
Gene Expression Profiling in Coeliac Disease Confirmed the Key Role of the Immune System and Revealed a Molecular Overlap with Non-Celiac Gluten Sensitivity
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Results
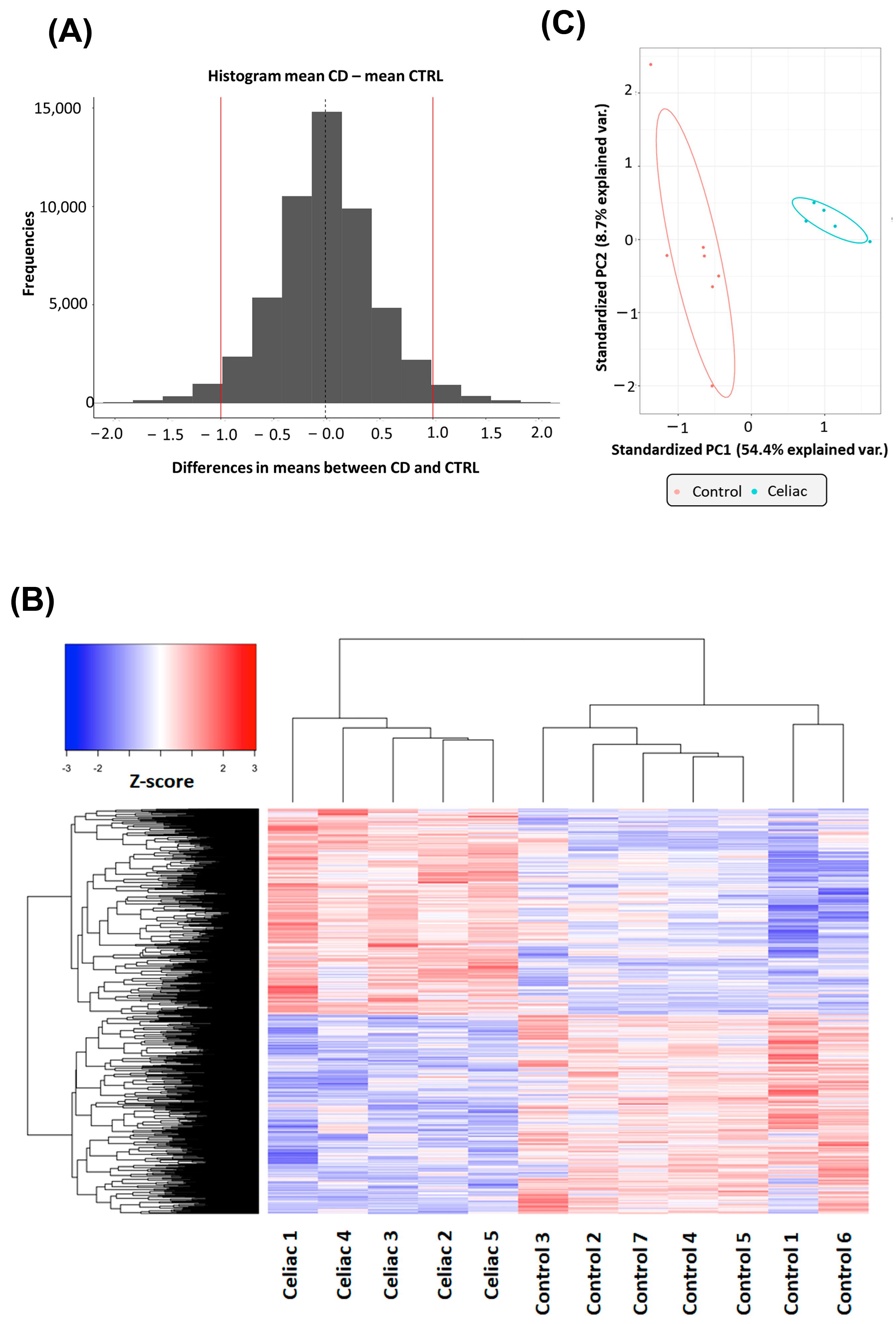
2.1. Gene Expression Changes in Intestinal Biopsies of Coeliac Patients
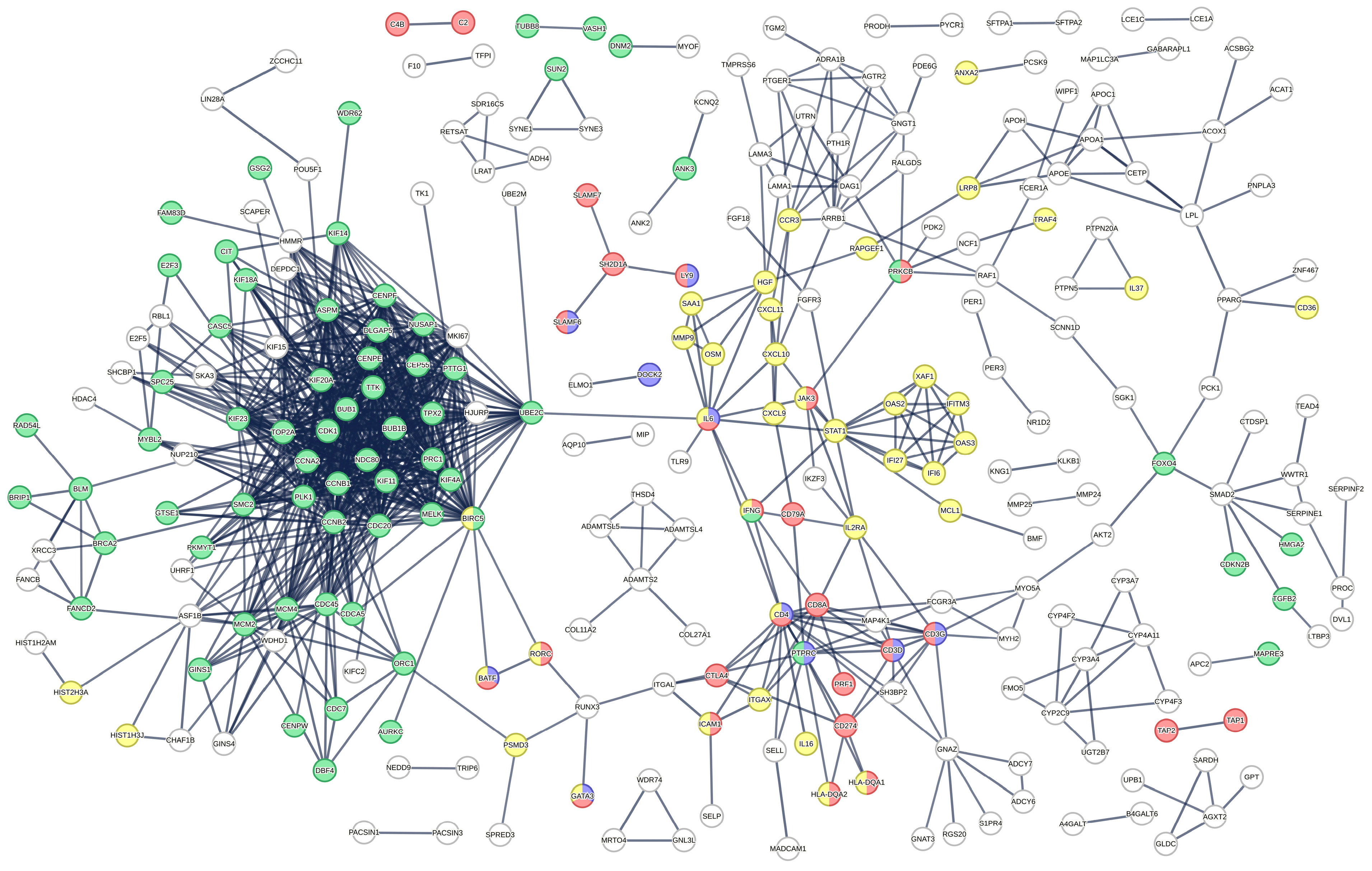
2.2. Functional Analysis of DE Gene in Intestinal Biopsies of Coeliac Patients
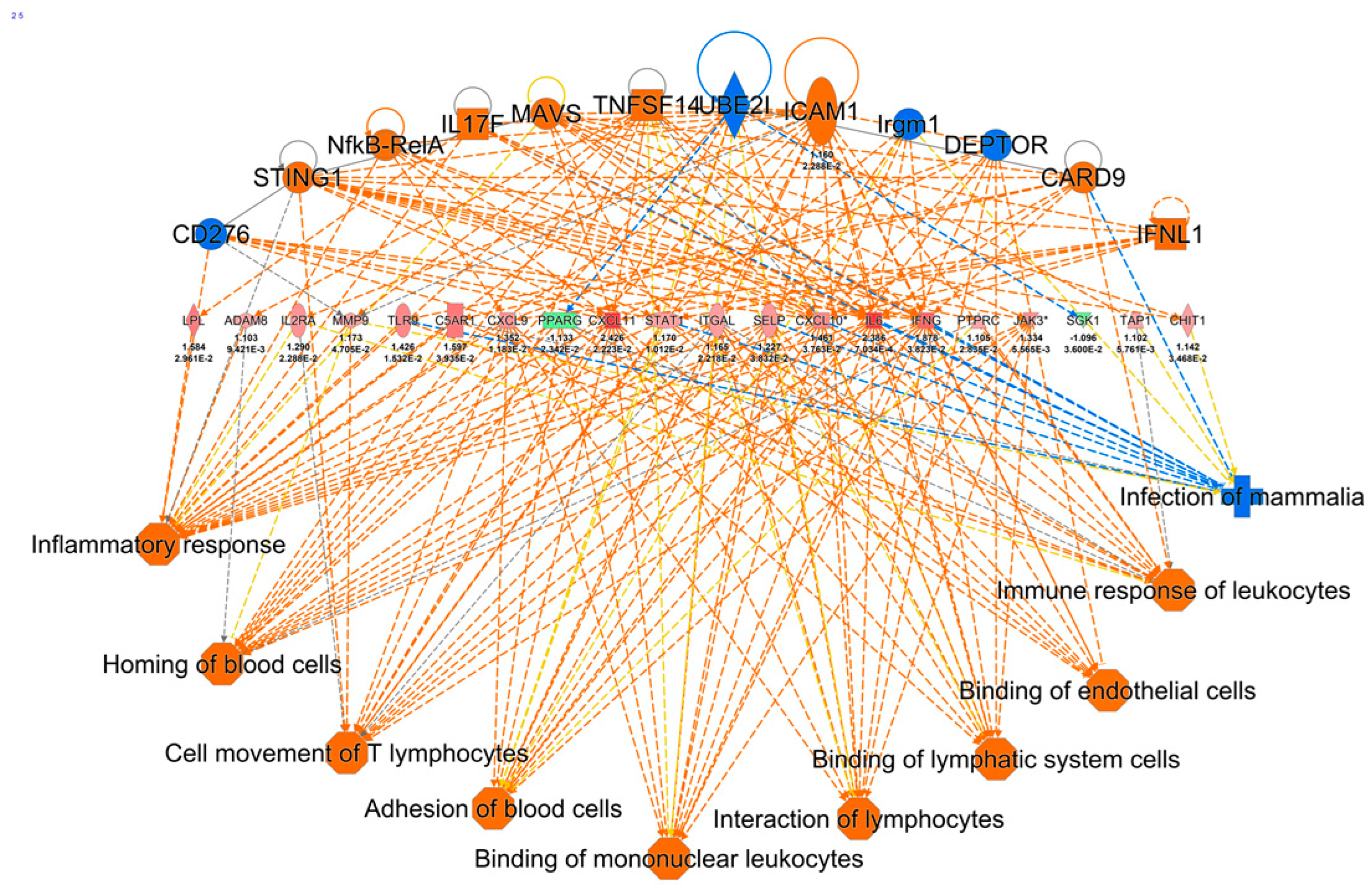
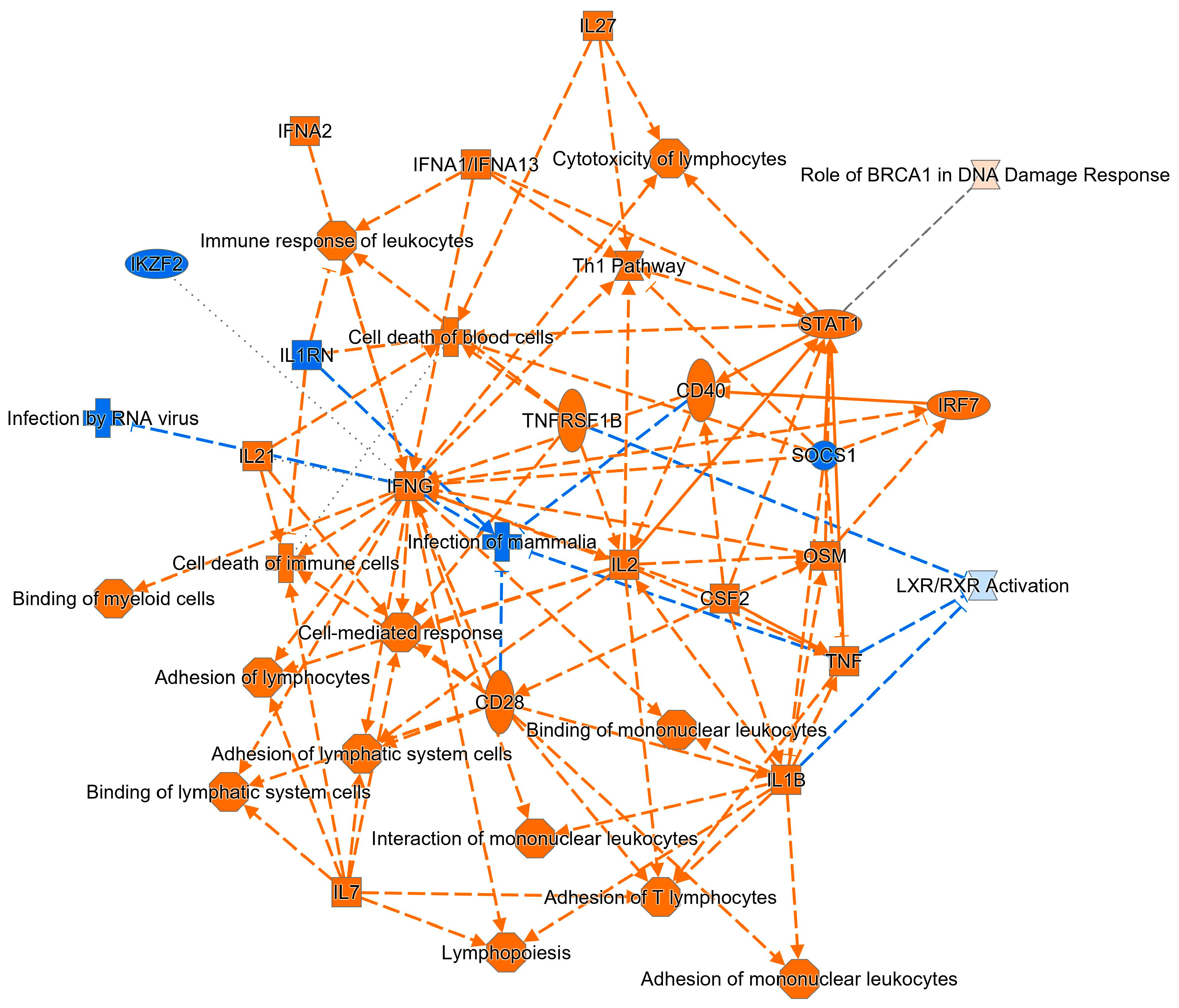
2.3. Ingenuity Pathway Analysis of DE Genes
2.4. Comparative Analysis of DE Genes in Celiac Disease and NCGS
3. Discussion
4. Materials and Methods
4.1. Patients Participating in the Study
4.2. RNA Extraction, Microarray Analysis, Statistical Testing and Functional Analysis of DE Genes
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sallese, M.; Lopetuso, L.R.; Efthymakis, K.; Neri, M. Beyond the HLA Genes in Gluten-Related Disorders. Front. Nutr. 2020, 7, 575844. [Google Scholar] [CrossRef] [PubMed]
- Catassi, C.; Verdu, E.F.; Bai, J.C.; Lionetti, E. Coeliac disease. Lancet 2022, 399, 2413–2426. [Google Scholar] [CrossRef] [PubMed]
- Green, P.H.; Cellier, C. Celiac disease. N. Engl. J. Med. 2007, 357, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Caio, G.; Volta, U.; Sapone, A.; Leffler, D.A.; De Giorgio, R.; Catassi, C.; Fasano, A. Celiac disease: A comprehensive current review. BMC Med. 2019, 17, 142. [Google Scholar] [CrossRef]
- Mayassi, T.; Ladell, K.; Gudjonson, H.; McLaren, J.E.; Shaw, D.G.; Tran, M.T.; Rokicka, J.J.; Lawrence, I.; Grenier, J.C.; van Unen, V.; et al. Chronic Inflammation Permanently Reshapes Tissue-Resident Immunity in Celiac Disease. Cell 2019, 176, 967–981.e19. [Google Scholar] [CrossRef]
- Marsh, M.N.; Hinde, J. Inflammatory component of celiac sprue mucosa. I. Mast cells, basophils, and eosinophils. Gastroenterology 1985, 89, 92–101. [Google Scholar] [CrossRef]
