

Dialog beyond the Grave: Necrosis in the Tumor Microenvironment and Its Contribution to Tumor Growth

Abstract
:1. Introduction
2. Tumor Necrosis in Human Solid Tumors: Clinical and Pathological Aspects
2.1. Necrosis Assessment by Medical Imaging
2.1.1. Conventional Magnetic Resonance (MR) Imaging
2.1.2. Functional Imaging Techniques
2.1.3. Molecular Imaging Techniques
2.2. Necrosis Assessment by Histopathological Examination
2.3. Circulating Biomarkers—Biological Assays for Necrosis Assessment
3. Biology of Tumor Necrosis and Extra-Cellular Necrotic Products
3.1. Stages in Necrosis
3.2. Relationships with Hypoxia—Metabolic Reprogramming (Warburg Effect)
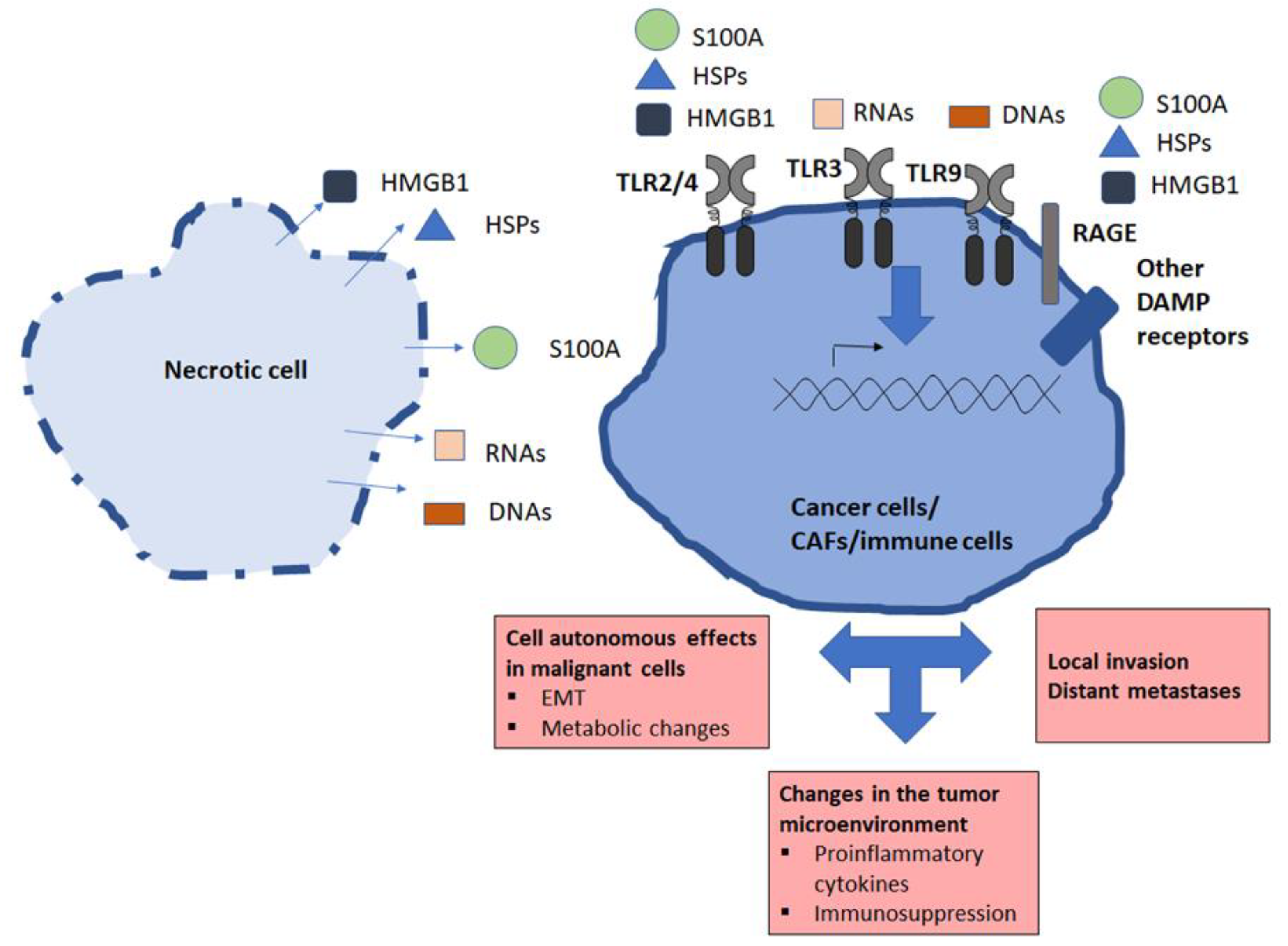
3.3. General Features of Extracellular Products Released by Necrotic Cells: Damage-Associated Molecular Patterns (DAMPs)
3.3.1. HMGB1 and RAGE Receptor
3.3.2. Other Proteins Released by Necrotic Cells
Histones
S100
Heat Shock Proteins (HSPs)
Annexin A1/FPR1
3.3.3. Lipids and Carbohydrates Released by Necrotic Cells
3.3.4. Metabolite-Related DAMPs
ATP
Uric Acid
3.3.5. Nucleic Acids Released by Necrotic Cells
3.4. Necrotic Products and Tumor Microenvironment
4. Direct Impact of Necrotic Products on Malignant Cells—Role of TLR Ligands
4.1. Role of TLR Ligands, Especially TLR3 Ligands
4.2. HMGB1 and RAGE
4.3. Other Product and Receptors
5. Impacts of Necrotic Products on the Other Components of the Tumor Microenvironment
5.1. Recruitment and Action of Immune Cells
5.2. Deleterious Effects of Neutrophils
5.3. Immunosuppressive Effects of Necrotic Products
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kumar, V.; Abbas, A.K.; Aster, J.C. Robbins and Cotran Pathologic Basis of Disease, 9th ed.; Elsevier/Saunders: Philadelphia, PA, USA, 2015; pp. 31–68. [Google Scholar]
- Wimmer, K.; Sachet, M.; Oehler, R. Circulating biomarkers of cell death. Clin. Chim. Acta 2020, 500, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.M.; Li, Z.X.; Lin, R.H.; Shan, J.Q.; Yu, Q.W.; Wang, R.X.; Liao, L.S.; Yan, W.T.; Wang, Z.; Shang, L.; et al. Guidelines for Regulated Cell Death Assays: A Systematic Summary, A Categorical Comparison, A Prospective. Front. Cell Dev. Biol. 2021, 9, 634690. [Google Scholar] [CrossRef]
- Proskuryakov, S.Y.; Gabai, V.L. Mechanisms of tumor cell necrosis. Curr. Pharm. Des. 2010, 16, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Ju, M.K.; Jeon, H.M.; Jeong, E.K.; Lee, Y.J.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Regulation of Tumor Progression by Programmed Necrosis. Oxid. Med. Cell Longev. 2018, 2018, 3537471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.M. Tumor microenvironment and the response to anticancer therapy. Cancer Biol. Ther. 2002, 1, 453–458. [Google Scholar] [CrossRef] [Green Version]
- Khatun, S.; Appidi, T.; Rengan, A.K. The role played by bacterial infections in the onset and metastasis of cancer. Curr. Res. Microb. Sci. 2021, 2, 100078. [Google Scholar] [CrossRef]
- Cummins, J.; Tangney, M. Bacteria and tumours: Causative agents or opportunistic inhabitants? Infect. Agent Cancer 2013, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Karsch-Bluman, A.; Feiglin, A.; Arbib, E.; Stern, T.; Shoval, H.; Schwob, O.; Berger, M.; Benny, O. Tissue necrosis and its role in cancer progression. Oncogene 2019, 38, 1920–1935. [Google Scholar] [CrossRef]
- Richards, C.H.; Mohammed, Z.; Qayyum, T.; Horgan, P.G.; McMillan, D.C. The prognostic value of histological tumor necrosis in solid organ malignant disease: A systematic review. Future Oncol. 2011, 7, 1223–1235. [Google Scholar] [CrossRef]
- Gkogkou, C.; Frangia, K.; Saif, M.W.; Trigidou, R.; Syrigos, K. Necrosis and apoptotic index as prognostic factors in non-small cell lung carcinoma: A review. Springerplus 2014, 3, 120. [Google Scholar] [CrossRef] [Green Version]
- Ling, Y.H.; Chen, J.W.; Wen, S.H.; Huang, C.Y.; Li, P.; Lu, L.H.; Mei, J.; Li, S.H.; Wei, W.; Cai, M.Y.; et al. Tumor necrosis as a poor prognostic predictor on postoperative survival of patients with solitary small hepatocellular carcinoma. BMC Cancer 2020, 20, 607. [Google Scholar] [CrossRef] [PubMed]
