
Prospective Roles of Tumor Necrosis Factor-Alpha (TNF-α) in COVID-19: Prognosis, Therapeutic and Management

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Inflammation in SARS-CoV-2 Infection
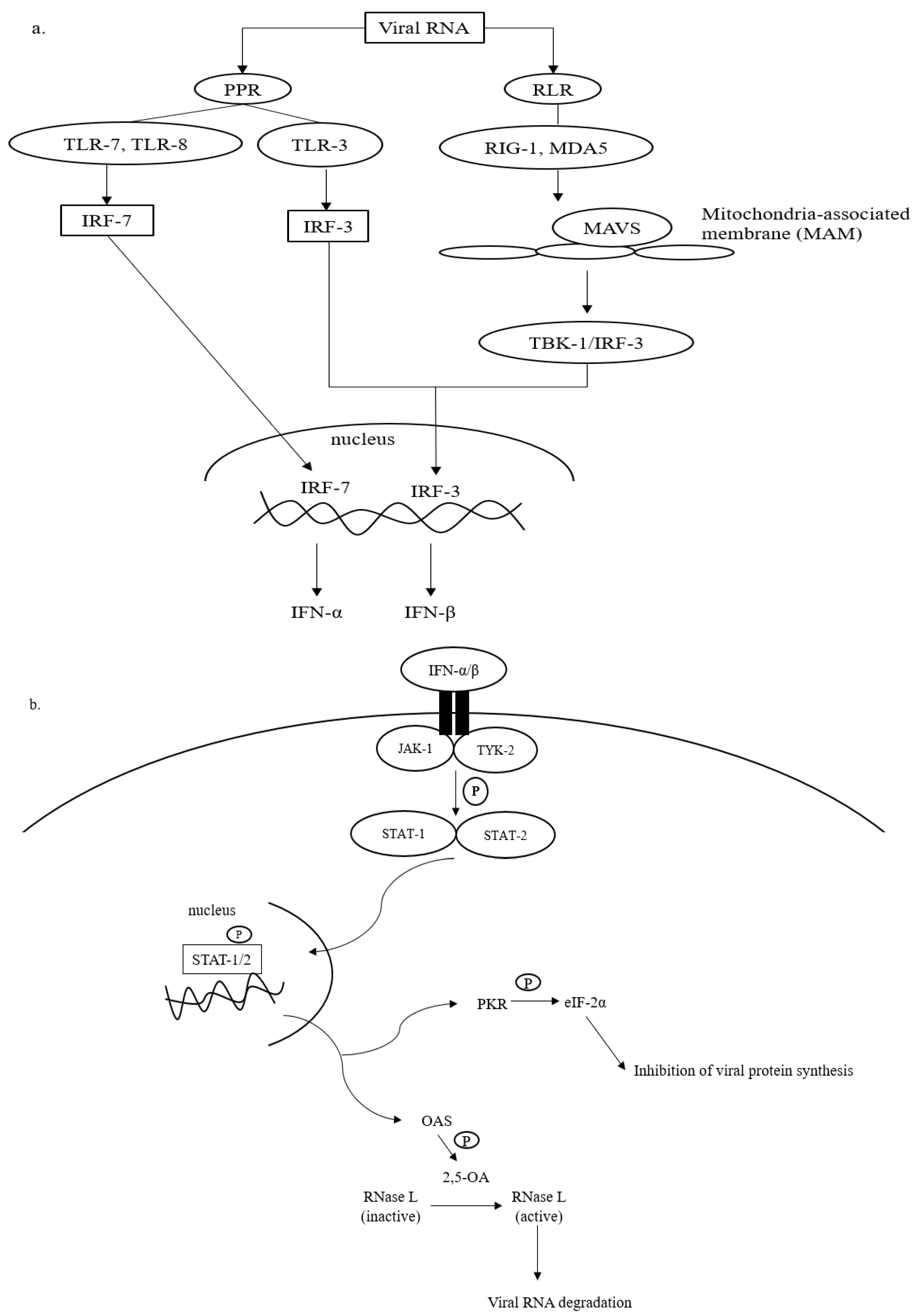
2.1. Activation of Innate Immune Response by SARS-CoV-2
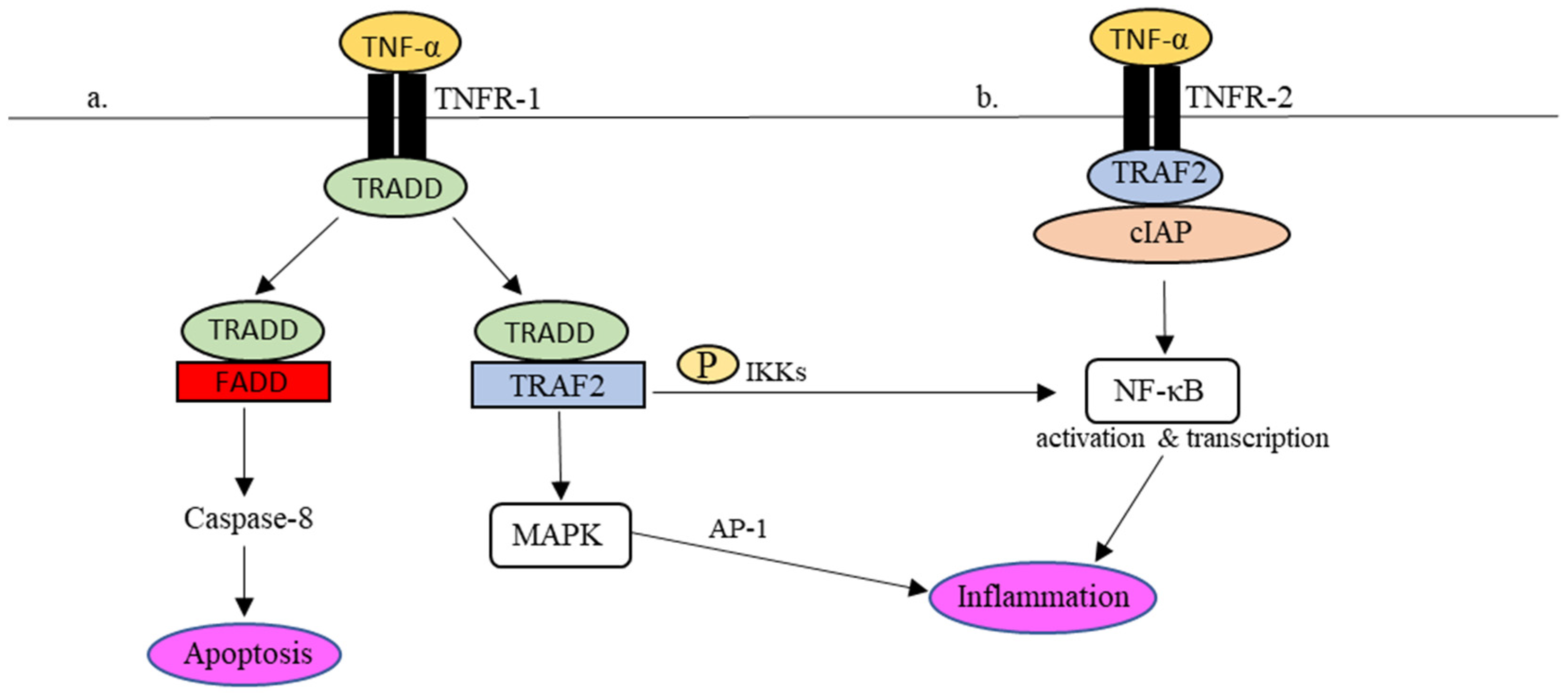
2.2. Activation of TNF-α Signaling Pathway by SARS-CoV-2 and Its Potential in Virus Containment
3. Implication of TNF-α Signaling in COVID-19 Mortality and Morbidity
4. Potential and Prospective Applications of TNF-α in COVID-19 Management
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ludwig, S.; Zarbock, A. Coronaviruses and SARS-CoV-2: A Brief Overview. Anesth. Analg. 2020, 131, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Mohandas, S.; Yadav, P.D.; Shete, A.; Nyayanit, D.; Sapkal, G.; Lole, K.; Gupta, N. SARS-CoV-2 Delta Variant Pathogenesis and Host Response in Syrian Hamsters. Viruses 2021, 13, 1773. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Pascarella, G.; Strumia, A.; Piliego, C.; Bruno, F.; Del Buono, R.; Costa, F.; Scarlata, S.; Agrò, F.E. COVID-19 diagnosis and management: A comprehensive review. J. Intern. Med. 2020, 288, 192–206. [Google Scholar] [CrossRef]
- Rivas, M.N.; Porritt, R.A.; Cheng, M.H.; Bahar, I.; Arditi, M. COVID-19–associated multisystem inflammatory syndrome in children (MIS-C): A novel disease that mimics toxic shock syndrome—The superantigen hypothesis. J. Allergy Clin. Immunol. 2020, 147, 57–59. [Google Scholar] [CrossRef]
- Abdelmoaty, M.M.; Yeapuri, P.; Machhi, J.; Olson, K.E.; Shahjin, F.; Kumar, V.; Pandey, K. Defining the Innate Immune Responses for SARS-CoV-2-Human Macrophage Interactions. Front. Immunol. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Grant, R.A.; Morales-Nebreda, L.; Markov, N.S.; Swaminathan, S.; Querrey, M.; Guzman, E.R.; Abbott, D.A.; Donnelly, H.K.; Donayre, A.; Goldberg, I.A.; et al. Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature 2021, 590, 635–641. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Qu, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, J.; Zhang, D.; Xu, Z.; Ji, J.; Wen, C. Cytokine Storm in COVID-19: The Current Evidence and Treatment Strategies. Front. Immunol. 2020, 11, 1708. [Google Scholar] [CrossRef]
- Ragab, D.; Eldin, H.S.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 Cytokine Storm; What We Know So Far. Front. Immunol. 2020, 11, 1446. [Google Scholar] [CrossRef]
- Que, Y.; Hu, C.; Wan, K.; Hu, P.; Wang, R.; Luo, J.; Li, T.; Ping, R.; Hu, Q.; Sun, Y.; et al. Cytokine release syndrome in COVID-19: A major mechanism of morbidity and mortality. Int. Rev. Immunol. 2021, 41, 217–230. [Google Scholar] [CrossRef]
