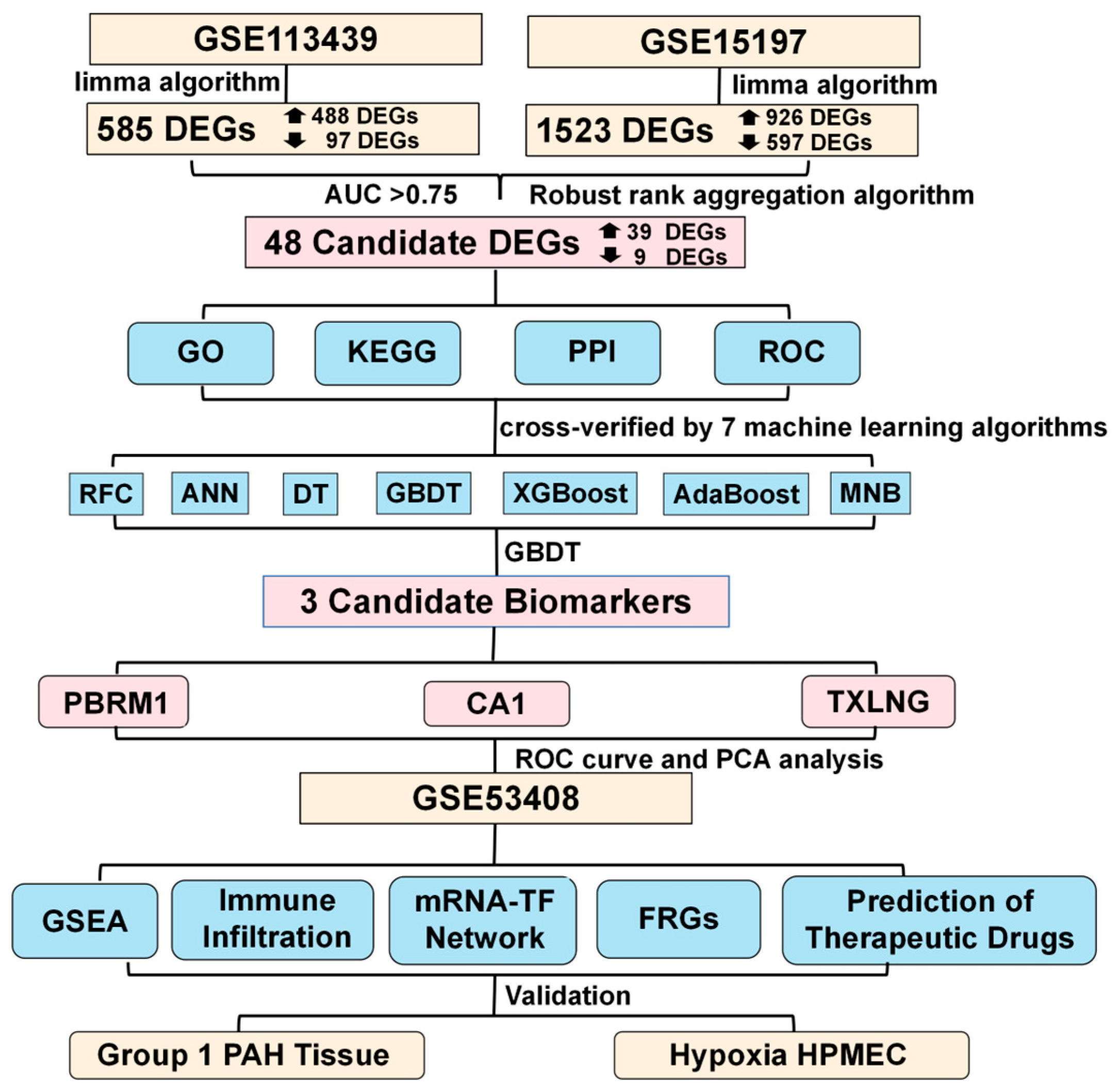
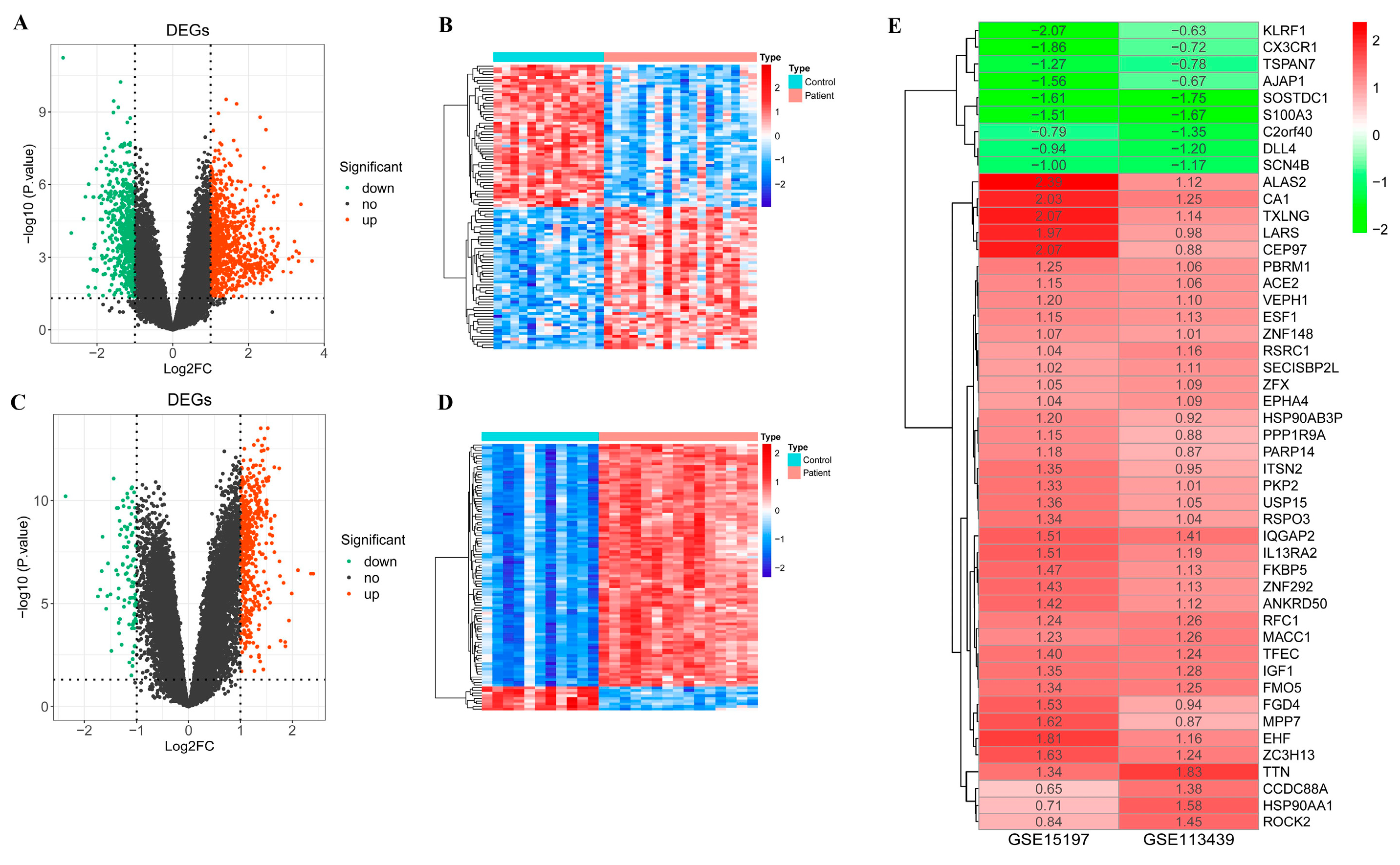
2.1. Differentially Expressed Genes between PAH Patient and Control Samples
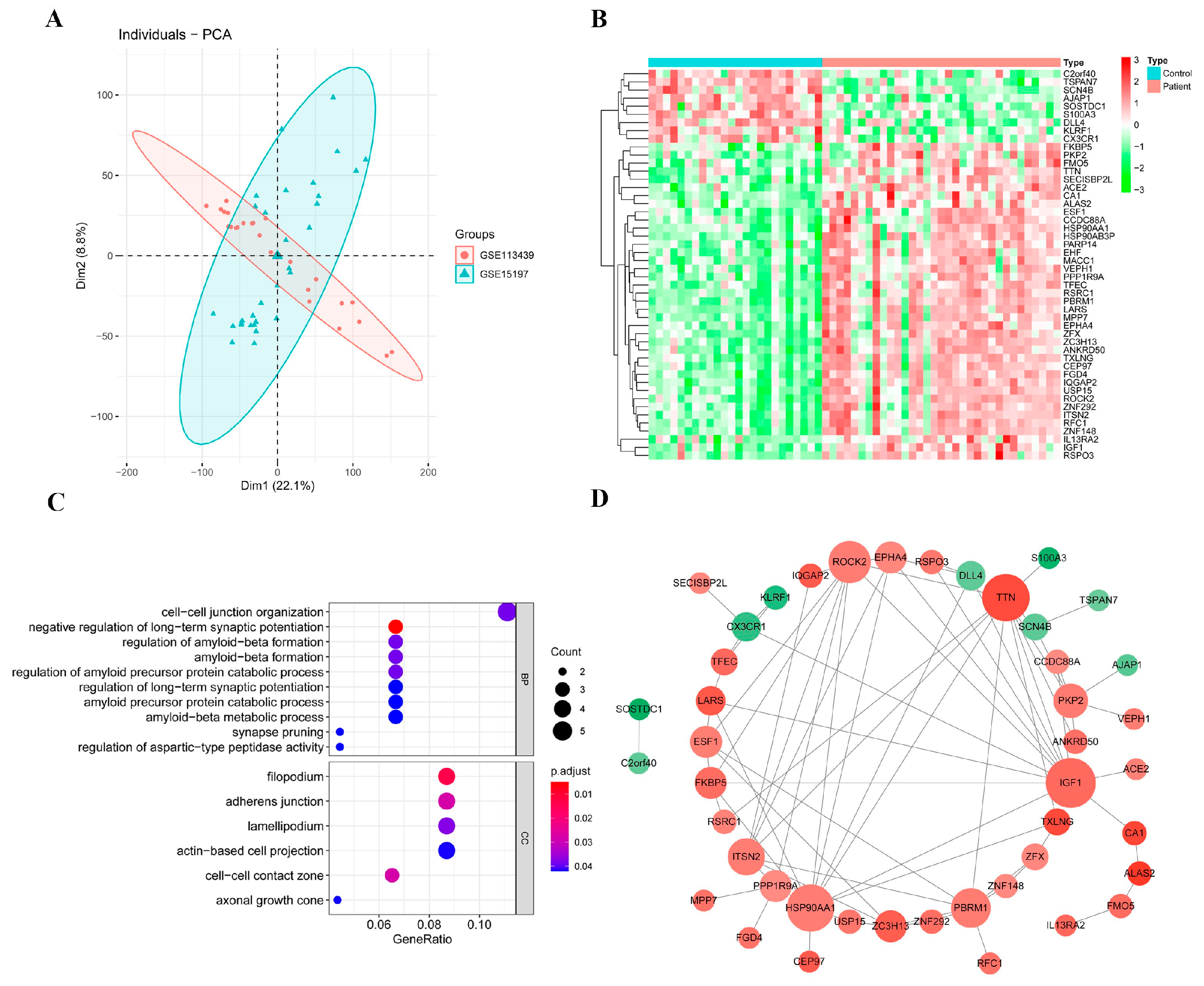
2.2. Candidate DEGs
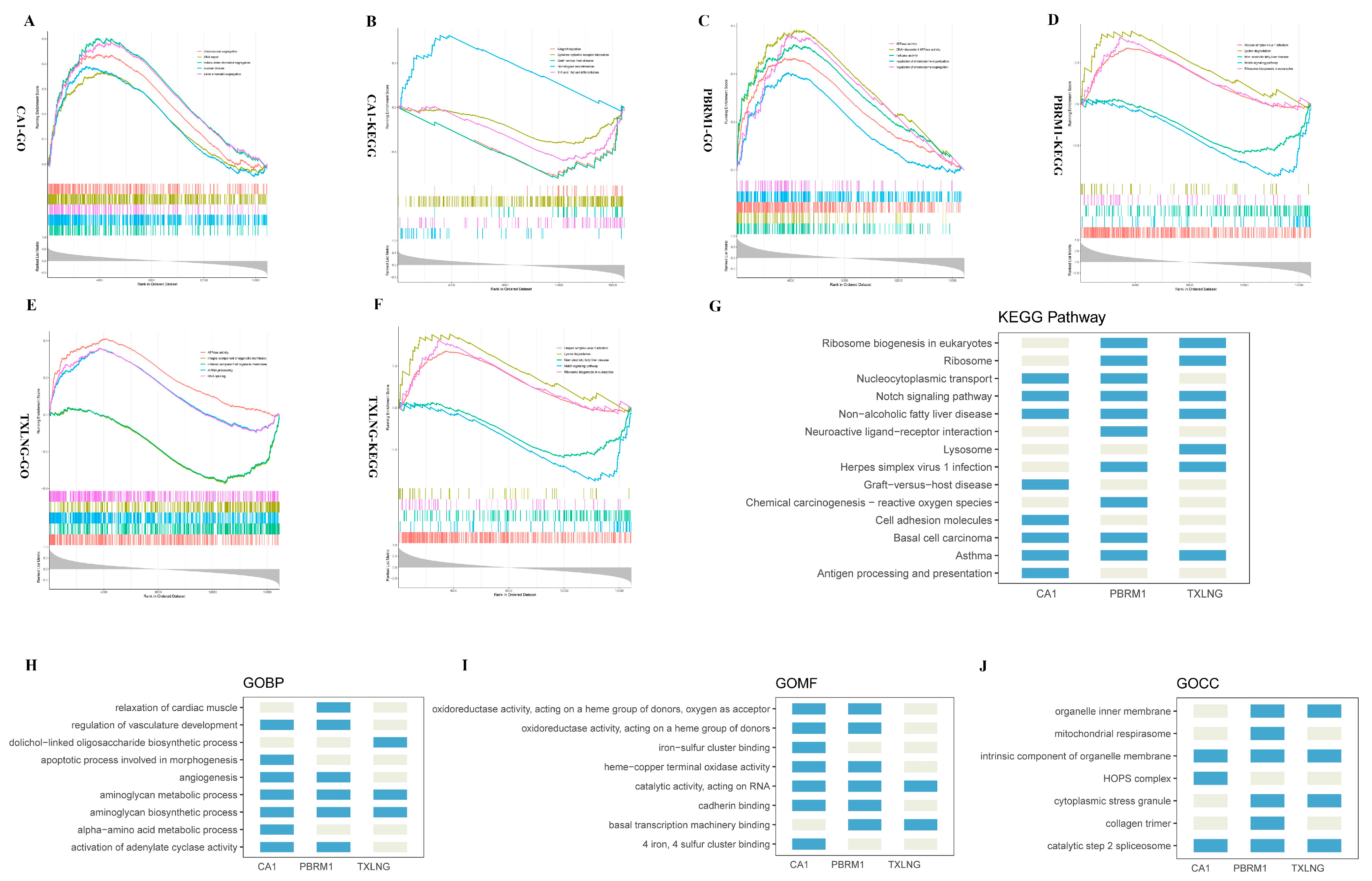
2.3. Functional Enrichment Analysis of Candidate DEGs
2.4. Protein–Protein Interaction (PPI) Network of Candidate DEGs
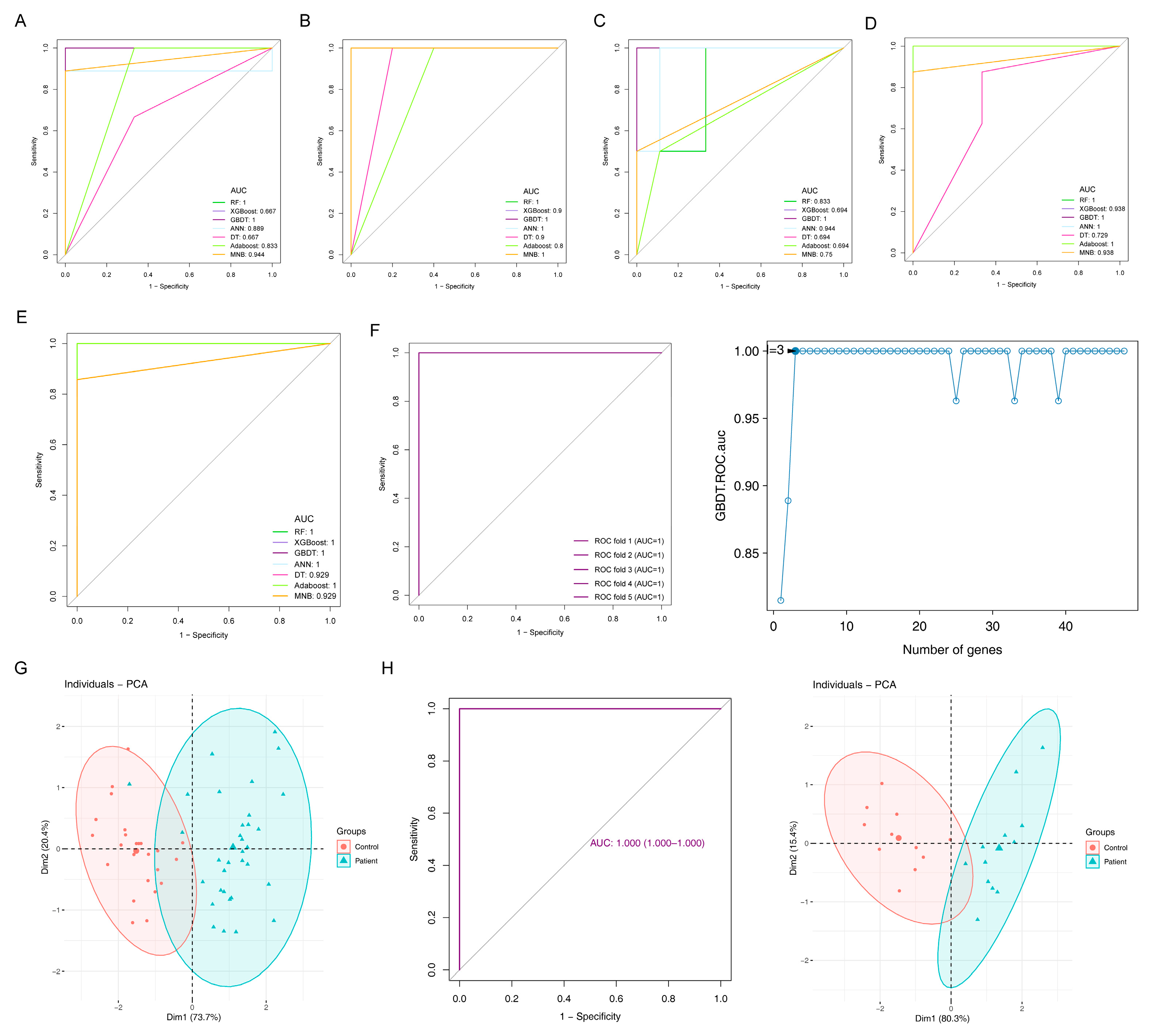
2.5. Diagnostic Models and Potential Biomarkers
2.6. Functional Enrichment Analysis of the Three Biomarkers
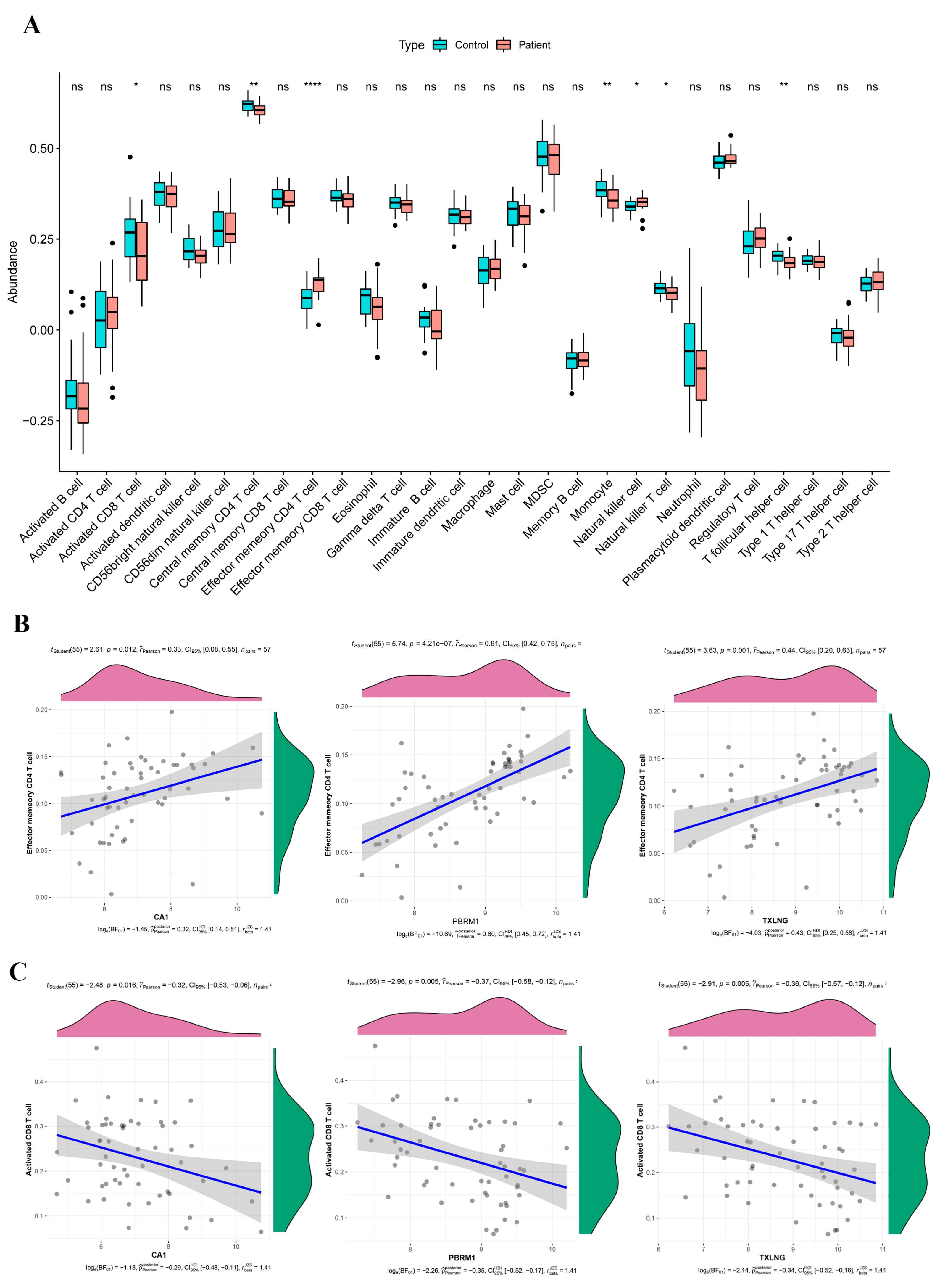
2.7. Immune Infiltration and Its Relation to the Biomarkers
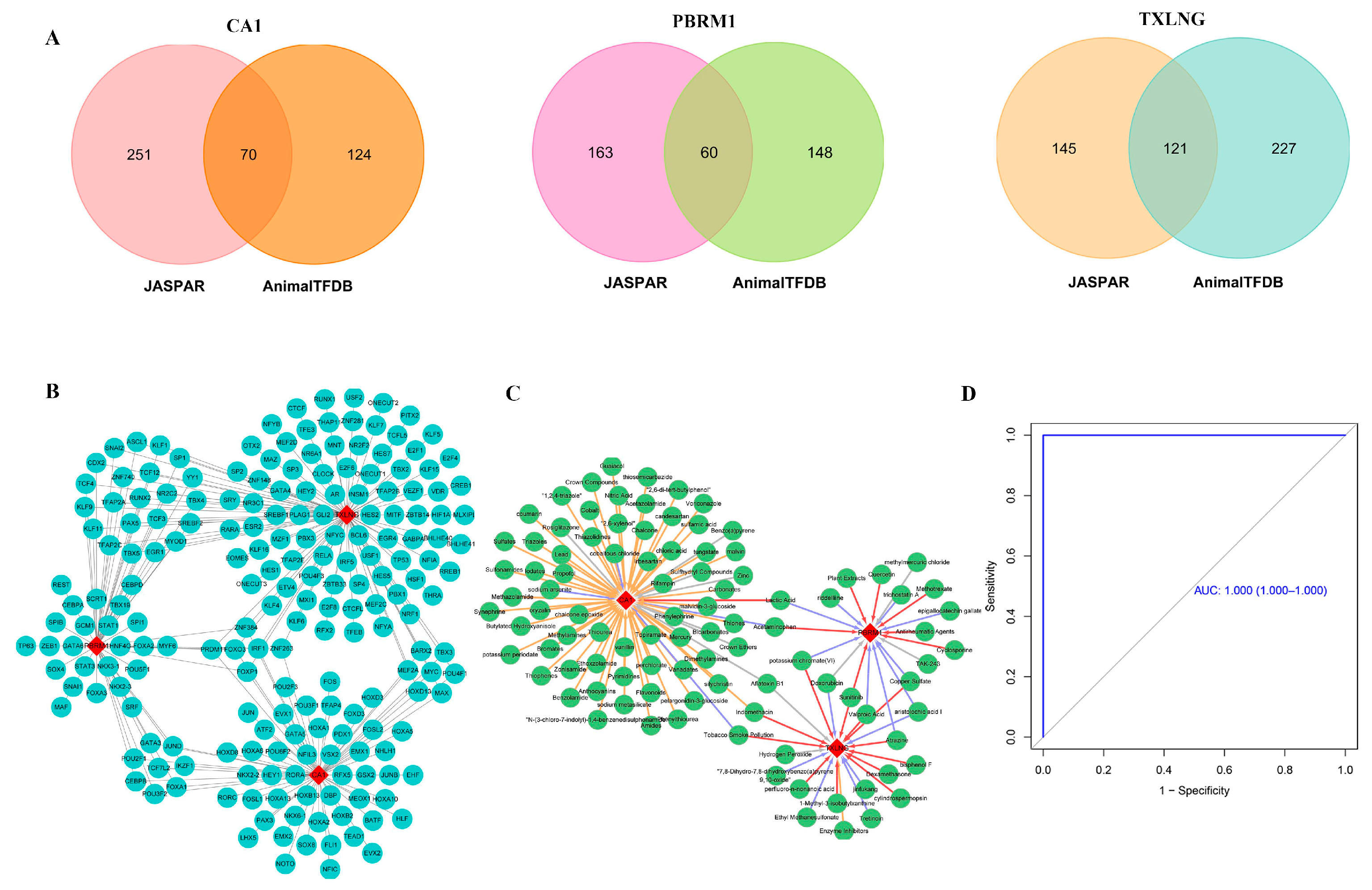
2.8. Regulatory Network of Biomarker-TFs and Predicted Therapeutic Drugs
2.9. Correlation between Biomarkers and FRGs
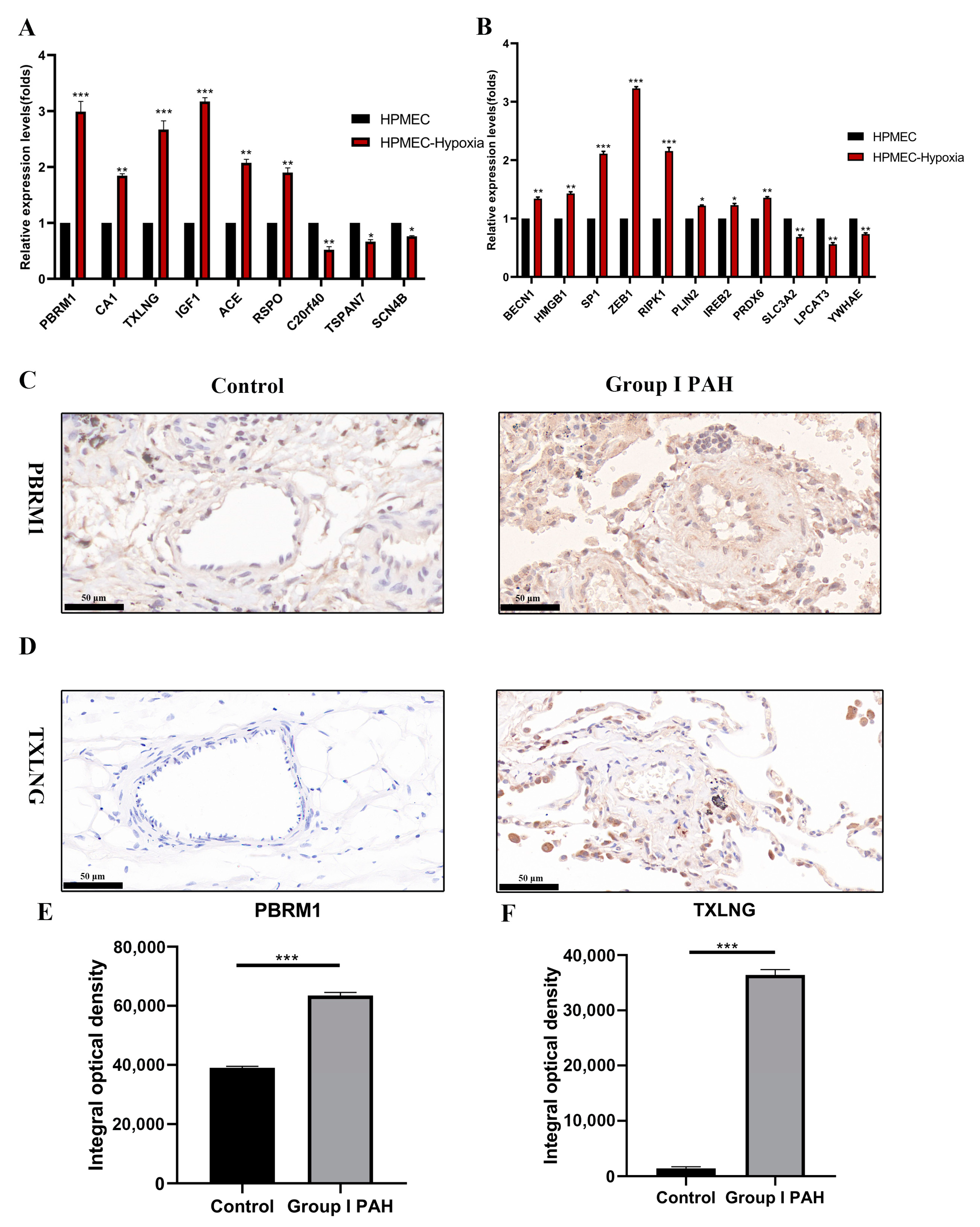
2.10. Differential Expressions of Potential Biomarkers and FRGs in PAH Were Experimentally Confirmed
3. Discussion
4. Methods
4.1. Gene Expression Data
4.2. Screening for Differentially Expressed Genes (DEGs)
4.3. Functional Enrichment Analysis and Protein–Protein Interaction (PPI) Network Construction
4.4. Identification of Candidate DEGs with Diagnostic Value
4.5. Construction and Validation of Diagnostic Model to Screen for Potential Biomarkers
4.6. Exploration for the Potential Regulatory Mechanisms of Potential Biomarkers through a Series of Bioinformatic Analyses
4.6.1. Single-Gene Gene Set Enrichment Analysis (GSEA)
4.6.2. Analysis of Immune Cell Infiltration
4.6.3. Construction of Biomarker-Transcription Factor Regulatory Network
4.6.4. Prediction of Therapeutic Drugs Targeting the Potential Biomarkers
4.7. Verifying the Expression of Potential Biomarkers Using in Vivo and In Vitro Experiments
4.7.1. Cell Culture and Hypoxia Culture Conditions
4.7.2. Quantitative Reverse Transcription PCR (qRT-PCR)
4.7.3. Human Lung Tissue Samples
4.7.4. Immunohistochemistry (IHC)
4.8. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barst, R.J.; McGoon, M.; Torbicki, A.; Sitbon, O.; Krowka, M.J.; Olschewski, H.; Gaine, S. Diagnosis and differential assessment of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, 40S–47S. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, S.; Feldman, J.; McLaughlin, V.V.; Rischard, F.; Lange, T.J.; White, R.J.; Peacock, A.J.; Gerhardt, F.; Ebrahimi, R.; Brooks, G.; et al. Selonsertib in adults with pulmonary arterial hypertension (ARROW): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Respir. Med. 2022, 10, 35–46. [Google Scholar] [CrossRef]
- Sahay, S. Evaluation and classification of pulmonary arterial hypertension. J. Thorac. Dis. 2019, 11, S1789–S1799. [Google Scholar] [CrossRef] [PubMed]
- Nies, M.K.; Yang, J.; Griffiths, M.; Damico, R.; Zhu, J.; Vaydia, D.; Fu, Z.; Brandal, S.; Austin, E.D.; Ivy, D.D.; et al. Proteomics discovery of pulmonary hypertension biomarkers: Insulin-like growth factor binding proteins are associated with disease severity. Pulm. Circ. 2022, 12, e12039. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, C.; Lin, Y.; Zhang, P. Identification of Crucial Hub Genes and Differential T Cell Infiltration in Idiopathic Pulmonary Arterial Hypertension Using Bioinformatics Strategies. Front. Mol. Biosci. 2022, 9, 800888. [Google Scholar] [CrossRef]
- Kulasingam, V.; Diamandis, E.P. Strategies for discovering novel cancer biomarkers through utilization of emerging technologies. Nat. Clin. Pract. Oncol. 2008, 5, 588–599. [Google Scholar] [CrossRef]
- Ching, T.; Himmelstein, D.S.; Beaulieu-Jones, B.K.; Kalinin, A.A.; Do, B.T.; Way, G.P.; Ferrero, E.; Agapow, P.M.; Zietz, M.; Hoffman, M.M.; et al. Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface 2018, 15, 20170387. [Google Scholar] [CrossRef]
- Libbrecht, M.W.; Noble, W.S. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 2015, 16, 321–332. [Google Scholar] [CrossRef]