- Friebele, J.C.; Peck, J.; Pan, X.; Abdel-Rasoul, M.; Mayerson, J.L. Osteosarcoma: A Meta-Analysis and Review of the Literature. Am. J. Orthop. 2015, 44, 547–553. [Google Scholar]
- Choudhury, A.; West, C.M.; Porta, N.; Hall, E.; Denley, H.; Hendron, C.; Lewis, R.; Hussain, S.A.; Huddart, R.; James, N. The predictive and prognostic value of tumour necrosis in muscle invasive bladder cancer patients receiving radiotherapy with or without chemotherapy in the BC2001 trial (CRUK/01/004). Br. J. Cancer 2017, 116, 649–657. [Google Scholar] [CrossRef] [Green Version]
- Nael, K.; Bauer, A.H.; Hormigo, A.; Lemole, M.; Germano, I.M.; Puig, J.; Stea, B. Multiparametric MRI for Differentiation of Radiation Necrosis From Recurrent Tumor in Patients With Treated Glioblastoma. AJR Am. J. Roentgenol. 2018, 210, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Beddy, P.; Genega, E.M.; Ngo, L.; Hindman, N.; Wei, J.; Bullock, A.; Bhatt, R.S.; Atkins, M.B.; Pedrosa, I. Tumor necrosis on magnetic resonance imaging correlates with aggressive histology and disease progression in clear cell renal cell carcinoma. Clin. Genitourin. Cancer 2014, 12, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowosielski, M.; Gorlia, T.; Bromberg, J.E.C.; Sahm, F.; Harting, I.; Kickingereder, P.; Brandes, A.A.; Taphoorn, M.J.B.; Taal, W.; Domont, J.; et al. Imaging necrosis during treatment is associated with worse survival in EORTC 26101 study. Neurology 2019, 92, e2754–e2763. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.; Cowperthwaite, M.C.; Burnett, M.G.; Markey, M.K. Differentiating tumor recurrence from treatment necrosis: A review of neuro-oncologic imaging strategies. Neuro Oncol. 2013, 15, 515–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strauss, S.B.; Meng, A.; Ebani, E.J.; Chiang, G.C. Imaging Glioblastoma Posttreatment: Progression, Pseudoprogression, Pseudoresponse, Radiation Necrosis. Neuroimaging Clin. N. Am. 2021, 31, 103–120. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, M.; Jin, Q.; Ni, Y.; Zhang, J. Updated developments on molecular imaging and therapeutic strategies directed against necrosis. Acta Pharm. Sin. B 2019, 9, 455–468. [Google Scholar] [CrossRef]
- Stroet, M.C.M.; de Blois, E.; Haeck, J.; Seimbille, Y.; Mezzanotte, L.; de Jong, M.; Lowik, C.; Panth, K.M. In Vivo Evaluation of Gallium-68-Labeled IRDye800CW as a Necrosis Avid Contrast Agent in Solid Tumors. Contrast Media Mol. Imaging 2021, 2021, 2853522. [Google Scholar] [CrossRef]
- Zhang, D.; Jin, Q.; Ni, Y.; Zhang, J. Discovery of necrosis avidity of rhein and its applications in necrosis imaging. J. Drug Target 2020, 28, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Jin, Q.; Su, C.; Zhang, D.; Jiang, C.; Fish, A.F.; Feng, Y.; Ni, Y.; Zhang, J.; Yin, Z. Radiolabeled Rhein as Small-Molecule Necrosis Avid Agents for Imaging of Necrotic Myocardium. Anal. Chem. 2017, 89, 1260–1266. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Zhang, J.; Jin, Q.; Gao, M.; Zhang, D.; Zhang, L.; Feng, Y.; Ni, Y.; Yin, Z. Rhein-based necrosis-avid MRI contrast agents for early evaluation of tumor response to microwave ablation therapy. Magn. Reason. Med. 2019, 82, 2212–2224. [Google Scholar] [CrossRef] [PubMed]
- Bian, L.; Gao, M.; Zhang, D.; Ji, A.; Su, C.; Duan, X.; Luo, Q.; Huang, D.; Feng, Y.; Ni, Y.; et al. Synthesis and Biological Evaluation of Rhein-Based MRI Contrast Agents for in Vivo Visualization of Necrosis. Anal. Chem. 2018, 90, 13249–13256. [Google Scholar] [CrossRef]
- Zhang, A.; Wu, T.; Bian, L.; Li, P.; Liu, Q.; Zhang, D.; Jin, Q.; Zhang, J.; Huang, G.; Song, S. Synthesis and Evaluation of Ga-68-Labeled Rhein for Early Assessment of Treatment-Induced Tumor Necrosis. Mol. Imaging Biol. 2020, 22, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Zhai, X.; Lu, S.; Vuletic, I.; Wang, L.; Zhou, K.; Peng, Z.; Ren, Q.; Xie, Z. A Hybrid Imaging Platform(CT/PET/FMI) for Evaluating Tumor Necrosis and Apoptosis in Real-Time. Front. Oncol. 2022, 12, 772392. [Google Scholar] [CrossRef]
- Tonnus, W.; Meyer, C.; Paliege, A.; Belavgeni, A.; von Massenhausen, A.; Bornstein, S.R.; Hugo, C.; Becker, J.U.; Linkermann, A. The pathological features of regulated necrosis. J. Pathol. 2019, 247, 697–707. [Google Scholar] [CrossRef]