- Reyes, A.; Hu, K.; Teperman, J.; Muskardin, T.W.; Tardif, J.-C.; Shah, B.; Pillinger, M. Anti-inflammatory therapy for COVID-19 infection: The case for colchicine. Ann. Rheum. Dis. 2021, 80, 550–557. [Google Scholar] [CrossRef]
- Jang, D.-I.; Lee, A.-H.; Shin, H.-Y.; Song, H.-R.; Park, J.-H.; Kang, T.-B.; Lee, S.-R.; Yang, S.-H. The Role of Tumor Necrosis Factor Alpha (TNF-α) in Autoimmune Disease and Current TNF-α Inhibitors in Therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef]
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.; McDermott, M.F. Tumour necrosis factor signalling in health and disease. F1000Research 2019, 8, 111. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.-Y.; Chang, C.-Y.; Hsu, H.-J.; Shih, H.-J.; Huang, I.-T.; Patel, H.H.; Huang, C.-J. Tumor Necrosis Factor-α Mediates Lung Injury in the Early Phase of Endotoxemia. Pharmaceuticals 2022, 15, 287. [Google Scholar] [CrossRef]
- Sinha, P.; Ware, L.B. Selective tumour necrosis factor receptor-1 inhibition in acute lung injury: A new hope or a false dawn? Thorax 2018, 73, 699–701. [Google Scholar] [CrossRef]
- Weaver, A.L. Differentiating the new rheumatoid arthritis biologic thera-pies. JCR J. Clin. Rheumatol. 2003, 9, 99–114. [Google Scholar] [CrossRef]
- Barrera, P.; van Der Maas, A.; Van Ede, A.E.; Kiemeney BA, L.M.; Laan RF, J.M.; Van de Putte LB, A.; van Riel PL, C.M. Drug survival, efficacy and toxicity of monotherapy with a fully human anti-tumour necrosis factor-α anti-body compared with methotrexate in long-standing rheumatoid arthritis. Rheu-Matology 2002, 41, 430–439. [Google Scholar] [CrossRef] [Green Version]
- Borst, S.E. The role of TNF-α in insulin resistance. Endocrine 2004, 23, 177–182. [Google Scholar] [CrossRef]
- Viganò, M.; Degasperi, E.; Aghemo, A.; Lampertico, P.; Colombo, M. Anti-TNF drugs in patients with hepatitis B or C virus infection: Safety and clinical management. Expert Opin. Biol. Ther. 2011, 12, 193–207. [Google Scholar] [CrossRef] [PubMed]
- McDermott, J.E.; Mitchell, H.D.; Gralinski, L.E.; Eisfeld, A.J.; Josset, L.; Bankhead, A.; Neumann, G.; Tilton, S.C.; Schäfer, A.; Li, C.; et al. The effect of inhibition of PP1 and TNFα signaling on pathogenesis of SARS coronavirus. BMC Syst. Biol. 2016, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussell, T.; Pennycook, A.; Openshaw, P.J.M. Inhibition of tumor necrosis factor reduces the severity of virus-specific lung immunopathology. Eur. J. Immunol. 2001, 31, 2566–2573. [Google Scholar] [CrossRef] [PubMed]
- Gianfrancesco, M.; Yazdany, J.; Robinson, P.C. Epidemiology and outcomes of novel coronavirus 2019 in patients with immune-mediated inflammatory diseases. Curr. Opin. Rheumatol. 2020, 32, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.S.; Warrington, R.; Watson, W.; Kim, H.L. An introduction to immunology and immunopathology. Allergy Asthma Clin. Immunol. 2018, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.-D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef]