- Bi, Q.; Goodman, K.E.; Kaminsky, J.; Lessler, J. What is Machine Learning? A Primer for the Epidemiologist. Am. J. Epidemiol. 2019, 188, 2222–2239. [Google Scholar] [CrossRef]
- Xiao, Y.; Wu, J.; Lin, Z.; Zhao, X. A deep learning-based multi-model ensemble method for cancer prediction. Comput. Methods Programs Biomed. 2018, 153, 1–9. [Google Scholar] [CrossRef]
- Liu, P.; Tseng, G.; Wang, Z.; Huang, Y.; Randhawa, P. Diagnosis of T-cell-mediated kidney rejection in formalin-fixed, paraffin-embedded tissues using RNA-Seq-based machine learning algorithms. Hum. Pathol. 2019, 84, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Alanni, R.; Hou, J.; Azzawi, H.; Xiang, Y. A novel gene selection algorithm for cancer classification using microarray datasets. BMC Med. Genom. 2019, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.; Hu, Z.; Kang, Z.; Zheng, Z. Based on multiple machine learning to identify the ENO2 as diagnosis biomarkers of glaucoma. BMC Ophthalmol. 2022, 22, 155. [Google Scholar] [CrossRef] [PubMed]
- Li, W.H.; Han, J.R.; Ren, P.P.; Xie, Y.; Jiang, D.Y. Exploration of the mechanism of Zisheng Shenqi decoction against gout arthritis using network pharmacology. Comput. Biol. Chem. 2021, 90, 107358. [Google Scholar] [CrossRef]
- Wang, W.; Jiang, Z.; Zhang, D.; Fu, L.; Wan, R.; Hong, K. Comparative Transcriptional Analysis of Pulmonary Arterial Hypertension Associated with Three Different Diseases. Front. Cell Dev. Biol. 2021, 9, 672159. [Google Scholar] [CrossRef]
- Ma, Y.; Chen, S.S.; Feng, Y.Y.; Wang, H.L. Identification of novel biomarkers involved in pulmonary arterial hypertension based on multiple-microarray analysis. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef]
- Duan, T.; Kuang, Z.; Wang, J.; Ma, Z. GBDTLRL2D Predicts LncRNA-Disease Associations Using MetaGraph2Vec and K-Means Based on Heterogeneous Network. Front. Cell Dev. Biol. 2021, 9, 753027. [Google Scholar] [CrossRef]
- Yang, Z.; Zhou, D.; Li, H.; Cai, X.; Liu, W.; Wang, L.; Chang, H.; Li, M.; Xiao, X. The genome-wide risk alleles for psychiatric disorders at 3p21.1 show convergent effects on mRNA expression, cognitive function, and mushroom dendritic spine. Mol. Psychiatry 2020, 25, 48–66. [Google Scholar] [CrossRef]
- Perez-Pena, J.; Paez, R.; Nieto-Jimenez, C.; Sanchez, V.C.; Galan-Moya, E.M.; Pandiella, A.; Gyorffy, B.; Ocana, A. Mapping Bromodomains in breast cancer and association with clinical outcome. Sci. Rep. 2019, 9, 5734. [Google Scholar] [CrossRef]
- Huang, X.; Gao, X.; Diaz-Trelles, R.; Ruiz-Lozano, P.; Wang, Z. Coronary development is regulated by ATP-dependent SWI/SNF chromatin remodeling component BAF180. Dev. Biol. 2008, 319, 258–266. [Google Scholar] [CrossRef]
- Magheru, C.; Magheru, S.; Coltau, M.; Hoza, A.; Moldovan, C.; Sachelarie, L.; Gradinaru, I.; Hurjui, L.L.; Marc, F.; Farcas, D.M. Antiepileptic Drugs and Their Dual Mechanism of Action on Carbonic Anhydrase. J. Clin. Med. 2022, 11, 2614. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Chen, H. Aberrance of Zinc Metalloenzymes-Induced Human Diseases and Its Potential Mechanisms. Nutrients 2021, 13, 4456. [Google Scholar] [CrossRef]
- Tunali, I.; Tan, Y.; Gray, J.E.; Katsoulakis, E.; Eschrich, S.A.; Saller, J.; Aerts, H.; Boyle, T.; Qi, J.; Guvenis, A.; et al. Hypoxia-Related Radiomics and Immunotherapy Response: A Multicohort Study of Non-Small Cell Lung Cancer. JNCI Cancer Spectr. 2021, 5, pkab048. [Google Scholar] [CrossRef] [PubMed]