- Chung, A.G.; Frye, J.B.; Zbesko, J.C.; Constantopoulos, E.; Hayes, M.; Figueroa, A.G.; Becktel, D.A.; Antony Day, W.; Konhilas, J.P.; McKay, B.S.; et al. Liquefaction of the Brain following Stroke Shares a Similar Molecular and Morphological Profile with Atherosclerosis and Mediates Secondary Neurodegeneration in an Osteopontin-Dependent Mechanism. eNeuro 2018, 5, ENEURO.0076-18.2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, F.K.; Moriwaki, K.; De Rosa, M.J. Detection of necrosis by release of lactate dehydrogenase activity. Methods Mol. Biol. 2013, 979, 65–70. [Google Scholar]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Omary, M.B.; Ku, N.O.; Strnad, P.; Hanada, S. Toward unraveling the complexity of simple epithelial keratins in human disease. J. Clin. Investig. 2009, 119, 1794–1805. [Google Scholar] [CrossRef] [Green Version]
- Kramer, G.; Erdal, H.; Mertens, H.J.; Nap, M.; Mauermann, J.; Steiner, G.; Marberger, M.; Biven, K.; Shoshan, M.C.; Linder, S. Differentiation between cell death modes using measurements of different soluble forms of extracellular cytokeratin 18. Cancer Res. 2004, 64, 1751–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulz, P.; Thallinger, G.G.; Auer, M.; Graf, R.; Kashofer, K.; Jahn, S.W.; Abete, L.; Pristauz, G.; Petru, E.; Geigl, J.B.; et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat. Genet. 2016, 48, 1273–1278. [Google Scholar] [CrossRef]
- Rostami, A.; Lambie, M.; Yu, C.W.; Stambolic, V.; Waldron, J.N.; Bratman, S.V. Senescence, Necrosis, and Apoptosis Govern Circulating Cell-free DNA Release Kinetics. Cell Rep. 2020, 31, 107830. [Google Scholar] [CrossRef]
- Jiang, P.; Chan, C.W.; Chan, K.C.; Cheng, S.H.; Wong, J.; Wong, V.W.; Wong, G.L.; Chan, S.L.; Mok, T.S.; Chan, H.L.; et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, E1317–E1325. [Google Scholar] [CrossRef] [Green Version]
- McCall, M.N.; Kim, M.S.; Adil, M.; Patil, A.H.; Lu, Y.; Mitchell, C.J.; Leal-Rojas, P.; Xu, J.; Kumar, M.; Dawson, V.L.; et al. Toward the human cellular microRNAome. Genome Res. 2017, 27, 1769–1781. [Google Scholar] [CrossRef] [Green Version]
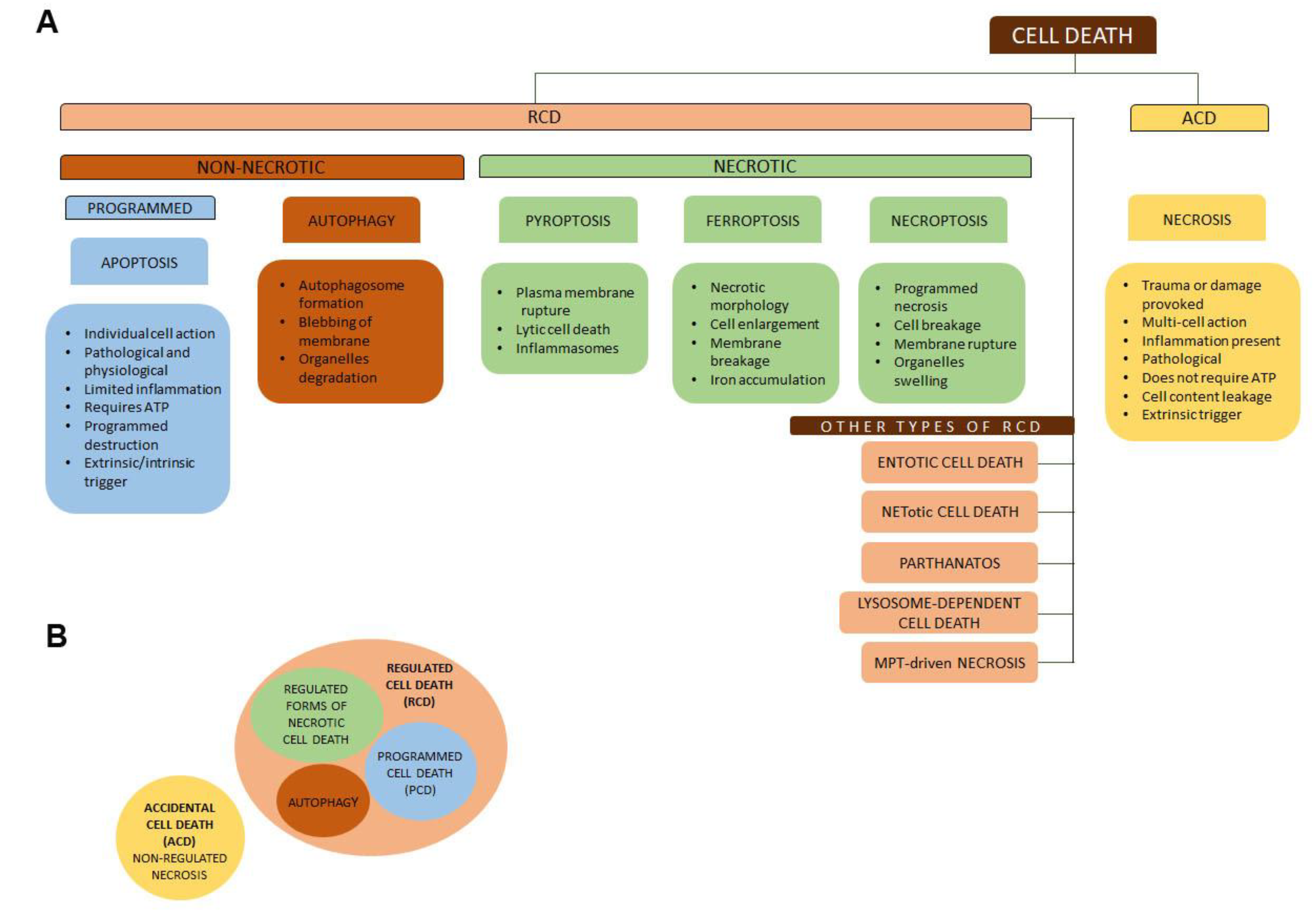
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Khoury, M.K.; Gupta, K.; Franco, S.R.; Liu, B. Necroptosis in the Pathophysiology of Disease. Am. J. Pathol. 2020, 190, 272–285. [Google Scholar] [CrossRef] [Green Version]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflamm. 2018, 15, 199. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Xie, Y.; Sun, X.; Zeh, H.J., 3rd; Kang, R.; Lotze, M.T.; Tang, D. DAMPs, ageing, and cancer: The ‘DAMP Hypothesis’. Ageing Res. Rev. 2015, 24 Pt A, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Zhang, Q.; Hou, W.; Yan, Z.; Chen, R.; Bonaroti, J.; Bansal, P.; Billiar, T.R.; Tsung, A.; Wang, Q.; et al. Intracellular Hmgb1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology 2014, 146, 1097–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Prasad, R.; Wilson, S.H. HMGB1: Roles in base excision repair and related function. Biochim. Biophys. Acta 2010, 1799, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Polanska, E.; Dobsakova, Z.; Dvorackova, M.; Fajkus, J.; Stros, M. HMGB1 gene knockout in mouse embryonic fibroblasts results in reduced telomerase activity and telomere dysfunction. Chromosoma 2012, 121, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Chen, K.; Xia, Z.; Chavez, M.; Pal, S.; Seol, J.H.; Chen, C.C.; Li, W.; Tyler, J.K. Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev. 2014, 28, 396–408. [Google Scholar] [CrossRef] [Green Version]