- Park, A.; Iwasaki, A. Type I and type III interferons–induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe 2020, 27, 870–878. [Google Scholar] [CrossRef]
- Ramasamy, S.; Subbian, S. Critical Determinants of Cytokine Storm and Type I Interferon Response in COVID-19 Pathogenesis. Clin. Microbiol. Rev. 2021, 34, e00299-20. [Google Scholar] [CrossRef]
- Amor, S.; Blanco, L.F.; Baker, D. Innate immunity during SARS-CoV-2: Evasion strategies and activation trigger hypoxia and vascular damage. Clin. Exp. Immunol. 2020, 202, 193–209. [Google Scholar] [CrossRef]
- Diamond, M.S.; Kanneganti, T.-D. Innate immunity: The first line of defense against SARS-CoV-2. Nat. Immunol. 2022, 23, 165–176. [Google Scholar] [CrossRef]
- Li, Y.; Renner, D.M.; Comar, C.E.; Whelan, J.N.; Reyes, H.M.; Cardenas-Diaz, F.L.; Weiss, S.R. SARS-CoV-2 induces double-stranded rna-mediated innate immune responses in respiratory epithelial-derived cells and cardiomyocytes. Proc. Natl. Acad. Sci. USA 2021, 118, e2022643118. [Google Scholar] [CrossRef]
- Schultze, J.L.; Aschenbrenner, A.C. COVID-19 and the human innate immune system. Cell 2021, 184, 1671–1692. [Google Scholar] [CrossRef]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef]
- Lu, Y.; Michel, H.A.; Wang, P.-H.; Smith, G.L. Manipulation of innate immune signaling pathways by SARS-CoV-2 non-structural proteins. Front. Microbiol. 2022, 13, 1027015. [Google Scholar] [CrossRef]
- Cesaro, T.; Michiels, T. Inhibition of PKR by Viruses. Front. Microbiol. 2021, 12, 757238. [Google Scholar] [CrossRef]
- Rabouw, H.H.; Langereis, M.A.; Knaap, R.C.M.; Dalebout, T.J.; Canton, J.; Sola, I.; Enjuanes, L.; Bredenbeek, P.J.; Kikkert, M.; de Groot, R.J.; et al. Middle East Respiratory Coronavirus Accessory Protein 4a Inhibits PKR-Mediated Antiviral Stress Responses. PLoS Pathog. 2016, 12, e1005982. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Xu, L.H.; Yamada, Y.; Liu, D.X. Coronavirus Spike Protein Inhibits Host Cell Translation by Interaction with eIF3f. PLoS ONE 2008, 3, e1494. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Shu, T.; Wu, D.; Mu, J.; Wang, C.; Huang, M.; Han, Y.; Zhang, X.-Y.; Zhou, W.; Qiu, Y.; et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell. Mol. Immunol. 2020, 17, 881–883. [Google Scholar] [CrossRef]
- Majumdar, P.; Niyogi, S. ORF3a mutation associated with higher mortality rate in SARS-CoV-2 infection. Epide-Miology Infect. 2020, 148, e262. [Google Scholar] [CrossRef]
- Faist, A.; Janowski, J.; Kumar, S.; Hinse, S.; Çalışkan, D.M.; Lange, J.; Ludwig, S.; Brunotte, L. Virus Infection and Systemic Inflammation: Lessons Learnt from COVID-19 and Beyond. Cells 2022, 11, 2198. [Google Scholar] [CrossRef]