- Said, M.F.; George, R.F.; Petreni, A.; Supuran, C.T.; Mohamed, N.M. Synthesis, molecular modelling and QSAR study of new N-phenylacetamide-2-oxoindole benzensulfonamide conjugates as carbonic anhydrase inhibitors with antiproliferative activity. J. Enzyme Inhib. Med. Chem. 2022, 37, 701–717. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic Anhydrase Inhibition and the Management of Hypoxic Tumors. Metabolites 2017, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Alim, Z. 1H-indazole molecules reduced the activity of human erythrocytes carbonic anhydrase I and II isoenzymes. J. Biochem. Mol. Toxicol. 2018, 32, e22194. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.B.; Lu, X.K.; Zhang, X.; Li, Z.G.; Li, C.X. Carbonic anhydrase 1 is a promising biomarker for early detection of non-small cell lung cancer. Tumour. Biol. 2016, 37, 553–559. [Google Scholar] [CrossRef]
- Nigro, E.; Imperlini, E.; Scudiero, O.; Monaco, M.L.; Polito, R.; Mazzarella, G.; Orru, S.; Bianco, A.; Daniele, A. Differentially expressed and activated proteins associated with non small cell lung cancer tissues. Respir. Res. 2015, 16, 74. [Google Scholar] [CrossRef]
- Hotokezaka, Y.; Katayama, I.; van Leyen, K.; Nakamura, T. GSK-3beta-dependent downregulation of gamma-taxilin and alphaNAC merge to regulate ER stress responses. Cell Death Dis. 2015, 6, e1719. [Google Scholar] [CrossRef]
- Hotokezaka, Y.; van Leyen, K.; Lo, E.H.; Beatrix, B.; Katayama, I.; Jin, G.; Nakamura, T. alphaNAC depletion as an initiator of ER stress-induced apoptosis in hypoxia. Cell Death Differ. 2009, 16, 1505–1514. [Google Scholar] [CrossRef] [PubMed]
- Voelkel, N.F.; Tamosiuniene, R.; Nicolls, M.R. Challenges and opportunities in treating inflammation associated with pulmonary hypertension. Expert Rev. Cardiovasc. Ther. 2016, 14, 939–951. [Google Scholar] [CrossRef] [PubMed]
- McKinney, E.F.; Smith, K.G. T cell exhaustion and immune-mediated disease-the potential for therapeutic exhaustion. Curr. Opin. Immunol. 2016, 43, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Austin, E.D.; Rock, M.T.; Mosse, C.A.; Vnencak-Jones, C.L.; Yoder, S.M.; Robbins, I.M.; Loyd, J.E.; Meyrick, B.O. T lymphocyte subset abnormalities in the blood and lung in pulmonary arterial hypertension. Respir. Med. 2010, 104, 454–462. [Google Scholar] [CrossRef]
- Maston, L.D.; Jones, D.T.; Giermakowska, W.; Howard, T.A.; Cannon, J.L.; Wang, W.; Wei, Y.; Xuan, W.; Resta, T.C.; Gonzalez Bosc, L.V. Central role of T helper 17 cells in chronic hypoxia-induced pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L609–L624. [Google Scholar] [CrossRef]
- Hawke, S.; Zinger, A.; Juillard, P.G.; Holdaway, K.; Byrne, S.N.; Grau, G.E. Selective modulation of trans-endothelial migration of lymphocyte subsets in multiple sclerosis patients under fingolimod treatment. J. Neuroimmunol. 2020, 349, 577392. [Google Scholar] [CrossRef]
- Wang, T.; Wang, W.; Li, W.; Duan, H.; Xu, C.; Tian, X.; Zhang, D. Genome-wide DNA methylation analysis of pulmonary function in middle and old-aged Chinese monozygotic twins. Respir. Res. 2021, 22, 300. [Google Scholar] [CrossRef]
- Yang, J.Y.; Hung, M.C. Deciphering the role of forkhead transcription factors in cancer therapy. Curr. Drug Targets 2011, 12, 1284–1290. [Google Scholar] [CrossRef]
- Kim, J.H.; Hwang, J.; Jung, J.H.; Lee, H.J.; Lee, D.Y.; Kim, S.H. Molecular networks of FOXP family: Dual biologic functions, interplay with other molecules and clinical implications in cancer progression. Mol. Cancer 2019, 18, 180. [Google Scholar] [CrossRef]
- Stenmark, K.R.; Hu, C.J.; Pullamsetti, S.S. How Many FOXs Are There on The Road to Pulmonary Hypertension? Am. J. Respir. Crit. Care Med. 2018, 198, 704–707. [Google Scholar] [CrossRef]
- Grobs, Y.; Awada, C.; Lemay, S.E.; Romanet, C.; Bourgeois, A.; Toro, V.; Nadeau, V.; Shimauchi, K.; Orcholski, M.; Breuils-Bonnet, S.; et al. Preclinical Investigation of Trifluoperazine as a Novel Therapeutic Agent for the Treatment of Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2021, 22, 2919. [Google Scholar] [CrossRef] [PubMed]
