

Renal Denervation Helps Preserve the Ejection Fraction by Preserving Endocardial-Endothelial Function during Heart Failure
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
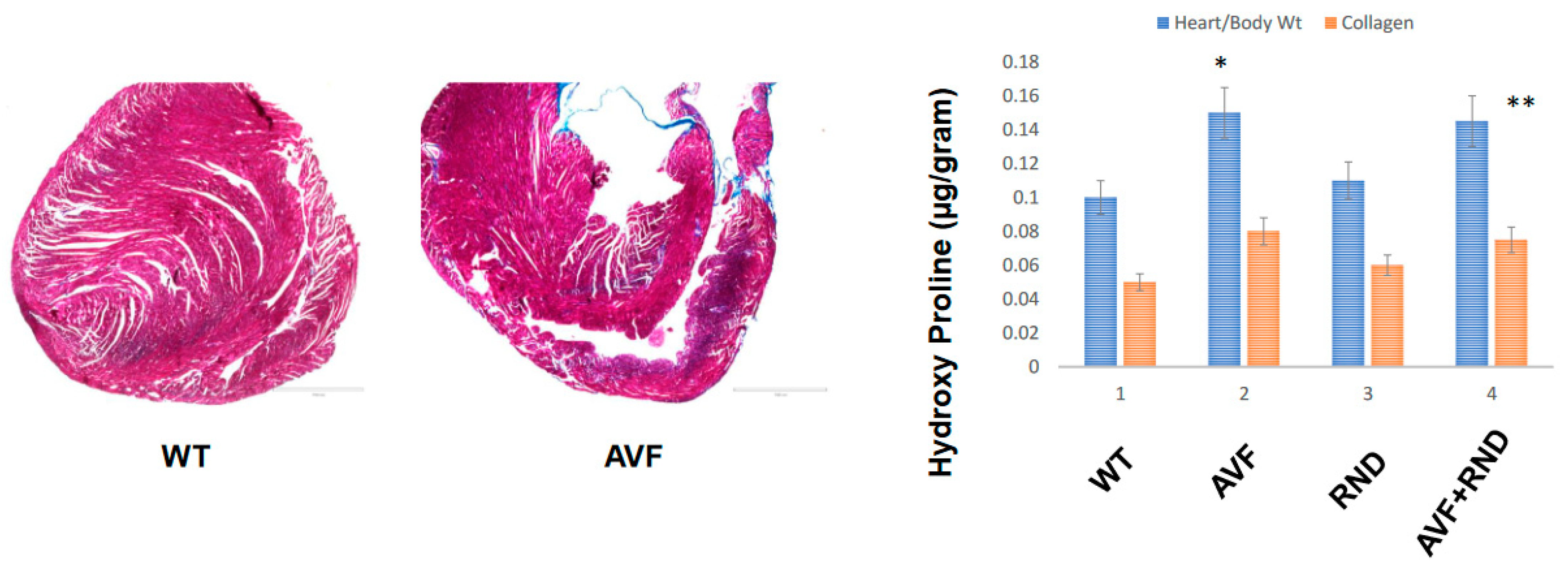
2. Results
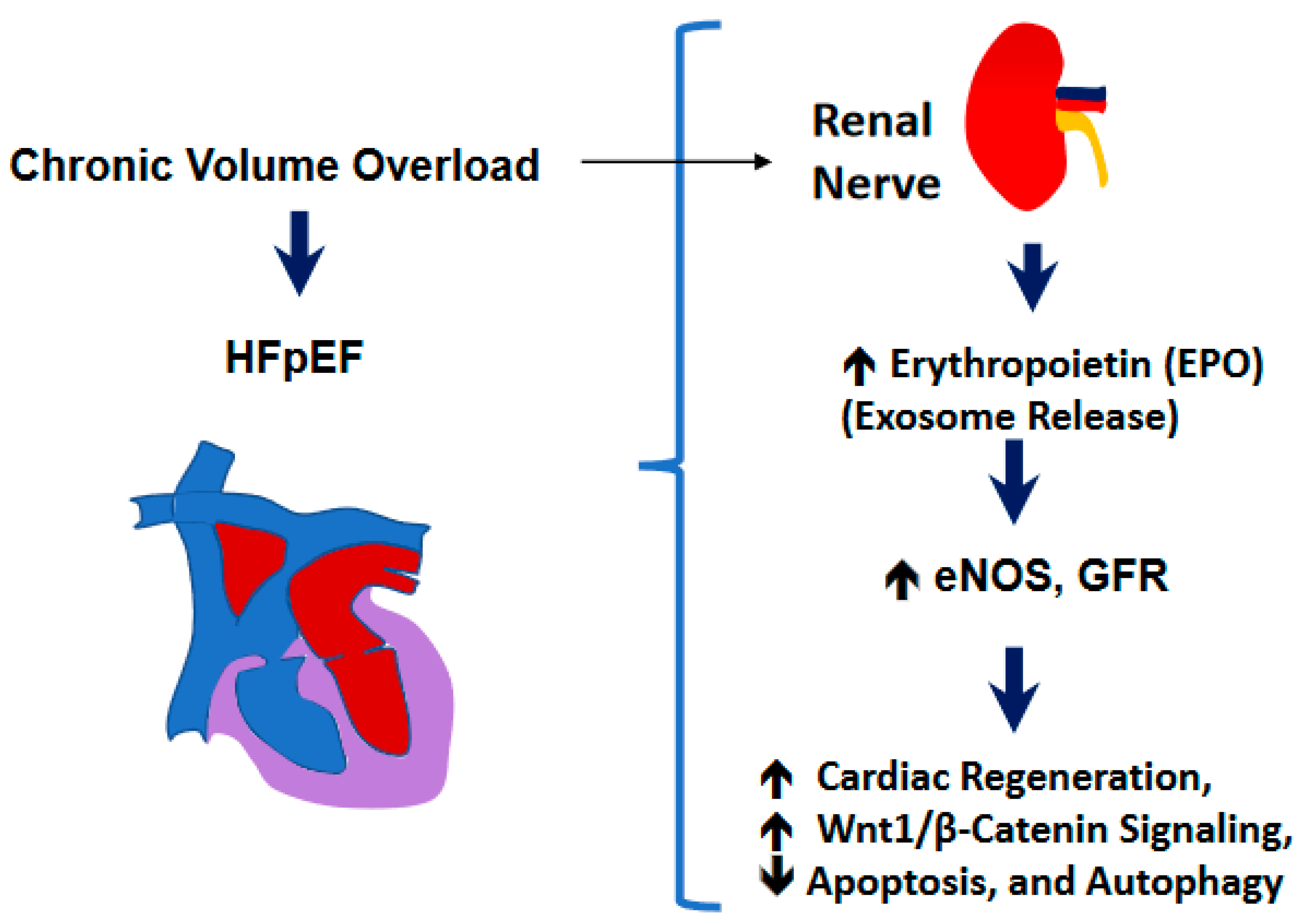
3. Discussion
4. Materials and Methods
4.1. Creation of the Aorta Vena Cava Fistula (AVF) in a Mouse Model
4.2. Serial and Longitudinal Echocardiograms (ECHOs) in Mice
4.3. Measurement of Renal and Blood Exosomal Levels of EPO
4.4. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vasan, R.S.; Benjamin, E.J.; Levy, D. Prevalence, clinical features, and prognosis of diastolic heart failure: An epidemiologic perspective. J. Am. Coll. Cardiol. 1995, 26, 1565–1574. [Google Scholar] [CrossRef] [Green Version]
- Kupari, M.; Lindroos, M.; Iivanainen, A.M.; Heikkilä, J.; Tilvis, R. Congestive heart failure in old age: Prevalence, mechanisms and 4-year prognosis in the Helsinki Ageing Study. J. Intern. Med. 1997, 241, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Vasan, R.S.; Larson, S.M.G.; Benjamin, E.J.; Evans, J.C.; Reiss, C.K.; Levy, D. Congestive heart failure in subjects with normal versus reduced left ventricular ejection fraction: Prevalence and mortality in a population-based cohort. J. Am. Coll. Cardiol. 1999, 33, 1948–1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devereux, R.B.; Roman, M.J.; Liu, J.E.; Welty, T.K.; Lee, E.T.; Rodeheffer, R.; Fabsitz, R.R.; Howard, B.V. Congestive heart failure despite normal left ventricular systolic function in a population-based sample: The Strong Heart Study. Am. J. Cardiol. 2000, 86, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Kitzman, D.W.; Gardin, J.M.; Gottdiener, J.S.; Arnold, A.; Boineau, R.; Aurigemma, G.; Marino, E.K.; Lyles, M.; Cushman, M.; Enright, P.L. Importance of heart failure with preserved systolic function in patients ≥65 years of age. Am. J. Cardiol. 2001, 87, 413–419. [Google Scholar] [CrossRef]
- Gottdiener, J.S.; McClelland, R.L.; Marshall, R.; Shemanski, L.; Furberg, C.D.; Kitzman, D.W.; Cushman, M.; Polak, J.; Gardin, J.M.; Gersh, B.J.; et al. Outcome of Congestive Heart Failure in Elderly Persons: Influence of Left Ventricular Systolic Function: The Cardiovascular Health Study. Ann. Intern. Med. 2002, 137, 631–639. [Google Scholar] [CrossRef]