- Gebhardt, C.; Riehl, A.; Durchdewald, M.; Nemeth, J.; Furstenberger, G.; Muller-Decker, K.; Enk, A.; Arnold, B.; Bierhaus, A.; Nawroth, P.P.; et al. RAGE signaling sustains inflammation and promotes tumor development. J. Exp. Med. 2008, 205, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Bettum, I.J.; Vasiliauskaite, K.; Nygaard, V.; Clancy, T.; Pettersen, S.J.; Tenstad, E.; Maelandsmo, G.M.; Prasmickaite, L. Metastasis-associated protein S100A4 induces a network of inflammatory cytokines that activate stromal cells to acquire pro-tumorigenic properties. Cancer Lett. 2014, 344, 28–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Xie, M.; Yang, M.; Yu, Y.; Zhu, S.; Hou, W.; Kang, R.; Lotze, M.T.; Billiar, T.R.; Wang, H.; et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat. Commun. 2014, 5, 4436. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Cai, L.; Guo, X.; Li, Z.; Liao, X.; Zhang, X.; Huang, L.; He, J. HMGB1-activated fibroblasts promote breast cancer cells metastasis via RAGE/aerobic glycolysis. Neoplasma 2021, 68, 71–78. [Google Scholar] [CrossRef]
- Calogero, S.; Grassi, F.; Aguzzi, A.; Voigtlander, T.; Ferrier, P.; Ferrari, S.; Bianchi, M.E. The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nat. Genet. 1999, 22, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Bertheloot, D.; Latz, E. HMGB1, IL-1alpha, IL-33 and S100 proteins: Dual-function alarmins. Cell Mol. Immunol. 2017, 14, 43–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Zhang, Q.; Zeh, H.J., 3rd; Lotze, M.T.; Tang, D. HMGB1 in cancer: Good, bad, or both? Clin. Cancer Res. 2013, 19, 4046–4057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Shi, Y.; Kang, R.; Li, T.; Xiao, W.; Wang, H.; Xiao, X. Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J. Leukoc. Biol. 2007, 81, 741–747. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R.; Xiao, W.; Zhang, H.; Lotze, M.T.; Wang, H.; Xiao, X. Quercetin prevents LPS-induced high-mobility group box 1 release and proinflammatory function. Am. J. Respir. Cell Mol. Biol. 2009, 41, 651–660. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Lu, Y.; Yao, J.; Li, Z.; Chen, Z.; Wang, G.; Jing, H.; Zhang, X.; Li, M.; Peng, J.; et al. Novel role of resveratrol: Suppression of high-mobility group protein box 1 nucleocytoplasmic translocation by the upregulation of sirtuin 1 in sepsis-induced liver injury. Shock 2014, 42, 440–447. [Google Scholar] [CrossRef]
- Kim, Y.H.; Kwak, M.S.; Park, J.B.; Lee, S.A.; Choi, J.E.; Cho, H.S.; Shin, J.S. N-linked glycosylation plays a crucial role in the secretion of HMGB1. J. Cell Sci. 2016, 129, 29–38. [Google Scholar]
- Ito, I.; Fukazawa, J.; Yoshida, M. Post-translational methylation of high mobility group box 1 (HMGB1) causes its cytoplasmic localization in neutrophils. J. Biol. Chem. 2007, 282, 16336–16344. [Google Scholar] [CrossRef] [Green Version]
- Bonaldi, T.; Talamo, F.; Scaffidi, P.; Ferrera, D.; Porto, A.; Bachi, A.; Rubartelli, A.; Agresti, A.; Bianchi, M.E. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003, 22, 5551–5560. [Google Scholar] [CrossRef] [Green Version]
- Nystrom, S.; Antoine, D.J.; Lundback, P.; Lock, J.G.; Nita, A.F.; Hogstrand, K.; Grandien, A.; Erlandsson-Harris, H.; Andersson, U.; Applequist, S.E. TLR activation regulates damage-associated molecular pattern isoforms released during pyroptosis. EMBO J. 2013, 32, 86–99. [Google Scholar] [CrossRef] [Green Version]
- Bell, C.W.; Jiang, W.; Reich, C.F., 3rd; Pisetsky, D.S. The extracellular release of HMGB1 during apoptotic cell death. Am. J. Physiol. Cell Physiol. 2006, 291, C1318–C1325. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.W.; Farkas, A.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J., 3rd; et al. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef]
- Jiao, Y.; Wang, H.C.; Fan, S.J. Growth suppression and radiosensitivity increase by HMGB1 in breast cancer. Acta Pharmacol. Sin. 2007, 28, 1957–1967. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Pellegrini, L.; Napolitano, A.; Giorgi, C.; Jube, S.; Preti, A.; Jennings, C.J.; De Marchis, F.; Flores, E.G.; Larson, D.; et al. Aspirin delays mesothelioma growth by inhibiting HMGB1-mediated tumor progression. Cell Death Dis. 2015, 6, e1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Tang, D.; Schapiro, N.E.; Loux, T.; Livesey, K.M.; Billiar, T.R.; Wang, H.; Van Houten, B.; Lotze, M.T.; Zeh, H.J. The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Oncogene 2014, 33, 567–577. [Google Scholar] [CrossRef] [Green Version]