- Montazersaheb, S.; Khatibi, S.M.H.; Hejazi, M.S.; Tarhriz, V.; Farjami, A.; Sorbeni, F.G.; Farahzadi, R.; Ghasemnejad, T. COVID-19 infection: An overview on cytokine storm and related interventions. Virol. J. 2022, 19, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Manik, M.; Singh, R.K. Role of toll-like receptors in modulation of cytokine storm signaling in SARS-CoV-2-induced COVID-19. J. Med. Virol. 2021, 94, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Lan, Z.; Ye, J.; Pang, L.; Liu, Y.; Wu, W.; Qin, X.; Guo, Y.; Zhang, P. Cytokine Storm: The Primary Determinant for the Pathophysiological Evolution of COVID-19 Deterioration. Front. Immunol. 2021, 12, 589095. [Google Scholar] [CrossRef] [PubMed]
- Udomsinprasert, W.; Jittikoon, J.; Sangroongruangsri, S.; Chaikledkaew, U. Circulating Levels of Interleukin-6 and Interleukin-10, But Not Tumor Necrosis Factor-Alpha, as Potential Biomarkers of Severity and Mortality for COVID-19: Systematic Review with Meta-analysis. J. Clin. Immunol. 2020, 41, 11–22. [Google Scholar] [CrossRef]
- Tan, L.Y.; Komarasamy, T.V.; Balasubramaniam, V.R. Hyperinflammatory Immune Response and COVID-19: A Double Edged Sword. Front. Immunol. 2021, 12, 742941. [Google Scholar] [CrossRef]
- Bacci, M.; Leme, R.; Zing, N.P.C.; Murad, N.; Adami, F.; Hinnig, P.; Feder, D.; Chagas, A.; Fonseca, F. IL-6 and TNF-α serum levels are associated with early death in community-acquired pneumonia patients. Braz. J. Med Biol. Res. 2015, 48, 427–432. [Google Scholar] [CrossRef] [Green Version]
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.-H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A.; et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020, 26, 1636–1643. [Google Scholar] [CrossRef]
- Mortaz, E.; Tabarsi, P.; Jamaati, H.; Dalil Roofchayee, N.; Dezfuli, N.K.; Hashemian, S.M.; Moniri, A.; Marjani, M.; Malekmohammad, M.; Mansouri, D.; et al. Increased Serum Levels of Soluble TNF-α Receptor Is Asso-ci-ated With ICU Mortality in COVID-19 Patients. Front Immunol. 2021, 12, 592727. [Google Scholar] [CrossRef]
- Mokhtari, T.; Hassani, F.; Ghaffari, N.; Ebrahimi, B.; Yarahmadi, A.; Hassanzadeh, G. COVID-19 and multiorgan failure: A narrative review on potential mechanisms. Histochem. J. 2020, 51, 613–628. [Google Scholar] [CrossRef]
- Liu, K.; Yang, T.; Peng, X.; Lv, S.; Ye, X.; Zhao, T.; Li, J.; Shao, Z.; Lu, Q.; Li, J.; et al. A systematic meta-analysis of immune signatures in patients with COVID-19. Rev. Med. Virol. 2020, 31, e2195. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Yang, S.; Wang, J.; Brand, D.D.; Zheng, S.G. Role of TNF–TNF Receptor 2 Signal in Regulatory T Cells and Its Therapeutic Implications. Front. Immunol. 2018, 9, 784. [Google Scholar] [CrossRef] [Green Version]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef] [Green Version]