- Tribouilloy, C.; Rusinaru, D.; Mahjoub, H.; Soulière, V.; Lévy, F.; Peltier, M.; Slama, M.; Massy, Z. Prognosis of heart failure with preserved ejection fraction: A 5-year prospective population-based study. Eur. Heart J. 2007, 29, 339–347. [Google Scholar] [CrossRef]
- Lenzen, M.; Reimer, W.S.O.; Boersma, E.; Vantrimpont, P.; Follath, F.; Swedberg, K.; Cleland, J.; Komajda, M. Differences between patients with a preserved and a depressed left ventricular function: A report from the EuroHeart Failure Survey. Eur. Heart J. 2004, 25, 1214–1220. [Google Scholar] [CrossRef] [Green Version]
- Yancy, C.W.; Lopatin, M.; Stevenson, L.W.; De Marco, T.; Fonarow, G.C. Clinical presentation, management, and in-hospital outcomes of patients admitted with acute decompensated heart failure with preserved systolic function: A report from the Acute Decompensated Heart Failure National Registry (ADHERE) Database. J. Am. Coll. Cardiol. 2006, 47, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Fonarow, G.C.; Stough, W.G.; Abraham, W.T.; Albert, N.M.; Gheorghiade, M.; Greenberg, B.H.; O’Connor, C.M.; Sun, J.L.; Yancy, C.W.; Young, J.B.; et al. Characteristics, treatments, and outcomes of patients with preserved systolic function hospitalized for heart failure: A report from the OPTIMIZE-HF Registry. J. Am. Coll. Cardiol. 2007, 50, 768–777. [Google Scholar] [CrossRef] [Green Version]
- Owan, T.E.; Hodge, D.O.; Herges, R.M.; Jacobsen, S.J.; Roger, V.L.; Redfield, M.M. Trends in Prevalence and Outcome of Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2006, 355, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K.; Kass, D.A. Heart failure with preserved ejection fraction: Mechanisms, clinical features, and therapies. Circ. Res. 2014, 115, 79–96. [Google Scholar] [CrossRef] [Green Version]
- Dryer, K.; Gajjar, M.; Narang, N.; Lee, M.; Paul, J.; Shah, A.P.; Nathan, S.; Butler, J.; Davidson, C.J.; Fearon, W.F.; et al. Coronary microvascular dysfunction in patients with heart failure with preserved ejection fraction. Am. J. Physiol. Circ. Physiol. 2018, 314, H1033–H1042. [Google Scholar] [CrossRef]
- Shah, S.J.; Borlaug, B.A.; Kitzman, D.W.; McCulloch, A.D.; Blaxall, B.C.; Agarwal, R.; Chirinos, J.A.; Collins, S.; Deo, R.C.; Gladwin, M.T.; et al. Research Priorities for Heart Failure with Preserved Ejection Fraction: National Heart, Lung, and Blood Institute Working Group Summary. Circulation 2020, 141, 1001–1026. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.W.; Lyass, A.; Enserro, D.; Larson, M.G.; Ho, J.E.; Kizer, J.R.; Gottdiener, J.S.; Psaty, B.M.; Vasan, R.S. Temporal Trends in the Incidence of and Mortality Associated with Heart Failure with Preserved and Reduced Ejection Fraction. JACC: Heart Fail. 2018, 6, 678–685. [Google Scholar] [CrossRef]