- Parker, K.H.; Sinha, P.; Horn, L.A.; Clements, V.K.; Yang, H.; Li, J.; Tracey, K.J.; Ostrand-Rosenberg, S. HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of myeloid-derived suppressor cells. Cancer Res. 2014, 74, 5723–5733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, A.; Kuwata, T.; Yamauchi, C.; Higuchi, Y.; Ochiai, A. High Mobility Group Box1 (HMGB1) released from cancer cells induces the expression of pro-inflammatory cytokines in peritoneal fibroblasts. Pathol. Int. 2014, 64, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.X.; Wu, H.P.; Zhang, H.L.; Ashton, C.; Tong, C.; Wu, H.; Qian, Q.J.; Wang, H.Y.; Ying, Q.L. p53 promotes inflammation-associated hepatocarcinogenesis by inducing HMGB1 release. J. Hepatol. 2013, 59, 762–768. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Xu, L.; Yang, T.; Wang, F. High-mobility group box-1 and its role in angiogenesis. J. Leukoc. Biol. 2014, 95, 563–574. [Google Scholar] [CrossRef]
- Huber, R.; Meier, B.; Otsuka, A.; Fenini, G.; Satoh, T.; Gehrke, S.; Widmer, D.; Levesque, M.P.; Mangana, J.; Kerl, K.; et al. Tumour hypoxia promotes melanoma growth and metastasis via High Mobility Group Box-1 and M2-like macrophages. Sci. Rep. 2016, 6, 29914. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Zhang, X.; Du, X.; Zou, N.; Shen, W.; Ma, M.; Wang, Y.; Zhu, S. HMGB1 overexpression promotes a malignant phenotype and radioresistance in ESCC. J. Cancer 2022, 13, 2717–2726. [Google Scholar] [CrossRef] [PubMed]
- Amornsupak, K.; Thongchot, S.; Thinyakul, C.; Box, C.; Hedayat, S.; Thuwajit, P.; Eccles, S.A.; Thuwajit, C. HMGB1 mediates invasion and PD-L1 expression through RAGE-PI3K/AKT signaling pathway in MDA-MB-231 breast cancer cells. BMC Cancer 2022, 22, 578. [Google Scholar] [CrossRef] [PubMed]
- Jung, A.R.; Kim, G.E.; Kim, M.Y.; Ha, U.S.; Hong, S.H.; Lee, J.Y.; Kim, S.W.; Park, Y.H. HMGB1 promotes tumor progression and invasion through HMGB1/TNFR1/NF-kappaB axis in castration-resistant prostate cancer. Am. J. Cancer Res. 2021, 11, 2215–2227. [Google Scholar] [PubMed]
- Song, Y.; Zou, X.; Zhang, D.; Liu, S.; Duan, Z.; Liu, L. Self-enforcing HMGB1/NF-kappaB/HIF-1alpha Feedback Loop Promotes Cisplatin Resistance in Hepatocellular Carcinoma Cells. J. Cancer 2020, 11, 3893–3902. [Google Scholar] [CrossRef]
- Ma, H.; Zheng, S.; Zhang, X.; Gong, T.; Lv, X.; Fu, S.; Zhang, S.; Yin, X.; Hao, J.; Shan, C.; et al. High mobility group box 1 promotes radioresistance in esophageal squamous cell carcinoma cell lines by modulating autophagy. Cell Death Dis. 2019, 10, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ren, H.; Wang, J.; Zhang, P.; Shi, X. Extracellular HMGB1 promotes CD44 expression in hepatocellular carcinoma via regulating miR-21. Aging 2021, 13, 8380–8395. [Google Scholar] [CrossRef] [PubMed]
- Tesarova, P.; Cabinakova, M.; Mikulova, V.; Zima, T.; Kalousova, M. RAGE and its ligands in cancer-culprits, biomarkers, or therapeutic targets? Neoplasma 2015, 62, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Fritz, G. RAGE: A single receptor fits multiple ligands. Trends Biochem. Sci. 2011, 36, 625–632. [Google Scholar] [CrossRef]
- Mizumoto, S.; Takahashi, J.; Sugahara, K. Receptor for advanced glycation end products (RAGE) functions as receptor for specific sulfated glycosaminoglycans, and anti-RAGE antibody or sulfated glycosaminoglycans delivered in vivo inhibit pulmonary metastasis of tumor cells. J. Biol. Chem. 2012, 287, 18985–18994. [Google Scholar] [CrossRef] [Green Version]
- Rai, V.; Toure, F.; Chitayat, S.; Pei, R.; Song, F.; Li, Q.; Zhang, J.; Rosario, R.; Ramasamy, R.; Chazin, W.J.; et al. Lysophosphatidic acid targets vascular and oncogenic pathways via RAGE signaling. J. Exp. Med. 2012, 209, 2339–2350. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Mendez, J.D.; Mendez-Valenzuela, V.; Aguilar-Hernandez, M.M. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell. Signal 2013, 25, 2185–2197. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Vazzana, N.; Bucciarelli, L.G.; Davi, G. Soluble forms of RAGE in human diseases: Clinical and therapeutical implications. Curr. Med. Chem. 2009, 16, 940–952. [Google Scholar] [CrossRef] [PubMed]