- Andreas, A.; Maloy, A.; Nyunoya, T.; Zhang, Y.; Chandra, D. The FoxP1 gene regulates lung function, production of matrix metalloproteinases and inflammatory mediators, and viability of lung epithelia. Respir. Res. 2022, 23, 281. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Lockett, A.D.; Gomes, M.T.; Stearman, R.S.; Machado, R.F. Sphingosine Kinase 1 Regulates the Pulmonary Vascular Immune Response. Cell Biochem. Biophys. 2021, 79, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, K.M.; Qu, C.; Gao, Q.; Iacobucci, I.; Gu, Z.; Yoshihara, H.; Backhaus, E.A.; Chang, Y.; Janke, L.J.; Xu, B.; et al. ZNF384 Fusion Oncoproteins Drive Lineage Aberrancy in Acute Leukemia. Blood Cancer Discov. 2022, 3, 240–263. [Google Scholar] [CrossRef] [PubMed]
- Zaliova, M.; Winkowska, L.; Stuchly, J.; Fiser, K.; Triska, P.; Zwyrtkova, M.; Hrusak, O.; Starkova, J.; Sramkova, L.; Stary, J.; et al. A novel class of ZNF384 aberrations in acute leukemia. Blood Adv. 2021, 5, 4393–4397. [Google Scholar] [CrossRef]
- Puscas, I.; Ifrim, M.; Maghiar, T.; Coltau, M.; Domuta, G.; Baican, M.; Hecht, A. Indomethacin activates carbonic anhydrase and antagonizes the effect of the specific carbonic anhydrase inhibitor acetazolamide, by a direct mechanism of action. Int. J. Clin. Pharmacol. Ther. 2001, 39, 265–270. [Google Scholar] [CrossRef]
- Nishimori, I.; Minakuchi, T.; Onishi, S.; Vullo, D.; Cecchi, A.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Cloning, characterization, and inhibition studies of the cytosolic isozyme III with sulfonamides. Bioorg. Med. Chem. 2007, 15, 7229–7236. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, J.; Zhang, E.; Wu, Q.; Wu, X.; Zhang, D.; Liu, Y.; Wang, R.; Li, W. Bisphenol A affects ovarian development in adolescent mice caused by genes expression change. Gene 2020, 740, 144535. [Google Scholar] [CrossRef] [PubMed]
- Verbanck, M.; Canouil, M.; Leloire, A.; Dhennin, V.; Coumoul, X.; Yengo, L.; Froguel, P.; Poulain-Godefroy, O. Low-dose exposure to bisphenols A, F and S of human primary adipocyte impacts coding and non-coding RNA profiles. PLoS ONE 2017, 12, e0179583. [Google Scholar] [CrossRef]
- Krug, A.K.; Kolde, R.; Gaspar, J.A.; Rempel, E.; Balmer, N.V.; Meganathan, K.; Vojnits, K.; Baquie, M.; Waldmann, T.; Ensenat-Waser, R.; et al. Human embryonic stem cell-derived test systems for developmental neurotoxicity: A transcriptomics approach. Arch. Toxicol. 2013, 87, 123–143. [Google Scholar] [CrossRef]
- Waldmann, T.; Grinberg, M.; Konig, A.; Rempel, E.; Schildknecht, S.; Henry, M.; Holzer, A.K.; Dreser, N.; Shinde, V.; Sachinidis, A.; et al. Stem Cell Transcriptome Responses and Corresponding Biomarkers That Indicate the Transition from Adaptive Responses to Cytotoxicity. Chem. Res. Toxicol. 2017, 30, 905–922. [Google Scholar] [CrossRef]
- van Breda, S.G.J.; Claessen, S.M.H.; van Herwijnen, M.; Theunissen, D.H.J.; Jennen, D.G.J.; de Kok, T.; Kleinjans, J.C.S. Integrative omics data analyses of repeated dose toxicity of valproic acid in vitro reveal new mechanisms of steatosis induction. Toxicology 2018, 393, 160–170. [Google Scholar] [CrossRef] [PubMed]
- van de Wouw, J.; Steenhorst, J.J.; Sorop, O.; van Drie, R.W.A.; Wielopolski, P.A.; Kleinjan, A.; Hirsch, A.; Duncker, D.J.; Merkus, D. Impaired pulmonary vasomotor control in exercising swine with multiple comorbidities. Basic Res. Cardiol. 2021, 116, 51. [Google Scholar] [CrossRef] [PubMed]
- Gorbunov, N.V.; Atkins, J.L.; Gurusamy, N.; Pitt, B.R. Iron-induced remodeling in cultured rat pulmonary artery endothelial cells. Biometals 2012, 25, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Mura, M.; Cecchini, M.J.; Joseph, M.; Granton, J.T. Osteopontin lung gene expression is a marker of disease severity in pulmonary arterial hypertension. Respirology 2019, 24, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Peng, J.; Lu, C.; Hsin, M.; Mura, M.; Wu, L.; Chu, L.; Zamel, R.; Machuca, T.; Waddell, T.; et al. Metabolomic heterogeneity of pulmonary arterial hypertension. PLoS ONE 2014, 9, e88727. [Google Scholar] [CrossRef]