- Dhesi, I.; Marsh, M.N.; Kelly, C.; Crowe, P. Morphometric analysis of small intestinal mucosa. II. Determination of lamina propria volumes; plasma cell and neutrophil populations within control and coeliac disease mucosae. Virchows Arch. A Pathol. Anat. Histopathol. 1984, 403, 173–180. [Google Scholar] [CrossRef]
- Vader, L.W.; de Ru, A.; van der Wal, Y.; Kooy, Y.M.; Benckhuijsen, W.; Mearin, M.L.; Drijfhout, J.W.; van Veelen, P.; Koning, F. Specificity of tissue transglutaminase explains cereal toxicity in celiac disease. J. Exp. Med. 2002, 195, 643–649. [Google Scholar] [CrossRef]
- Dunne, M.R.; Byrne, G.; Chirdo, F.G.; Feighery, C. Coeliac Disease Pathogenesis: The Uncertainties of a Well-Known Immune Mediated Disorder. Front. Immunol. 2020, 11, 1374. [Google Scholar] [CrossRef]
- Mazzarella, G. Effector and suppressor T cells in celiac disease. World J. Gastroenterol. 2015, 21, 7349–7356. [Google Scholar] [CrossRef]
- Nilsen, E.M.; Lundin, K.E.A.; Krajci, P.; Scott, H.; Sollid, L.M.; Brandtzaeg, P. Gluten Specific, Hla-Dq Restricted T-Cells from Celiac Mucosa Produce Cytokines with Thl or Tho Profile Dominated by Interferon-Gamma. Gut 1995, 37, 766–776. [Google Scholar] [CrossRef]
- Nanayakkara, M.; Lania, G.; Maglio, M.; Auricchio, R.; De Musis, C.; Discepolo, V.; Miele, E.; Jabri, B.; Troncone, R.; Auricchio, S.; et al. P31–43, an undigested gliadin peptide, mimics and enhances the innate immune response to viruses and interferes with endocytic trafficking: A role in celiac disease. Sci. Rep. 2018, 8, 10821. [Google Scholar] [CrossRef] [PubMed]
- Abadie, V.; Jabri, B. IL-15: A central regulator of celiac disease immunopathology. Immunol. Rev. 2014, 260, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef]
- Costantino, A.; Aversano, G.M.; Lasagni, G.; Smania, V.; Doneda, L.; Vecchi, M.; Roncoroni, L.; Pastorello, E.A.; Elli, L. Diagnostic management of patients reporting symptoms after wheat ingestion. Front. Nutr. 2022, 9, 1007007. [Google Scholar] [CrossRef]