- Marsman, G.; Zeerleder, S.; Luken, B.M. Extracellular histones, cell-free DNA, or nucleosomes: Differences in immunostimulation. Cell Death Dis. 2016, 7, e2518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Kang, R.; Fan, X.G.; Tang, D. Release and activity of histone in diseases. Cell Death Dis. 2014, 5, e1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Liu, T.; Rios, Z.; Mei, Q.; Lin, X.; Cao, S. Heat Shock Proteins and Cancer. Trends Pharmacol. Sci. 2017, 38, 226–256. [Google Scholar] [CrossRef]
- He, R.; Sang, H.; Ye, R.D. Serum amyloid A induces IL-8 secretion through a G protein-coupled receptor, FPRL1/LXA4R. Blood 2003, 101, 1572–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bours, M.J.; Swennen, E.L.; Di Virgilio, F.; Cronstein, B.N.; Dagnelie, P.C. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol. Ther. 2006, 112, 358–404. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.J.; Coban, C.; Kato, H.; Takahashi, K.; Torii, Y.; Takeshita, F.; Ludwig, H.; Sutter, G.; Suzuki, K.; Hemmi, H.; et al. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat. Immunol. 2006, 7, 40–48. [Google Scholar] [CrossRef]
- Mathew, A.; Lindsley, T.A.; Sheridan, A.; Bhoiwala, D.L.; Hushmendy, S.F.; Yager, E.J.; Ruggiero, E.A.; Crawford, D.R. Degraded mitochondrial DNA is a newly identified subtype of the damage associated molecular pattern (DAMP) family and possible trigger of neurodegeneration. J. Alzheimers Dis. 2012, 30, 617–627. [Google Scholar] [CrossRef]
- Sharma, S.; DeOliveira, R.B.; Kalantari, P.; Parroche, P.; Goutagny, N.; Jiang, Z.; Chan, J.; Bartholomeu, D.C.; Lauw, F.; Hall, J.P.; et al. Innate immune recognition of an AT-rich stem-loop DNA motif in the Plasmodium falciparum genome. Immunity 2011, 35, 194–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kariko, K.; Ni, H.; Capodici, J.; Lamphier, M.; Weissman, D. mRNA is an endogenous ligand for Toll-like receptor 3. J. Biol. Chem. 2004, 279, 12542–12550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, J.J.; Cowing-Zitron, C.; Nakatsuji, T.; Muehleisen, B.; Muto, J.; Borkowski, A.W.; Martinez, L.; Greidinger, E.L.; Yu, B.D.; Gallo, R.L. Ultraviolet radiation damages self noncoding RNA and is detected by TLR3. Nat. Med. 2012, 18, 1286–1290. [Google Scholar] [CrossRef]
- Ren, Y.; Cao, L.; Wang, L.; Zheng, S.; Zhang, Q.; Guo, X.; Li, X.; Chen, M.; Wu, X.; Furlong, F.; et al. Autophagic secretion of HMGB1 from cancer-associated fibroblasts promotes metastatic potential of non-small cell lung cancer cells via NFkappaB signaling. Cell Death Dis. 2021, 12, 858. [Google Scholar] [CrossRef] [PubMed]
- Flis, E.; Barber, G.; Nulty, C.; Keogh, B.; McGuirk, P.; Anand, A.; O’Sullivan, J.; Quante, M.; Creagh, E.M. Identification of TLR2 Signalling Mechanisms Which Contribute to Barrett’s and Oesophageal Adenocarcinoma Disease Progression. Cancers 2021, 13, 2065. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.D.; Wang, Y.Y.; Lin, S.Y.; Chang, C.Y.; Li, J.R.; Huang, S.W.; Chen, W.Y.; Liao, S.L.; Chen, C.J. Exosomal HMGB1 Promoted Cancer Malignancy. Cancers 2021, 13, 877. [Google Scholar] [CrossRef]
- Gao, W.; He, R.; Ren, J.; Zhang, W.; Wang, K.; Zhu, L.; Liang, T. Exosomal HMGB1 derived from hypoxia-conditioned bone marrow mesenchymal stem cells increases angiogenesis via the JNK/HIF-1alpha pathway. FEBS Open Bio 2021, 11, 1364–1373. [Google Scholar] [CrossRef]
- Hance, M.W.; Dole, K.; Gopal, U.; Bohonowych, J.E.; Jezierska-Drutel, A.; Neumann, C.A.; Liu, H.; Garraway, I.P.; Isaacs, J.S. Secreted Hsp90 is a novel regulator of the epithelial to mesenchymal transition (EMT) in prostate cancer. J. Biol. Chem. 2012, 287, 37732–37744. [Google Scholar] [CrossRef] [Green Version]
- Sims, J.D.; McCready, J.; Jay, D.G. Extracellular heat shock protein (Hsp)70 and Hsp90alpha assist in matrix metalloproteinase-2 activation and breast cancer cell migration and invasion. PLoS ONE 2011, 6, e18848. [Google Scholar] [CrossRef] [Green Version]
- Diao, J.; Yang, X.; Song, X.; Chen, S.; He, Y.; Wang, Q.; Chen, G.; Luo, C.; Wu, X.; Zhang, Y. Exosomal Hsp70 mediates immunosuppressive activity of the myeloid-derived suppressor cells via phosphorylation of Stat3. Med. Oncol. 2015, 32, 453. [Google Scholar] [CrossRef]