- Beale, A.L.; Nanayakkara, S.; Kaye, D.M. Impact of Sex on Ventricular-Vascular Stiffness and Long-Term Outcomes in Heart Failure with Preserved Ejection Fraction: TOPCAT Trial Substudy. J. Am. Heart Assoc. 2019, 8, e012190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, P.; Paul, T.; Almarzooq, Z.I.; Peterson, J.C.; Krishnan, U.; Swaminathan, R.V.; Feldman, D.N.; Wells, M.T.; Karas, M.G.; Sobol, I.; et al. Sex- and Race-Related Differences in Characteristics and Outcomes of Hospitalizations for Heart Failure with Preserved Ejection Fraction. J. Am. Heart Assoc. 2017, 6, e003330. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K.; Mok, Y.; Kwak, L.; Agarwal, S.K.; Chang, P.P.; Deswal, A.; Shah, A.M.; Kitzman, D.W.; Wruck, L.M.; Loehr, L.R.; et al. Predictors of Mortality by Sex and Race in Heart Failure with Preserved Ejection Fraction: ARIC Community Surveillance Study. J. Am. Heart Assoc. 2020, 9, e014669. [Google Scholar] [CrossRef]
- Sotomi, Y.; Hikoso, S.; Nakatani, D.; Mizuno, H.; Okada, K.; Dohi, T.; Kitamura, T.; Sunaga, A.; Kida, H.; Oeun, B.; et al. Sex Differences in Heart Failure with Preserved Ejection Fraction. J. Am. Heart Assoc. 2021, 10, e018574. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.A.; Shah, A.M.; Borlaug, B.A. Heart Failure with Preserved Ejection Fraction In Perspective. Circ. Res. 2019, 124, 1598–1617. [Google Scholar] [CrossRef]
- Heusch, G.; Bøtker, H.E.; Przyklenk, K.; Redington, A.; Yellon, D. Remote ischemic conditioning. J. Am. Coll. Cardiol. 2015, 65, 177–195. [Google Scholar] [CrossRef] [Green Version]
- Ciccarelli, M.; Dawson, D.; Falcao-Pires, I.; Giacca, M.; Hamdani, N.; Heymans, S.; Hooghiemstra, A.; Leeuwis, A.; Hermkens, D.; Tocchetti, C.G.; et al. Reciprocal organ interactions during heart failure: A position paper from the ESC Working Group on Myocardial Function. Cardiovasc. Res. 2021, 117, 2416–2433. [Google Scholar] [CrossRef] [PubMed]
- Málek, F.; Gajewski, P.; Zymliński, R.; Janczak, D.; Chabowski, M.; Fudim, M.; Martinca, T.; Neužil, P.; Biegus, J.; Mates, M.; et al. Surgical ablation of the right greater splanchnic nerve for the treatment of heart failure with preserved ejection fraction: First-in-human clinical trial. Eur. J. Heart Fail. 2021, 23, 1134–1143. [Google Scholar] [CrossRef]