- Darif, D.; Hammi, I.; Kihel, A.; Saik, I.E.I.; Guessous, F.; Akarid, K. The pro-inflammatory cytokines in COVID-19 pathogenesis: What goes wrong? Microb. Pathog. 2021, 153, 104799. [Google Scholar] [CrossRef]
- Wang, W.; Ye, L.; Ye, L.; Li, B.; Gao, B.; Zeng, Y.; Kong, L.; Fang, X.; Zheng, H.; Wu, Z. Up-regulation of IL-6 and TNF-α induced by SARS-coronavirus spike protein in murine macrophages via NF-κB pathway. Virus Res. 2007, 128, 1–8. [Google Scholar] [CrossRef]
- Hojyo, S.; Uchida, M.; Tanaka, K.; Hasebe, R.; Tanaka, Y.; Murakami, M.; Hirano, T. How COVID-19 induces cyto-kine storm with high mortality. Inflamm. Regen. 2020, 40, 1–7. [Google Scholar] [CrossRef]
- Leisman, D.E.; Ronner, L.; Pinotti, R.; Taylor, M.D.; Sinha, P.; Calfee, C.S.; Deutschman, C.S. Cytokine eleva-tion in severe and critical COVID-19: A rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir. Med. 2020, 8, 1233–1244. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Tanabe, K.; Takai, S.; Matsushima-Nishiwaki, R.; Adachi, S.; Iida, H.; Kozawa, O.; Dohi, S. Involvement of Rho-kinase in tumor necrosis factor-α-induced interleukin-6 release from C6 glioma cells. Neurochem. Int. 2009, 55, 438–445. [Google Scholar] [CrossRef]
- Tanabe, K.; Matsushima-Nishiwaki, R.; Yamaguchi, S.; Iida, H.; Dohi, S.; Kozawa, O. Mechanisms of tumor necrosis factor-α-induced interleukin-6 synthesis in glioma cells. J. Neuroinflammation 2010, 7, 16. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.J.; Thorburn, J.; Thomas, L.; Maxwell, T.; Brothman, A.R. An apoptosis signaling pathway induced by the death domain of FADD selectively kills normal but not cancerous prostate epithelial cells. Cell Death Differ. 2001, 8, 696–705. [Google Scholar] [CrossRef] [Green Version]
- Attiq, A.; Yao, L.J.; Afzal, S.; Khan, M.A. The triumvirate of NF-κB, inflammation and cytokine storm in COVID-19. Int. Immunopharmacol. 2021, 101, 108255. [Google Scholar] [CrossRef] [PubMed]
- Yapasert, R.; Khaw-On, P.; Banjerdpongchai, R. Coronavirus Infection-Associated Cell Death Signaling and Potential Therapeutic Targets. Molecules 2021, 26, 7459. [Google Scholar] [CrossRef] [PubMed]
- Birra, D.; Benucci, M.; Landolfi, L.; Merchionda, A.; Loi, G.; Amato, P.; Licata, G.; Quartuccio, L.; Triggiani, M.; Moscato, P. COVID 19: A clue from innate immunity. Immunol. Res. 2020, 68, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Dorrington, M.G.; Fraser, I.D. NF-κB signaling in macrophages: Dynamics, crosstalk, and signal integration. Front. Immunol. 2019, 10, 705. [Google Scholar] [CrossRef]
- Domm, S.; Cinatl, J.; Mrowietz, U. The impact of treatment with tumour necrosis factor-α antagonists on the course of chronic viral infections: A review of the literature. Br. J. Dermatol. 2008, 159, 1217–1228. [Google Scholar] [CrossRef]