- Ignacio, R.M.C.; Gibbs, C.R.; Kim, S.; Lee, E.S.; Adunyah, S.E.; Son, D.S. Serum amyloid A predisposes inflammatory tumor microenvironment in triple negative breast cancer. Oncotarget 2019, 10, 511–526. [Google Scholar] [CrossRef] [Green Version]
- Fourie, C.; Shridas, P.; Davis, T.; de Villiers, W.J.S.; Engelbrecht, A.M. Serum amyloid A and inflammasome activation: A link to breast cancer progression? Cytokine Growth Factor Rev. 2021, 59, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Stone, M.L.; Porrett, P.M.; Thomas, S.K.; Komar, C.A.; Li, J.H.; Delman, D.; Graham, K.; Gladney, W.L.; Hua, X.; et al. Hepatocytes direct the formation of a pro-metastatic niche in the liver. Nature 2019, 567, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Kim, E.K.; Seo, H.; Jeon, I.; Chae, M.J.; Park, Y.J.; Song, B.; Kim, Y.S.; Kim, Y.J.; Ko, H.J.; et al. Serum amyloid A3 exacerbates cancer by enhancing the suppressive capacity of myeloid-derived suppressor cells via TLR2-dependent STAT3 activation. Eur. J. Immunol. 2014, 44, 1672–1684. [Google Scholar] [CrossRef]
- Miller-Ocuin, J.L.; Liang, X.; Boone, B.A.; Doerfler, W.R.; Singhi, A.D.; Tang, D.; Kang, R.; Lotze, M.T.; Zeh, H.J., 3rd. DNA released from neutrophil extracellular traps (NETs) activates pancreatic stellate cells and enhances pancreatic tumor growth. Oncoimmunology 2019, 8, e1605822. [Google Scholar] [CrossRef] [PubMed]
- Singel, K.L.; Grzankowski, K.S.; Khan, A.; Grimm, M.J.; D’Auria, A.C.; Morrell, K.; Eng, K.H.; Hylander, B.; Mayor, P.C.; Emmons, T.R.; et al. Mitochondrial DNA in the tumour microenvironment activates neutrophils and is associated with worse outcomes in patients with advanced epithelial ovarian cancer. Br. J. Cancer 2019, 120, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Anunobi, R.; Boone, B.A.; Cheh, N.; Tang, D.; Kang, R.; Loux, T.; Lotze, M.T.; Zeh, H.J. Extracellular DNA promotes colorectal tumor cell survival after cytotoxic chemotherapy. J. Surg. Res. 2018, 226, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Abu, N.; Rus Bakarurraini, N.A.A.; Nasir, S.N. Extracellular Vesicles and DAMPs in Cancer: A Mini-Review. Front. Immunol. 2021, 12, 740548. [Google Scholar] [CrossRef]
- Nabet, B.Y.; Qiu, Y.; Shabason, J.E.; Wu, T.J.; Yoon, T.; Kim, B.C.; Benci, J.L.; DeMichele, A.M.; Tchou, J.; Marcotrigiano, J.; et al. Exosome RNA Unshielding Couples Stromal Activation to Pattern Recognition Receptor Signaling in Cancer. Cell 2017, 170, 352–366.e13. [Google Scholar] [CrossRef] [Green Version]
- Rakoff-Nahoum, S.; Medzhitov, R. Toll-like receptors and cancer. Nat. Rev. Cancer 2009, 9, 57–63. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, Y.; Han, Y.; Zhang, Q.; Jiang, Z.; Zhang, X.; Huang, B.; Xu, X.; Zheng, J.; Cao, X. Tumor Exosomal RNAs Promote Lung Pre-metastatic Niche Formation by Activating Alveolar Epithelial TLR3 to Recruit Neutrophils. Cancer Cell 2016, 30, 243–256. [Google Scholar] [CrossRef] [Green Version]
- Thuringer, D.; Jego, G.; Wettstein, G.; Terrier, O.; Cronier, L.; Yousfi, N.; Hebrard, S.; Bouchot, A.; Hazoume, A.; Joly, A.L.; et al. Extracellular HSP27 mediates angiogenesis through Toll-like receptor 3. FASEB J. 2013, 27, 4169–4183. [Google Scholar] [CrossRef]
- Veyrat, M.; Durand, S.; Classe, M.; Glavan, T.M.; Oker, N.; Kapetanakis, N.I.; Jiang, X.; Gelin, A.; Herman, P.; Casiraghi, O.; et al. Stimulation of the toll-like receptor 3 promotes metabolic reprogramming in head and neck carcinoma cells. Oncotarget 2016, 7, 82580–82593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matijevic Glavan, T.; Cipak Gasparovic, A.; Verillaud, B.; Busson, P.; Pavelic, J. Toll-like receptor 3 stimulation triggers metabolic reprogramming in pharyngeal cancer cell line through Myc, MAPK, and HIF. Mol. Carcinog. 2016, 56, 1214–1226. [Google Scholar] [CrossRef] [PubMed]
- Muresan, X.M.; Slabakova, E.; Prochazkova, J.; Drapela, S.; Fedr, R.; Pickova, M.; Vacek, O.; Vichova, R.; Suchankova, T.; Bouchal, J.; et al. Toll-Like Receptor 3 Overexpression Induces Invasion of Prostate Cancer Cells, whereas Its Activation Triggers Apoptosis. Am. J. Pathol. 2022, 192, 1321–1335. [Google Scholar] [CrossRef] [PubMed]