- Rostami, K.; Ensari, A.; Marsh, M.N.; Srivastava, A.; Villanacci, V.; Carroccio, A.; Asadzadeh Aghdaei, H.; Bai, J.C.; Bassotti, G.; Becheanu, G.; et al. Gluten Induces Subtle Histological Changes in Duodenal Mucosa of Patients with Non-Coeliac Gluten Sensitivity: A Multicentre Study. Nutrients 2022, 14, 2487. [Google Scholar] [CrossRef] [PubMed]
- Corazza, G.R.; Villanacci, V.; Zambelli, C.; Milione, M.; Luinetti, O.; Vindigni, C.; Chioda, C.; Albarello, L.; Bartolini, D.; Donato, F. Comparison of the interobserver reproducibility with different histologic criteria used in celiac disease. Clin. Gastroenterol. Hepatol. 2007, 5, 838–843. [Google Scholar] [CrossRef]
- Leonard, M.M.; Sapone, A.; Catassi, C.; Fasano, A. Celiac Disease and Nonceliac Gluten Sensitivity: A Review. JAMA 2017, 318, 647–656. [Google Scholar] [CrossRef]
- Catassi, C.; Elli, L.; Bonaz, B.; Bouma, G.; Carroccio, A.; Castillejo, G.; Cellier, C.; Cristofori, F.; de Magistris, L.; Dolinsek, J.; et al. Diagnosis of Non-Celiac Gluten Sensitivity (NCGS): The Salerno Experts’ Criteria. Nutrients 2015, 7, 4966–4977. [Google Scholar] [CrossRef]
- Sapone, A.; Lammers, K.M.; Casolaro, V.; Cammarota, M.; Giuliano, M.T.; De Rosa, M.; Stefanile, R.; Mazzarella, G.; Tolone, C.; Russo, M.I.; et al. Divergence of gut permeability and mucosal immune gene expression in two gluten-associated conditions: Celiac disease and gluten sensitivity. BMC Med. 2011, 9, 23. [Google Scholar] [CrossRef]
- Valerii, M.C.; Ricci, C.; Spisni, E.; Di Silvestro, R.; De Fazio, L.; Cavazza, E.; Lanzini, A.; Campieri, M.; Dalpiaz, A.; Pavan, B.; et al. Responses of peripheral blood mononucleated cells from non-celiac gluten sensitive patients to various cereal sources. Food Chem. 2015, 176, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Sazzini, M.; De Fanti, S.; Cherubini, A.; Quagliariello, A.; Profiti, G.; Martelli, P.L.; Casadio, R.; Ricci, C.; Campieri, M.; Lanzini, A.; et al. Ancient pathogen-driven adaptation triggers increased susceptibility to non-celiac wheat sensitivity in present-day European populations. Genes. Nutr. 2016, 11, 15. [Google Scholar] [CrossRef] [PubMed]