- Rajkumar, R.; Konishi, K.; Richards, T.J.; Ishizawar, D.C.; Wiechert, A.C.; Kaminski, N.; Ahmad, F. Genomewide RNA expression profiling in lung identifies distinct signatures in idiopathic pulmonary arterial hypertension and secondary pulmonary hypertension. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1235–H1248. [Google Scholar] [CrossRef]
- Zhou, N.; Bao, J. FerrDb: A manually curated resource for regulators and markers of ferroptosis and ferroptosis-disease associations. Database 2020, 2020, baaa021. [Google Scholar] [CrossRef]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3. [Google Scholar] [CrossRef]
- Kolde, R.; Laur, S.; Adler, P.; Vilo, J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics 2012, 28, 573–580. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef] [PubMed]
- David, C.C.; Jacobs, D.J. Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. 2014, 1084, 193–226. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Muller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Zhang, J.; Li, C.; Yin, Y.; Zhang, J.; Grzegorzek, M. Applications of artificial neural networks in microorganism image analysis: A comprehensive review from conventional multilayer perceptron to popular convolutional neural network and potential visual transformer. Artif. Intell. Rev. 2023, 56, 1013–1070. [Google Scholar] [CrossRef] [PubMed]
- Quan, B.; Li, M.; Lu, S.; Li, J.; Liu, W.; Zhang, F.; Chen, R.; Ren, Z.; Yin, X. Predicting Disease-Specific Survival for Patients With Primary Cholangiocarcinoma Undergoing Curative Resection by Using a Decision Tree Model. Front. Oncol. 2022, 12, 824541. [Google Scholar] [CrossRef] [PubMed]
- Brito, M.P.; Stevenson, M.; Bravo, C. Subjective machines: Probabilistic risk assessment based on deep learning of soft information. Risk Anal. 2022, 43, 516–529. [Google Scholar] [CrossRef]
- Adnan, M.; Alarood, A.A.S.; Uddin, M.I.; Ur Rehman, I. Utilizing grid search cross-validation with adaptive boosting for augmenting performance of machine learning models. Peer J. Comput. Sci. 2022, 8, e803. [Google Scholar] [CrossRef] [PubMed]
- Nafees, A.; Khan, S.; Javed, M.F.; Alrowais, R.; Mohamed, A.M.; Mohamed, A.; Vatin, N.I. Forecasting the Mechanical Properties of Plastic Concrete Employing Experimental Data Using Machine Learning Algorithms: DT, MLPNN, SVM, and RF. Polymers 2022, 14, 1583. [Google Scholar] [CrossRef] [PubMed]
- Johannesdottir, K.B.; Kehlet, H.; Petersen, P.B.; Aasvang, E.K.; Sorensen, H.B.D.; Jorgensen, C.C.; Centre for Fast-track, H.; Knee Replacement Collaborative, G. Machine learning classifiers do not improve prediction of hospitalization > 2 days after fast-track hip and knee arthroplasty compared with a classical statistical risk model. Acta Orthop. 2022, 93, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Tamosiuniene, R.; Tian, W.; Dhillon, G.; Wang, L.; Sung, Y.K.; Gera, L.; Patterson, A.J.; Agrawal, R.; Rabinovitch, M.; Ambler, K.; et al. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ. Res. 2011, 109, 867–879. [Google Scholar] [CrossRef]
- Ru, B.; Wong, C.N.; Tong, Y.; Zhong, J.Y.; Zhong, S.S.W.; Wu, W.C.; Chu, K.C.; Wong, C.Y.; Lau, C.Y.; Chen, I.; et al. TISIDB: An integrated repository portal for tumor-immune system interactions. Bioinformatics 2019, 35, 4200–4202. [Google Scholar] [CrossRef] [PubMed]
- Toby, I.T.; Chicoine, L.G.; Cui, H.; Chen, B.; Nelin, L.D. Hypoxia-induced proliferation of human pulmonary microvascular endothelial cells depends on epidermal growth factor receptor tyrosine kinase activation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L600–L606. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Candidate Gene | Regulation | logFC | p-Value | AUC | AUC CI |
|---|---|---|---|---|---|
| KLRF1 | down | −1.349514767 | 0.11706773 | 0.783 | 0.664–0.902 |