- D’Assante, R.; Arcopinto, M.; Rengo, G.; Salzano, A.; Walser, M.; Gambino, G.; Monti, M.G.; Bencivenga, L.; Marra, A.M.; Åberg, D.N.; et al. Myocardial expression of somatotropic axis, adrenergic signalling, and calcium handling genes in heart failure with preserved ejection fraction and heart failure with reduced ejection fraction. ESC Heart Fail. 2021, 8, 1681–1686. [Google Scholar] [CrossRef] [PubMed]
- Wolfien, M.; Klatt, D.; Salybekov, A.A.; Ii, M.; Komatsu-Horii, M.; Gaebel, R.; Philippou-Massier, J.; Schrinner, E.; Akimaru, H.; Akimaru, E.; et al. Hematopoietic stem-cell senescence and myocardial repair—Coronary artery disease genotype/phenotype analysis of post-MI myocardial regeneration response induced by CABG/CD133+ bone marrow hematopoietic stem cell treatment in RCT PERFECT Phase 3. Ebiomedicine 2020, 57, 102862. [Google Scholar] [CrossRef]
- Subbotina, E.; Sierra, A.; Zhu, Z.; Gao, Z.; Koganti, S.R.; Reyes, S.; Stepniak, E.; Walsh, S.A.; Acevedo, M.R.; Perez-Terzic, C.M.; et al. Musclin is an activity-stimulated myokine that enhances physical endurance. Proc. Natl. Acad. Sci. USA 2015, 112, 16042–16047. [Google Scholar] [CrossRef] [Green Version]
- Homme, R.P.; Zheng, Y.; Smolenkova, I.; Singh, M.; Tyagi, S.C. Remote Hind-Limb Ischemia Mechanism of Preserved Ejection Fraction During Heart Failure. Front. Physiol. 2021, 12, 745328. [Google Scholar] [CrossRef]
- Joslin, J.R.; Lioudaki, E.; Androulakis, E. Interrelation between heart failure with preserved ejection fraction and renal impairment. Rev. Cardiovasc. Med. 2022, 23, 69. [Google Scholar] [CrossRef] [PubMed]
- Nejima, J.; Uemura, N.; Vatner, D.E.; Homcy, C.J.; Hintze, T.H.; Vatner, S.F. Role of intact cardiac nerves and reflex mechanisms in desensitization to catecholamines in conscious dogs. J. Clin. Investig. 1990, 86, 2046–2053. [Google Scholar] [CrossRef] [Green Version]
- Maranon, R.O.; Lima, R.; Mathbout, M.; Carmo, J.M.D.; Hall, J.E.; Roman, R.J.; Reckelhoff, J.F. Postmenopausal hypertension: Role of the sympathetic nervous system in an animal model. Am. J. Physiol. Integr. Comp. Physiol. 2014, 306, R248–R256. [Google Scholar] [CrossRef] [Green Version]
- Moreno, C.; Mistry, M.; Roman, R.J. Renal effects of glucagon-like peptide in rats. Eur. J. Pharmacol. 2001, 434, 163–167. [Google Scholar] [CrossRef]