- Carroccio, A.; Giannone, G.; Mansueto, P.; Soresi, M.; La Blasca, F.; Fayer, F.; Iacobucci, R.; Porcasi, R.; Catalano, T.; Geraci, G.; et al. Duodenal and Rectal Mucosa Inflammation in Patients with Non-celiac Wheat Sensitivity. Clin. Gastroenterol. Hepatol. 2019, 17, 682–690.e3. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Kim, J.J.; Chen, B.; Zhang, Y.; Ren, H. Gene Polymorphisms and Susceptibility to Functional Dyspepsia: A Systematic Review and Meta-Analysis. Gastroenterol. Res. Pract. 2019, 2019, 3420548. [Google Scholar] [CrossRef] [PubMed]
- Mokha, J.S.; Hyams, J.S.; Glidden, N.C.; Balarezo, F.; Young, E. Characterizing clinical features and location-specific gene expression profiles associated with pain burden in children with functional dyspepsia. Neurogastroenterol. Motil. 2021, 33, e14185. [Google Scholar] [CrossRef] [PubMed]
- Efthymakis, K.; Clemente, E.; Marchioni, M.; Di Nicola, M.; Neri, M.; Sallese, M. An Exploratory Gene Expression Study of the Intestinal Mucosa of Patients with Non-Celiac Wheat Sensitivity. Int. J. Mol. Sci. 2020, 21, 1969. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Wiesner, J.; Vilcinskas, A. Antimicrobial peptides: The ancient arm of the human immune system. Virulence 2010, 1, 440–464. [Google Scholar] [CrossRef]
- Sarikaya, M.; Dogan, Z.; Ergul, B.; Filik, L. Neutrophil-to-lymphocyte ratio as a sensitive marker in diagnosis of celiac disease. Ann. Gastroenterol. 2014, 27, 431–432. [Google Scholar]
- Gao, K.; Lin, Z.; Wen, S.; Jiang, Y. Potassium channels and epilepsy. Acta Neurol. Scand. 2022, 146, 699–707. [Google Scholar] [CrossRef]
- Giuffre, M.; Gazzin, S.; Zoratti, C.; Llido, J.P.; Lanza, G.; Tiribelli, C.; Moretti, R. Celiac Disease and Neurological Manifestations: From Gluten to Neuroinflammation. Int. J. Mol. Sci. 2022, 23, 15564. [Google Scholar] [CrossRef] [PubMed]