| CX3CR1 | down | −1.287895847 | 0.022492566 | 0.783 | 0.658–0.908 |
| TSPAN7 | down | −1.022431101 | 0.012950506 | 0.926 | 0.857–0.994 |
| AJAP1 | down | −1.118195957 | 0.036788191 | 0.848 | 0.744–0.953 |
| SOSTDC1 | down | −1.680530516 | 0.000676038 | 0.862 | 0.755–0.969 |
| S100A3 | down | −1.590859215 | 0.0016257 | 0.905 | 0.829–0.982 |
| C2orf40 | down | −1.066587667 | 0.019138646 | 0.838 | 0.727–0.95 |
| DLL4 | down | −1.070844547 | 0.03607353 | 0.879 | 0.79–0.907 |
| SCN4B | down | −1.088543027 | 0.043459681 | 0.895 | 0.813–0.978 |
| ALAS2 | up | 1.755557936 | 0.004588542 | 0.903 | 0.824–0.981 |
| CA1 | up | 1.639222475 | 0.001211991 | 0.931 | 0.866–0.995 |
| TXLNG | up | 1.606123262 | 0.003413892 | 0.937 | 0.869–1 |
| LARS | up | 1.478386085 | 0.16211777 | 0.922 | 0.85–0.994 |
| CEP97 | up | 1.473332937 | 0.037459459 | 0.893 | 0.802–0.984 |
| PBRM1 | up | 1.156107034 | 0.015174985 | 0.966 | 0.923–1 |
| ACE2 | up | 1.107381186 | 0.024049503 | 0.919 | 0.84–0.998 |
| VEPH1 | up | 1.148259545 | 0.019732353 | 0.891 | 0.81–0.972 |
| ESF1 | up | 1.139501155 | 0.023748417 | 0.908 | 0.825–0.99 |
| ZNF148 | up | 1.040895681 | 0.033516829 | 0.937 | 0.876–0.998 |
| RSRC1 | up | 1.103381776 | 0.03927137 | 0.96 | 0.916–1 |
| SECISBP2L | up | 1.067323553 | 0.044551819 | 0.903 | 0.822–0.984 |
| ZFX | up | 1.066420893 | 0.038982434 | 0.938 | 0.88–0.996 |
| EPHA4 | up | 1.067115797 | 0.039949703 | 0.905 | 0.817–0.994 |
| HSP90AB3P | up | 1.145135459 | 0.01306175 | 0.92 | 0.844–0.997 |
| PPP1R9A | up | 1.013500448 | 0.039078627 | 0.924 | 0.861–0.987 |
| PARP14 | up | 1.024736403 | 0.040732053 | 0.847 | 0.747–0.948 |
| ITSN2 | up | 1.147534029 | 0.022053977 | 0.908 | 0.829–0.987 |
| PKP2 | up | 1.167878389 | 0.013092511 | 0.824 | 0.72–0.929 |
| USP15 | up | 1.206258609 | 0.009817843 | 0.933 | 0.868–0.998 |
| RSPO3 | up | 1.189539709 | 0.010857124 | 0.881 | 0.78–0.982 |
| IQGAP2 | up | 1.458016279 | 0.00580652 | 0.819 | 0.698–0.941 |
| IL13RA2 | up | 1.350594967 | 0.00580652 | 0.819 | 0.698–0.941 |
| FKBP5 | up | 1.296433874 | 0.006691228 | 0.827 | 0.718–0.936 |
| ZNF292 | up | 1.278994973 | 0.00751151 | 0.881 | 0.791–0.971 |
| ANKRD50 | up | 1.271971317 | 0.007723989 | 0.908 | 0.83–0.986 |
| RFC1 | up | 1.247043964 | 0.016211777 | 0.946 | 0.893–0.998 |
| MACC1 | up | 1.246789565 | 0.017027733 | 0.885 | 0.801–0.969 |
| TFEC | up | 1.32215869 | 0.008379207 | 0.918 | 0.843–0.982 |
| IGF1 | up | 1.318199859 | 0.010011771 | 0.902 | 0.815–0.988 |
| FMO5 | up | 1.2962632 | 0.010504888 | 0.792 | 0.674–0.909 |
| FGD4 | up | 1.23373543 | 0.023974054 | 0.948 | 0.896–1 |
| MPP7 | up | 1.245878219 | 0.04033993 | 0.902 | 0.817–0.986 |
| EHF | up | 1.485013582 | 0.002842598 | 0.919 | 0.845–0.993 |
| ZC3H13 | up | 1.433143528 | 0.004006581 | 0.934 | 0.871–0.998 |
| TTN | up | 1.585014364 | 0.0055049 | 0.856 | 0.756–0.956 |
| CCDC88A | up | 1.014479213 | 0.045188545 | 0.866 | 0.775–0.957 |
| HSP90AA1 | up | 1.145135459 | 0.01306175 | 0.889 | 0.804–0.974 |
| ROCK2 | up | 1.145759039 | 0.0032232649 | 0.939 | 0.881–0.998 |
| Nfold | 1 | 2 | 3 | 4 | 5 | Mean | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AUC | Accuracy Rate | AUC | Accuracy Rate | AUC | Accuracy Rate | AUC | Accuracy Rate | AUC | Accuracy Rate | AUC | Accuracy Rate | |
| RF | 1.00 | 0.83 | 1.00 | 1.00 | 0.83 | 0.82 | 1.00 | 0.91 | 1.00 | 1.00 | 0.97 | 0.91 |
| XGB | 0.67 | 0.67 | 0.90 | 0.92 | 0.69 | 0.82 | 0.94 | 0.91 | 1.00 | 1.00 | 0.84 | 0.86 |
| GBDT | 1.00 | 0.92 | 1.00 | 0.92 | 1.00 | 0.91 | 1.00 | 0.91 | 1.00 | 1.00 | 1.00 | 0.93 |
| ANN | 0.89 | 0.89 | 1.00 | 0.92 | 0.94 | 0.91 | 1.00 | 1.00 | 1.00 | 1.00 | 0.97 | 0.93 |
| DT | 0.67 | 0.67 | 0.90 | 0.92 | 0.69 | 0.82 | 0.73 | 0.82 | 0.93 | 0.91 | 0.78 | 0.83 |
| AdaBoost | 0.83 | 0.92 | 0.80 | 0.83 | 0.69 | 0.82 | 1.00 | 1.00 | 1.00 | 1.00 | 0.97 | 0.91 |
| MNB | 0.94 | 0.92 | 1.00 | 1.00 | 0.75 | 0.91 | 0.94 | 0.91 | 0.93 | 0.91 | 0.91 | 0.93 |