- Espín-Palazón, R.; Martínez-López, A.; Roca, F.J.; López-Muñoz, A.; Tyrkalska, S.D.; Candel, S.; García-Moreno, D.; Falco, A.; Meseguer, J.; Estepa, A.; et al. TNFα Impairs Rhabdoviral Clearance by Inhibiting the Host Autophagic Antiviral Response. PLoS Pathog. 2016, 12, e1005699. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Dolhnikoff, M.; Duarte-Neto, A.N.; de Almeida Monteiro, R.A.; Da Silva, L.F.F.; De Oliveira, E.P.; Saldiva, P.H.N.; Mauad, T.; Negri, E.M. Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J. Thromb. Haemost. 2020, 18, 1517–1519. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wang, Y.; Li, X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Mauad, T.; Duarte-Neto, A.N.; da Silva, L.F.F.; de Oliveira, E.P.; de Brito, J.M.; Nascimento, E.C.T.D.; Monteiro, R.A.D.A.; Ferreira, J.C.; de Carvalho, C.R.R.; Saldiva, P.H.D.N.; et al. Tracking the time course of pathological patterns of lung injury in severe COVID-19. Respir. Res. 2021, 22, 1–11. [Google Scholar] [CrossRef]
- Leija-Martínez, J.J.; Huang, F.; Del-Río-Navarro, B.E.; Sanchéz-Muñoz, F.; Muñoz-Hernández, O.; Giacoman-Martínez, A.; Hall-Mondragon, M.S.; Espinosa-Velazquez, D. IL-17A and TNF-α as potential biomarkers for acute respiratory distress syndrome and mortality in patients with obesity and COVID-19. Med. Hypotheses 2020, 144, 109935. [Google Scholar] [CrossRef]
- Savin, I.A.; Zenkova, M.A.; Sen’kova, A.V. Pulmonary Fibrosis as a Result of Acute Lung Inflammation: Molecular Mechanisms, Relevant In Vivo Models, Prognostic and Therapeutic Approaches. Int. J. Mol. Sci. 2022, 23, 14959. [Google Scholar] [CrossRef]
- Zheng, K.; Gao, F.; Wang, X.-B.; Sun, Q.-F.; Pan, K.-H.; Wang, T.-Y.; Ma, H.-L.; Chen, Y.-P.; Liu, W.-Y.; George, J.; et al. Obesity as a risk factor for greater severity of COVID-19 in patients with metabolic associated fatty liver disease. Metabolism 2020, 108, 154244. [Google Scholar] [CrossRef]
- Lighter, J.; Phillips, M.; Hochman, S.; Sterling, S.; Johnson, D.; Francois, F.; Stachel, A. Obesity in patients younger than 60 years is a risk factor for COVID-19 hospital admission. Clin. Infect. Dis. 2020, 71, 896–897. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; McGoogan, J.M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: Summary of a report of 72 314 cases from the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef]
- Wang, E.Y.; Mao, T.; Klein, J.; Dai, Y.; Huck, J.D.; Jaycox, J.R.; Liu, F.; Zhou, T.; Israelow, B.; Wong, P.; et al. Diverse functional autoantibodies in patients with COVID-19. Nature 2021, 595, 283–288. [Google Scholar] [CrossRef]