- Ludvigsson, J.F.; Zingone, F.; Tomson, T.; Ekbom, A.; Ciacci, C. Increased risk of epilepsy in biopsy-verified celiac disease: A population-based cohort study. Neurology 2012, 78, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Casciato, S.; Morano, A.; Albini, M.; Fanella, M.; Lapenta, L.; Fattouch, J.; Carni, M.; Colonnese, C.; Manfredi, M.; Giallonardo, A.T.; et al. Cryptogenic focal epilepsy and "hidden" celiac disease in adulthood: A causal or accidental link? Int. J. Neurosci. 2015, 125, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.R.; Sanders, D.S.; Logan, R.F.; Fleming, K.M.; Hubbard, R.B.; West, J. Cholesterol profile in people with newly diagnosed coeliac disease: A comparison with the general population and changes following treatment. Br. J. Nutr. 2009, 102, 509–513. [Google Scholar] [CrossRef]
- Rezaei-Tavirani, S.; Rostami-Nejad, M.; Vafaee, R.; Khalkhal, E.; Keramatinia, A.; Ehsani-Ardakani, M.J.; Razzaghi, M. Introducing tumor necrosis factor as a prominent player in celiac disease and type 1 diabetes mellitus. Gastroenterol. Hepatol. Bed Bench 2019, 12, S123–S129. [Google Scholar]
- Sur, S.; Steele, R.; Ko, B.C.B.; Zhang, J.; Ray, R.B. Long noncoding RNA ELDR promotes cell cycle progression in normal oral keratinocytes through induction of a CTCF-FOXM1-AURKA signaling axis. J. Biol. Chem. 2022, 298, 101895. [Google Scholar] [CrossRef]
- Broderick, L.; Hoffman, H.M. IL-1 and autoinflammatory disease: Biology, pathogenesis and therapeutic targeting. Nat. Rev. Rheumatol. 2022, 18, 448–463. [Google Scholar] [CrossRef]
- Bragde, H.; Jansson, U.; Jarlsfelt, I.; Soderman, J. Gene expression profiling of duodenal biopsies discriminates celiac disease mucosa from normal mucosa. Pediatr. Res. 2011, 69, 530–537. [Google Scholar] [CrossRef]
- Yoshida, H.; Hunter, C.A. The immunobiology of interleukin-27. Annu. Rev. Immunol. 2015, 33, 417–443. [Google Scholar] [CrossRef]
- Vorobjova, T.; Tagoma, A.; Oras, A.; Alnek, K.; Kisand, K.; Talja, I.; Uibo, O.; Uibo, R. Celiac Disease in Children, Particularly with Accompanying Type 1 Diabetes, Is Characterized by Substantial Changes in the Blood Cytokine Balance, Which May Reflect Inflammatory Processes in the Small Intestinal Mucosa. J. Immunol. Res. 2019, 2019, 6179243. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Giuffrida, P.; Fornasa, G.; Salvatore, C.; Vanoli, A.; Naviglio, S.; De Leo, L.; Pasini, A.; De Amici, M.; Alvisi, C.; et al. Innate and adaptive immunity in self-reported nonceliac gluten sensitivity versus celiac disease. Dig. Liver Dis. 2016, 48, 745–752. [Google Scholar] [CrossRef]