| Immune Cells | CA1 | PBRM1 | TXLNG | |||
|---|---|---|---|---|---|---|
| Correlation R | p-Value | Correlation R | p-Value | Correlation R | p-Value | |
| Activated CD8 T cell | −0.32 | 0.01611 | −0.37 | 0.00459 | −0.36 | 0.00526 |
| Central memory CD4 T cell | −0.26 | 0.05130 | −0.32 | 0.01490 | −0.35 | 0.00840 |
| Effector memory CD4 T cell | 0.33 | 0.01170 | 0.61 | 4.21 × 10−7 | 0.44 | 0.00061 |
| Monocyte | −0.18 | 0.18123 | −0.41 | 0.00147 | −0.46 | 0.00035 |
| Natural killer cell | −0.03 | 0.80401 | 0.19 | 0.15172 | 0.18 | 0.17243 |
| Natural killer T cell | −0.25 | 0.05918 | −0.31 | 0.01999 | −0.29 | 0.03127 |
| T follicular helper cell | −0.26 | 0.04738 | −0.32 | 0.01399 | −0.36 | 0.00554 |
| Biomarker Gene Symbol | Chemical | Interaction Action | Biomarker Gene Symbol | Chemical | Interaction Action |
|---|---|---|---|---|---|
| PBRM1 | Acetaminophen | Increases | TXLNG | 1-Methyl-3-isobutylxanthine | Increases |
| PBRM1 | Antirheumatic agents | Increases | TXLNG | Aristolochic acid I | Decreases |
| PBRM1 | Aristolochic acid I | Decreases | TXLNG | Aflatoxin B1 | Decreases |
| PBRM1 | Atrazine | Decreases | TXLNG | Atrazine | Other |
| PBRM1 | Copper sulfate | Decreases | TXLNG | Bisphenol F | Increases |
| PBRM1 | Cyclosporine | Increases | TXLNG | Copper sulfate | Increases |
| PBRM1 | Doxorubicin | Other | TXLNG | Cylindrospermopsin | Increases |
| PBRM1 | Epigallocatechin gallate | Decreases | TXLNG | Dexamethasone | Increases |
| PBRM1 | Lactic acid | Decreases | TXLNG | Doxorubicin | Decreases |
| PBRM1 | Methotrexate | Increases | TXLNG | Valproic acid | Increases |
| PBRM1 | Methylmercuric chloride | Other | TXLNG | Ethyl methanesulfonate | Decreases |
| PBRM1 | Plant extracts | Increases | TXLNG | Hydrogen peroxide | Other |
| PBRM1 | Potassium chromate | Decreases | TXLNG | Indomethacin | Increases |
| PBRM1 | Quercetin | Increases | TXLNG | Jinfukang | Decreases |
| PBRM1 | Riddelliine | Decreases | TXLNG | Perfluoro-n-nonanoic acid | Increases |
| PBRM1 | Sunitinib | Increases | TXLNG | Potassium chromate (VI) | Increases |
| PBRM1 | Trichostatin A | Decreases | TXLNG | Sunitinib | Increases |
| PBRM1 | TAK-243 | Other | TXLNG | Tobacco smoke pollution | Increases |
| PBRM1 | Valproic acid | Decreases | TXLNG | Tretinoin | Decreases |
| CA1 | Acetaminophen | Decreases | CA1 | Bicarbonates | Other |
| CA1 | Acetazolamide | Decreases | CA1 | Bromates | Decreases |
| CA1 | Aflatoxin B1 | Other | CA1 | Butylated hydroxyanisole | Decreases |
| CA1 | Amides | Decreases | CA1 | Candesartan | Decreases |
| CA1 | Anthocyanins | Decreases | CA1 | Carbonates | Decreases |
| CA1 | Benzolamide | Decreases | CA1 | Chalcone | Decreases |
| CA1 | Chalcone epoxide | Decreases | CA1 | Crown ethers | Other |
| CA1 | Chloric acid | Decreases | CA1 | Dimethylamines | Decreases |
| CA1 | Cobalt | Decreases | CA1 | Ethoxzolamide | Decreases |
| CA1 | Cobaltous chloride | Decreases | CA1 | Flavonoids | Decreases |
| CA1 | Coumarin | Decreases | CA1 | Guaiacol | Decreases |
| CA1 | Crown compounds | Decreases | CA1 | Indomethacin | Decreases |
| CA1 | Iodates | Decreases | CA1 | Mercury | Decreases |
| CA1 | Irbesartan | Decreases | CA1 | Methazolamide | Decreases |
| CA1 | Lactic acid | Increases | CA1 | Methylamines | Decreases |
| CA1 | Lead | Decreases | CA1 | Nitric acid | Decreases |
| CA1 | Malvidin-3-glucoside | Decreases | CA1 | Oryzalin | Decreases |
| CA1 | Malvin | Decreases | CA1 | pelargonidin-3-glucoside | Decreases |
| CA1 | Perchlorate | Decreases | CA1 | Silychristin | Decreases |
| CA1 | Phenylephrine | Decreases | CA1 | Sodium arsenite | Decreases |
| CA1 | Phenylthiourea | Decreases | CA1 | Sodium metasilicate | Decreases |
| CA1 | Potassium periodate | Decreases | CA1 | Sulfamic acid | Decreases |
| CA1 | Propofol | Decreases | CA1 | Sulfates | Decreases |