- Mansour, S.G.; Bhatraju, P.K.; Coca, S.G.; Obeid, W.; Wilson, F.P.; Stanaway, I.B.; Jia, Y.; Thiessen-Philbrook, H.; Go, A.S.; Ikizler, T.A.; et al. Angiopoietins as Prognostic Markers for Future Kidney Disease and Heart Failure Events after Acute Kidney Injury. J. Am. Soc. Nephrol. 2022, 33, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Chedid, M.; Hanna, C.; Zaatari, G.; Mkhaimer, Y.; Reddy, P.; Rangel, L.; Zubidat, D.; Kaidbay, D.-H.N.; Irazabal, M.V.; Connolly, H.M.; et al. Congenital Heart Disease in Adults with Autosomal Dominant Polycystic Kidney Disease. Am. J. Nephrol. 2022, 53, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Hepokoski, M.; Singh, P. Mitochondria as mediators of systemic inflammation and organ cross talk in acute kidney injury. Am. J. Physiol. Physiol. 2022, 322, F589–F596. [Google Scholar] [CrossRef]
- Libby, A.E.; Jones, B.; Levi, M. Learning the ABCs of ATP release. J. Biol. Chem. 2020, 295, 5204–5205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Couto, G.; Mesquita, T.; Wu, X.; Rajewski, A.; Huang, F.; Akhmerov, A.; Na, N.; Wu, D.; Wang, Y.; Li, L.; et al. Cell therapy attenuates endothelial dysfunction in hypertensive rats with heart failure and preserved ejection fraction. Am. J. Physiol. Circ. Physiol. 2022, 323, H892–H903. [Google Scholar] [CrossRef]
- Fliser, D.; Bahlmann, F.H.; deGroot, K.; Haller, H. Mechanisms of disease: Erythropoietin—An old hormone with a new mission? Nature clinical practice. Cardiovasc. Med. 2006, 3, 563–572. [Google Scholar]
- Gok, M.G.; Paydas, S.; Boral, B.; Onan, E.; Kaya, B. Evaluation of eryptosis in patients with chronic kidney disease. Int. Urol. Nephrol. 2022, 54, 2919–2928. [Google Scholar] [CrossRef]
- Naing, P.; Forrester, D.; Kangaharan, N.; Muthumala, A.; Myint, S.M.; Playford, D. Heart failure with preserved ejection fraction: A growing global epidemic. Aust. J. Gen. Pract. 2019, 48, 465–471. [Google Scholar] [CrossRef]
- De Gasperi, R.; Hamidi, S.; Harlow, L.M.; Ksiezak-Reding, H.; Bauman, W.A.; Cardozo, C.P. Denervation-related alterations and biological activity of miRNAs contained in exosomes released by skeletal muscle fibers. Sci. Rep. 2017, 7, 12888. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Wang, K.; Wang, S.; Zhang, B.; Liu, Q.; Zhang, Q.; Geng, J.; Shan, Q. Beneficial effects of renal denervation on cardiac angiogenesis in rats with prolonged pressure overload. Acta Physiol. 2016, 220, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Shi, G.; Wang, X.; Tu, S.; Zhang, Z.; Zeng, L. [Renal sympathetic denervation can significantly reduce blood pressure and improve arterial stiffness in hypertensive beagles]. Nan Fang Yi Ke Da Xue Xue Bao = J. South. Med. Univ. 2021, 41, 1609–1615. [Google Scholar]