- Shahi, A.; Afzali, S.; Salehi, S.; Aslani, S.; Mahmoudi, M.; Jamshidi, A.; Amirzargar, A. IL-27 and autoimmune rheumatologic diseases: The good, the bad, and the ugly. Int. Immunopharmacol. 2020, 84, 106538. [Google Scholar] [CrossRef] [PubMed]
- Leonard, W.J.; Wan, C.K. IL-21 Signaling in Immunity. F1000Research 2016, 5, 224. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.M.; Lukacher, A.E.; Rahman, Z.S.M.; Olsen, N.J. New developments implicating IL-21 in autoimmune disease. J. Autoimmun. 2021, 122, 102689. [Google Scholar] [CrossRef] [PubMed]
- Iervasi, E.; Auricchio, R.; Strangio, A.; Greco, L.; Saverino, D. Serum IL-21 levels from celiac disease patients correlates with anti-tTG IgA autoantibodies and mucosal damage. Autoimmunity 2020, 53, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Tye-Din, J.A.; Daveson, A.J.M.; Ee, H.C.; Goel, G.; MacDougall, J.; Acaster, S.; Goldstein, K.E.; Dzuris, J.L.; Neff, K.M.; Truitt, K.E.; et al. Elevated serum interleukin-2 after gluten correlates with symptoms and is a potential diagnostic biomarker for coeliac disease. Aliment. Pharmacol. Ther. 2019, 50, 901–910. [Google Scholar] [CrossRef]
- Manavalan, J.S.; Hernandez, L.; Shah, J.G.; Konikkara, J.; Naiyer, A.J.; Lee, A.R.; Ciaccio, E.; Minaya, M.T.; Green, P.H.; Bhagat, G. Serum cytokine elevations in celiac disease: Association with disease presentation. Hum. Immunol. 2010, 71, 50–57. [Google Scholar] [CrossRef]
- Kaminsky, L.W.; Al-Sadi, R.; Ma, T.Y. IL-1beta and the Intestinal Epithelial Tight Junction Barrier. Front. Immunol. 2021, 12, 767456. [Google Scholar] [CrossRef]
- Marafini, I.; Monteleone, I.; Di Fusco, D.; Cupi, M.L.; Paoluzi, O.A.; Colantoni, A.; Ortenzi, A.; Izzo, R.; Vita, S.; De Luca, E.; et al. TNF-alpha Producing Innate Lymphoid Cells (ILCs) Are Increased in Active Celiac Disease and Contribute to Promote Intestinal Atrophy in Mice. PLoS ONE 2015, 10, e0126291. [Google Scholar] [CrossRef]