- Medina-Leyte, D.; Zepeda-García, O.; Domínguez-Pérez, M.; González-Garrido, A.; Villarreal-Molina, T.; Jacobo-Albavera, L. Endothelial Dysfunction, Inflammation and Coronary Artery Disease: Potential Biomarkers and Promising Therapeutical Approaches. Int. J. Mol. Sci. 2021, 22, 3850. [Google Scholar] [CrossRef]
- Re, V.L.; Dutcher, S.K.; Connolly, J.G.; Perez-Vilar, S.; Carbonari, D.M.; DeFor, T.A.; Djibo, D.A.; Harrington, L.B.; Hou, L.; Hennessy, S.; et al. Association of COVID-19 vs Influenza With Risk of Arterial and Venous Thrombotic Events Among Hospitalized Patients. JAMA 2022, 328, 637–651. [Google Scholar] [CrossRef]
- Katsoularis, I.; Fonseca-Rodríguez, O.; Farrington, P.; Jerndal, H.; Lundevaller, E.H.; Sund, M.; Lindmark, K.; Connolly, A.-M.F. Risks of deep vein thrombosis, pulmonary embolism, and bleeding after COVID-19: Nationwide self-controlled cases series and matched cohort study. BMJ 2022, 377, e069590. [Google Scholar] [CrossRef]
- Page, M.J.; Bester, J.; Pretorius, E. The inflammatory effects of TNF-α and complement component 3 on coagulation. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Kaptoge, S.; Seshasai, S.R.K.; Jørgensen, T.; Danesh, J.; Gao, P.; Freitag, D.F.; Butterworth, A.S.; Borglykke, A.; Di Angelantonio, E.; Gudnason, V.; et al. Inflammatory cytokines and risk of coronary heart disease: New prospective study and updated meta-analysis. Eur. Heart J. 2014, 35, 578–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelová, H.; Hošek, J. TNF-α signalling and inflammation: Interactions between old acquaintances. Inflamm. Res. 2013, 62, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Proal, A.D.; VanElzakker, M.B. Long COVID or Post-acute Sequelae of COVID-19 (PASC): An Overview of Biological Factors That May Contribute to Persistent Symptoms. Front. Microbiol. 2021, 12, 698169. [Google Scholar] [CrossRef] [PubMed]
- Peluso, M.J.; Lu, S.; Tang, A.F.; Durstenfeld, M.S.; Ho, H.-E.; Goldberg, S.A.; Forman, C.A.; Munter, S.E.; Hoh, R.; Tai, V.; et al. Markers of Immune Activation and Inflammation in Individuals With Postacute Sequelae of Severe Acute Respiratory Syndrome Coronavirus 2 Infection. J. Infect. Dis. 2021, 224, 1839–1848. [Google Scholar] [CrossRef] [PubMed]
- Schultheiß, C.; Willscher, E.; Paschold, L.; Gottschick, C.; Klee, B.; Henkes, S.-S.; Bosurgi, L.; Dutzmann, J.; Sedding, D.; Frese, T.; et al. The IL-1β, IL-6, and TNF cytokine triad is associated with post-acute sequelae of COVID-19. Cell Rep. Med. 2022, 3, 100663. [Google Scholar] [CrossRef]
- Taghiloo, S.; Soltanshahi, M.; Aliyali, M.; Abedi, S.; Mehravaran, H.; Ajami, A.; Asgarian-Omran, H. Cytokine profiling in Iranian patients with COVID-19; association with clinical severity. Iran. J. Immunol. 2021, 18, 54–64. [Google Scholar] [CrossRef]
- Palacios, Y.; Chavez-Galan, L. Immunosuppressant Therapies in COVID-19: Is the TNF Axis an Alternative? Pharmaceuticals 2022, 15, 616. [Google Scholar] [CrossRef]