| CA1 | Pyrimidines | Decreases | CA1 | Sulfonamides | Decreases |
| CA1 | Rifampin | Decreases | CA1 | Synephrine | Decreases |
| CA1 | Rosiglitazone | Other | CA1 | Thiazolidines | Decreases |
| CA1 | Thiones | Decreases | CA1 | Triazoles | Decreases |
| CA1 | Thiophenes | Decreases | CA1 | Tungstate | Decreases |
| CA1 | Thiosemicarbazide | Decreases | CA1 | Vanadates | Decreases |
| CA1 | Thiourea | Decreases | CA1 | Vanillin | Decreases |
| CA1 | Tobacco smoke pollution | Decreases | CA1 | Voriconazole | Decreases |
| CA1 | Topiramate | Decreases | CA1 | Zonisamide | Decreases |
| Biomarker Gene | Fer-Related Gene | Correlation R | p-Value | Biomarker Gene | Fer-Related Gene | Correlation R | p-Value |
|---|---|---|---|---|---|---|---|
| PBRM1 | BECN1 | 0.869423423 | 1.76 × 10−18 | TXLNG | ZEB1 | 0.773220309 | 1.81 × 10−12 |
| HMGB1 | 0.815007386 | 1.20 × 10−14 | BECN1 | 0.731996746 | 9.84 × 10−11 | ||
| SP1 | 0.757174898 | 9.40 × 10−12 | HMGB1 | 0.724502669 | 1.88 × 10−10 | ||
| ZEB1 | 0.728618544 | 1.32 × 10−10 | LPCAT3 | −0.705414565 | 8.97 × 10−10 | ||
| IREB2 | 0.707501483 | 7.60 × 10−10 | RIPK1 | 0.699642666 | 1.40 × 10−9 | ||
| PLIN2 | 0.657847236 | 2.70 × 10−8 | PRDX6 | 0.61934584 | 2.80 × 10−7 | ||
| SLC3A2 | −0.633598312 | 1.22 × 10−7 | SP1 | 0.60633034 | 5.77 × 10−7 | ||
| LPCAT3 | −0.61500838 | 3.58 × 10−7 | IREB2 | 0.604841956 | 6.26 × 10−7 | ||
| RIPK1 | 0.610161027 | 4.68 × 10−7 | YWHAE | −0.602218341 | 7.20 × 10−7 | ||
| CA1 | PLIN2 | 0.629426281 | 2.29 × 10−5 | CA1 | IREB2 | 0.696001081 | 8.73 × 10−5 |
| Gene Name | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| PBRM1 | AGGAGGAGACTTTCCAATCTTCC | CTTCGCTTTGGTGCCCTAATG |
| CA1 | CTGACAGCTACAGGCTCTTTC | CTACGTGAAGCTCGGCAGAAT |
| TXLNG | ATCCATCAAAGCGCCATCAAAGCG | ACAAATAAAGCAATAGCATCACAA |
| IGF1 | AAGCCTACAAAGTCAGCTGC | GGTCTTGTTTCCTGCACTT |
| ACE | TCCTATTCCCGCTCATCT | CCAGCCCTTCTGTACCATT |
| RSPO | CAGCCATAACTTCTGCACCA | AGAGCTGCTGCTTCTTGGAG |
| C2orf40 | GGTACCAGCAGTTTCTCTACATG | CAGCGTGTGGCAAGTCATGGTTAGT |
| TSPAN7 | CTCATCGGAACTGGCACCACTA | CCTGAAATGCCAGCTACGAGCT |
| SCN4B | CAACAGCAGTGACGCATTCAA | CTCCTTAGTAGAGCCTACCAGAG |
| IREB2 | GCGATTTCCAGGCTTGCTTA | GTTTAACACGCAGACCAGCT |
| LPCAT3 | AGCCTTAACAAGTTGGCGAC | TGCCGATAAAACAAAGCAAA |
| BECN1 | AGGAACTCACAGCTCCATTAC | AATGGCTCCTCTCCTGAGTT |
| ZEB1 | AAACTCGAGTACTTCAATTCCTCGGTATTG | AAATCTAGACACACTGTTCTACAGTCCAA |
| HMGB1 | ATATGGCAAAAGCGGACAAG | AGGCCAGGATGTTCTCCTTT |
| SLC3A2 | ACCCCTGTTTTCAGCTACGG | GGTCTTCACTCTGGCCCTTC |
| PLIN2 | CTGTCTACCAAGCTCTGCTC | CGATGCTTCTCTTCCACTCC |
| SP1 | GTGGAGCAACATCATTGCTG | GCCACTGGTACATTGGTCACAT |
| RIPK1 | AGGCTTTGGGAAGGTGTCTC | CGGAGTACTCATCTCGGCTTT |
| PRDX6 | TTTCAATAGACAGTGTTGAGGATCA | CGTGGGTGTTTCACCATTG |
| YWHAE | CTAACACTGGCGAGTCCAAGGT | GTAAGCCACG AGGCTGTTCTCT |
| GAPDH | CAATGACCCCTTCATTGACC | TTGATTTTGGAGGGATCTCG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, H.; Cai, J.; Qi, D.; Li, B.; Yu, L.; Wang, C.; Bajpai, A.K.; Huang, X.; Zhang, X.; Lu, L.; et al. Identification of Potential Biomarkers for Group I Pulmonary Hypertension Based on Machine Learning and Bioinformatics Analysis. Int. J. Mol. Sci. 2023, 24, 8050. https://doi.org/10.3390/ijms24098050
Hu H, Cai J, Qi D, Li B, Yu L, Wang C, Bajpai AK, Huang X, Zhang X, Lu L, et al. Identification of Potential Biomarkers for Group I Pulmonary Hypertension Based on Machine Learning and Bioinformatics Analysis. International Journal of Molecular Sciences. 2023; 24(9):8050. https://doi.org/10.3390/ijms24098050
Chicago/Turabian StyleHu, Hui, Jie Cai, Daoxi Qi, Boyu Li, Li Yu, Chen Wang, Akhilesh K. Bajpai, Xiaoqin Huang, Xiaokang Zhang, Lu Lu, and et al. 2023. "Identification of Potential Biomarkers for Group I Pulmonary Hypertension Based on Machine Learning and Bioinformatics Analysis" International Journal of Molecular Sciences 24, no. 9: 8050. https://doi.org/10.3390/ijms24098050