- Yang, H.; Madison, B.; Gumucio, D.L.; Teitelbaum, D.H. Specific overexpression of IL-7 in the intestinal mucosa: The role in intestinal intraepithelial lymphocyte development. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G1421–G1430. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Pickard, K.M.; Gordon, J.N.; Salvati, V.; Mazzarella, G.; Beattie, R.M.; Vossenkaemper, A.; Rovedatti, L.; Leakey, N.A.; Croft, N.M.; et al. Evidence for the role of interferon-alfa production by dendritic cells in the Th1 response in celiac disease. Gastroenterology 2007, 133, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Wapenaar, M.C.; van Belzen, M.J.; Fransen, J.H.; Sarasqueta, A.F.; Houwen, R.H.; Meijer, J.W.; Mulder, C.J.; Wijmenga, C. The interferon gamma gene in celiac disease: Augmented expression correlates with tissue damage but no evidence for genetic susceptibility. J. Autoimmun. 2004, 23, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Elli, L.; Branchi, F.; Tomba, C.; Villalta, D.; Norsa, L.; Ferretti, F.; Roncoroni, L.; Bardella, M.T. Diagnosis of gluten related disorders: Celiac disease, wheat allergy and non-celiac gluten sensitivity. World J. Gastroenterol. 2015, 21, 7110–7119. [Google Scholar] [CrossRef] [PubMed]
- Clemente, E.; Efthymakis, K.; Carletti, E.; Capone, V.; Sperduti, S.; Bologna, G.; Marchisio, M.; Di Nicola, M.; Neri, M.; Sallese, M. An explorative study identifies miRNA signatures for the diagnosis of non-celiac wheat sensitivity. PLoS ONE 2019, 14, e0226478. [Google Scholar] [CrossRef]
- Benjamini, Y.; Krieger, A.M.; Yekutieli, D. Adaptive linear step-up procedures that control the false discovery rate. Biometrika 2006, 93, 491–507. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | CTRL | Celiac | p-Value |
|---|---|---|---|
| Number of subjects | 7 | 5 | |
| Age (years), mean ± SD | 56.3 ± 22.8 | 45.8 ± 19.5 | 0.432 |