- Gao, S.; Cheng, Y.; Zhang, S.; Zheng, X.; Wu, J. A biolayer interferometry-based, aptamer–antibody receptor pair biosensor for real-time, sensitive, and specific detection of the disease biomarker TNF-α. Chem. Eng. J. 2022, 433, 133268. [Google Scholar] [CrossRef]
- Illahi, M.I.; Amjad, S.; Alam, S.M.; Ahmed, S.T.; Fatima, M.; Shahid, M.A. Serum Tumor Necrosis Factor-Alpha as a Competent Biomarker for Evaluation of Disease Activity in Early Rheumatoid Arthritis. Cureus 2021, 13, 15314. [Google Scholar] [CrossRef]
- Tian, T.; Wang, M.; Ma, D. TNF-α, a good or bad factor in hematological diseases? Stem Cell Investig. 2014, 1, 12. [Google Scholar] [CrossRef]
- Wu, B.; Zhao, T.V.; Jin, K.; Hu, Z.; Abdel, M.P.; Warrington, K.J.; Weyand, C.M. Mitochondrial aspartate regu-lates TNF biogenesis and autoimmune tissue inflammation. Nat. Immunol. 2021, 22, 1551–1562. [Google Scholar] [CrossRef]
- Izadi, Z.; Brenner, E.J.; Mahil, S.K.; Dand, N.; Yiu, Z.Z.N.; Yates, M.; Ungaro, R.C.; Zhang, X.; Agrawal, M.; Colombel, J.-F.; et al. Association Between Tumor Necrosis Factor Inhibitors and the Risk of Hospitalization or Death Among Patients With Immune-Mediated Inflammatory Disease and COVID-19. JAMA Netw. Open 2021, 4, e2129639. [Google Scholar] [CrossRef]
- Salesi, M.; Shojaie, B.; Farajzadegan, Z.; Salesi, N.; Mohammadi, E. TNF-α Blockers Showed Prophylactic Effects in Preventing COVID-19 in Patients with Rheumatoid Arthritis and Seronegative Spondyloarthropathies: A Case–Control Study. Rheumatol. Ther. 2021, 8, 1355–1370. [Google Scholar] [CrossRef]
- Arleo, T.; Tong, D.; Shabto, J.; O’Keefe, G.; Khosroshahi, A. Clinical course and outcomes of COVID-19 in rheumatic disease patients: A case cohort study with a diverse population. Clin. Rheumatol. 2021, 40, 2633–2642. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohd Zawawi, Z.; Kalyanasundram, J.; Mohd Zain, R.; Thayan, R.; Basri, D.F.; Yap, W.B. Prospective Roles of Tumor Necrosis Factor-Alpha (TNF-α) in COVID-19: Prognosis, Therapeutic and Management. Int. J. Mol. Sci. 2023, 24, 6142. https://doi.org/10.3390/ijms24076142
Mohd Zawawi Z, Kalyanasundram J, Mohd Zain R, Thayan R, Basri DF, Yap WB. Prospective Roles of Tumor Necrosis Factor-Alpha (TNF-α) in COVID-19: Prognosis, Therapeutic and Management. International Journal of Molecular Sciences. 2023; 24(7):6142. https://doi.org/10.3390/ijms24076142
Chicago/Turabian StyleMohd Zawawi, Zarina, Jeevanathan Kalyanasundram, Rozainanee Mohd Zain, Ravindran Thayan, Dayang Fredalina Basri, and Wei Boon Yap. 2023. "Prospective Roles of Tumor Necrosis Factor-Alpha (TNF-α) in COVID-19: Prognosis, Therapeutic and Management" International Journal of Molecular Sciences 24, no. 7: 6142. https://doi.org/10.3390/ijms24076142