- Wang, Y.; Rijal, B.; Xu, M.; Li, Z.; An, Y.; Zhang, F.; Lu, C. Renal denervation improves vascular endothelial dysfunction by inducing autophagy via AMPK/mTOR signaling activation in a rat model of type 2 diabetes mellitus with insulin resistance. Acta Diabetol. 2020, 57, 1227–1243. [Google Scholar] [CrossRef]
- Eikelis, N.; Hering, D.; Marusic, P.; Sari, C.; Walton, A.; Phillips, S.; Lambert, E.; Duval, J.; Krum, H.; Esler, M.; et al. The effect of renal denervation on endothelial function and inflammatory markers in patients with resistant hypertension. Int. J. Cardiol. 2015, 188, 96–98. [Google Scholar] [CrossRef]
- Doltra, A.; Messroghli, D.; Stawowy, P.; Hassel, J.; Gebker, R.; Leppänen, O.; Gräfe, M.; Schneeweis, C.; Schnackenburg, B.; Fleck, E.; et al. Potential Reduction of Interstitial Myocardial Fibrosis with Renal Denervation. J. Am. Heart Assoc. 2014, 3, e001353. [Google Scholar] [CrossRef] [Green Version]
- Xiao, B.; Liu, F.; Jin, Y.-H.; Jin, Y.-Q.; Wang, L.; Lu, J.-C.; Yang, X.-C. Renal sympathetic denervation attenuates left ventricle hypertrophy in spontaneously hypertensive rats by suppressing the Raf/MEK/ERK signaling pathway. Clin. Exp. Hypertens. 2020, 43, 142–150. [Google Scholar] [CrossRef]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA guideline for the management of heart failure: Executive summary: A report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013, 128, 1810–1852. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar]
- Yamada, S.; Fong, M.; Hsiao, Y.; Chang, S.; Tsai, Y.; Lo, L.; Chao, T.; Lin, Y.; Hu, Y.; Chung, F.; et al. Impact of Renal Denervation on Atrial Arrhythmogenic Substrate in Ischemic Model of Heart Failure. J. Am. Heart Assoc. 2018, 7, e007312. [Google Scholar] [CrossRef] [Green Version]
- Guyton, A.C.; Sagawa, K. Compensations of cardiac output and other circulatory functions in areflex dogs with large A-V fistulas. Am. J. Physiol. Content 1961, 200, 1157–1163. [Google Scholar] [CrossRef] [PubMed]
- Brower, G.L.; Henegar, J.R.; Janicki, J.S. Temporal evaluation of left ventricular remodeling and function in rats with chronic volume overload. Am. J. Physiol. Content 1996, 271, H2071–H2078. [Google Scholar] [CrossRef]