| Gender (M/F) | 1/6 | 0/5 | 0.583 |
| Marsh classification, n (%) | |||
| 1 | 1 (20%) | ||
| 2 | 0 (0%) | ||
| 3a | 1 (20%) | ||
| 3b | 2 (40%) | ||
| 3c | 1 (20%) | ||
| H. pylori positive, n (%) | 4 (57.1%) | 1 (20%) | 0.247 |
| tTG-IgA, (U/mL), mean ± SD | 1.86 ± 1.35 | 131.2 ± 91.8 | 0.016 |
| EMA positive | 0 (0%) | 4 (80%) | 0.01 |
| HLA-DQ2/8, n (%) | |||
| absent | 5 (71.4%) | 0 (0%) | |
| DQ2 | 2 (28.6%) | 4 (80%) | |
| DQ8 | 0 (0%) | 1 (20%) | |
| DQ2/8 (any) | 2 (28.6%) | 5 (100%) | 0.027 |
| ESR (mm/h), mean ± SD | 12.6 ± 4.3 | 20.8 ± 16.5 | 0.876 |
| CRP (mg/L), mean ± SD | 0.41 ± 0.21 | 0.54 ± 0.34 | 0.357 |
| Fecal calprotectin (μg/g), mean ± SD | 68 ± 35.5 | 153.2 ± 222.1 | 0.442 |
| Hb (g/dL), mean ± SD | 12.7 ± 0.8 | 11.6 ± 0.9 | 0.037 |
| Ferritin (ng/mL), mean ± SD | 24.2 ± 20.2 | 10.1 ± 6.2 | 0.053 |
| BMI (kg/m2), mean ± SD | 25.8 ± 4.0 | 22.6 ± 3.1 | 0.149 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sallese, M.; Efthymakis, K.; Marchioni, M.; Neri, B.; Dufrusine, B.; Dainese, E.; Di Nicola, M.; Neri, M. Gene Expression Profiling in Coeliac Disease Confirmed the Key Role of the Immune System and Revealed a Molecular Overlap with Non-Celiac Gluten Sensitivity. Int. J. Mol. Sci. 2023, 24, 7769. https://doi.org/10.3390/ijms24097769
Sallese M, Efthymakis K, Marchioni M, Neri B, Dufrusine B, Dainese E, Di Nicola M, Neri M. Gene Expression Profiling in Coeliac Disease Confirmed the Key Role of the Immune System and Revealed a Molecular Overlap with Non-Celiac Gluten Sensitivity. International Journal of Molecular Sciences. 2023; 24(9):7769. https://doi.org/10.3390/ijms24097769
Chicago/Turabian StyleSallese, Michele, Konstantinos Efthymakis, Michele Marchioni, Benedetto Neri, Beatrice Dufrusine, Enrico Dainese, Marta Di Nicola, and Matteo Neri. 2023. "Gene Expression Profiling in Coeliac Disease Confirmed the Key Role of the Immune System and Revealed a Molecular Overlap with Non-Celiac Gluten Sensitivity" International Journal of Molecular Sciences 24, no. 9: 7769. https://doi.org/10.3390/ijms24097769