- Cox, M.J.; Sood, H.S.; Hunt, M.J.; Chandler, D.; Henegar, J.R.; Aru, G.M.; Tyagi, S.C. Apoptosis in the left ventricle of chronic volume overload causes endocardial endothelial dysfunction in rats. Am. J. Physiol. Circ. Physiol. 2002, 282, H1197–H1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagi, S.C.; Kumar, S.G.; Haas, S.J.; Reddy, H.K.; Voelker, D.J.; Hayden, M.R.; Demmy, T.L.; Schmaltz, R.A.; Curtis, J.J. Post-transcriptional Regulation of Extracellular Matrix Metalloproteinase in Human Heart End-stage Failure Secondary to Ischemic Cardiomyopathy. J. Mol. Cell. Cardiol. 1996, 28, 1415–1428. [Google Scholar] [CrossRef]
- Lim, G.B. Sustained BP reduction with renal denervation. Nat. Rev. Cardiol. 2022, 19, 352. [Google Scholar] [CrossRef]
- Tyagi, S.C.; Kumar, S.; Voelker, D.J.; Reddy, H.K.; Janicki, J.S.; Curtis, J.J. Differential gene expression of extracellular matrix components in dilated cardiomyopathy. J. Cell. Biochem. 1996, 63, 185–198. [Google Scholar] [CrossRef]
- Passmore, J.C.; Joshua, I.G.; Rowell, P.P.; Tyagi, S.C.; Falcone, J.C. Reduced alpha adrenergic mediated contraction of renal preglomerular blood vessels as a function of gender and aging. J. Cell. Biochem. 2005, 96, 672–681. [Google Scholar] [CrossRef]
- Tarone, R.E. A modified Bonferroni method for discrete data. Biometrics 1990, 46, 515–522. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pushpakumar, S.; Singh, M.; Zheng, Y.; Akinterinwa, O.E.; Mokshagundam, S.P.L.; Sen, U.; Kalra, D.K.; Tyagi, S.C. Renal Denervation Helps Preserve the Ejection Fraction by Preserving Endocardial-Endothelial Function during Heart Failure. Int. J. Mol. Sci. 2023, 24, 7302. https://doi.org/10.3390/ijms24087302
Pushpakumar S, Singh M, Zheng Y, Akinterinwa OE, Mokshagundam SPL, Sen U, Kalra DK, Tyagi SC. Renal Denervation Helps Preserve the Ejection Fraction by Preserving Endocardial-Endothelial Function during Heart Failure. International Journal of Molecular Sciences. 2023; 24(8):7302. https://doi.org/10.3390/ijms24087302
Chicago/Turabian StylePushpakumar, Sathnur, Mahavir Singh, Yuting Zheng, Oluwaseun E. Akinterinwa, Sri Prakash L. Mokshagundam, Utpal Sen, Dinesh K. Kalra, and Suresh C. Tyagi. 2023. "Renal Denervation Helps Preserve the Ejection Fraction by Preserving Endocardial-Endothelial Function during Heart Failure" International Journal of Molecular Sciences 24, no. 8: 7302. https://doi.org/10.3390/ijms24087302