- Tavora, B.; Mederer, T.; Wessel, K.J.; Ruffing, S.; Sadjadi, M.; Missmahl, M.; Ostendorf, B.N.; Liu, X.; Kim, J.Y.; Olsen, O.; et al. Tumoural activation of TLR3-SLIT2 axis in endothelium drives metastasis. Nature 2020, 586, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Bugge, M.; Bergstrom, B.; Eide, O.K.; Solli, H.; Kjonstad, I.F.; Stenvik, J.; Espevik, T.; Nilsen, N.J. Surface Toll-like receptor 3 expression in metastatic intestinal epithelial cells induces inflammatory cytokine production and promotes invasiveness. J. Biol. Chem. 2017, 292, 15408–15425. [Google Scholar] [CrossRef] [Green Version]
- Hoste, E.; Arwert, E.N.; Lal, R.; South, A.P.; Salas-Alanis, J.C.; Murrell, D.F.; Donati, G.; Watt, F.M. Innate sensing of microbial products promotes wound-induced skin cancer. Nat. Commun. 2015, 6, 5932. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yan, W.; Tohme, S.; Chen, M.; Fu, Y.; Tian, D.; Lotze, M.; Tang, D.; Tsung, A. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through Toll-like receptor 9. J. Hepatol. 2015, 63, 114–121. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.Y.; Zang, J.; Zheng, M.H.; Zhang, Y.F.; Yue, K.Y.; Cao, X.L.; Cao, Y.; Li, X.X.; Han, H.; Jiang, X.F.; et al. Temozolomide Treatment Induces HMGB1 to Promote the Formation of Glioma Stem Cells via the TLR2/NEAT1/Wnt Pathway in Glioblastoma. Front. Cell Dev. Biol. 2021, 9, 620883. [Google Scholar] [CrossRef]
- Conti, L.; Lanzardo, S.; Arigoni, M.; Antonazzo, R.; Radaelli, E.; Cantarella, D.; Calogero, R.A.; Cavallo, F. The noninflammatory role of high mobility group box 1/Toll-like receptor 2 axis in the self-renewal of mammary cancer stem cells. FASEB J. 2013, 27, 4731–4744. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, M.; Zhou, L.; Feng, X.; Cheng, J.; Yu, Y.; Gong, Y.; Zhu, Y.; Li, C.; Tian, L.; et al. Increased HMGB1 and cleaved caspase-3 stimulate the proliferation of tumor cells and are correlated with the poor prognosis in colorectal cancer. J. Exp. Clin. Cancer Res. 2015, 34, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrivastava, S.; Mansure, J.J.; Almajed, W.; Cury, F.; Ferbeyre, G.; Popovic, M.; Seuntjens, J.; Kassouf, W. The Role of HMGB1 in Radioresistance of Bladder Cancer. Mol. Cancer Ther. 2016, 15, 471–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Ren, L.; Chen, Y.; Fang, J.; Ge, Z.; Li, X. Redox status of high-mobility group box 1 performs a dual role in angiogenesis of colorectal carcinoma. J. Cell Mol. Med. 2015, 19, 2128–2135. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cheng, F.; Liu, Y.; Zhang, L.; Song, L.; Cai, X.; You, T.; Fan, X.; Wang, D.; Gong, A.; et al. Toll-like receptor 2 and Toll-like receptor 4 exhibit distinct regulation of cancer cell stemness mediated by cell death-induced high-mobility group box 1. EBioMedicine 2019, 40, 135–150. [Google Scholar] [CrossRef]
- Teo Hansen Selno, A.; Schlichtner, S.; Yasinska, I.M.; Sakhnevych, S.S.; Fiedler, W.; Wellbrock, J.; Berger, S.M.; Klenova, E.; Gibbs, B.F.; Fasler-Kan, E.; et al. High Mobility Group Box 1 (HMGB1) Induces Toll-like Receptor 4-Mediated Production of the Immunosuppressive Protein Galectin-9 in Human Cancer Cells. Front. Immunol. 2021, 12, 675731. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, A.; Bolli, E.; Ruiu, R.; Ferrauto, G.; Di Gregorio, E.; Avalle, L.; Savino, A.; Poggio, P.; Merighi, I.F.; Riccardo, F.; et al. Toll-like receptor 2 promotes breast cancer progression and resistance to chemotherapy. Oncoimmunology 2022, 11, 2086752. [Google Scholar] [CrossRef]
- Deng, R.; Wu, H.; Ran, H.; Kong, X.; Hu, L.; Wang, X.; Su, Q. Glucose-derived AGEs promote migration and invasion of colorectal cancer by up-regulating Sp1 expression. Biochim. Biophys. Acta 2017, 1861 Pt A, 1065–1074. [Google Scholar] [CrossRef] [Green Version]
- Kwak, T.; Drews-Elger, K.; Ergonul, A.; Miller, P.C.; Braley, A.; Hwang, G.H.; Zhao, D.; Besser, A.; Yamamoto, Y.; Yamamoto, H.; et al. Targeting of RAGE-ligand signaling impairs breast cancer cell invasion and metastasis. Oncogene 2017, 36, 1559–1572. [Google Scholar] [CrossRef]
- Herwig, N.; Belter, B.; Wolf, S.; Haase-Kohn, C.; Pietzsch, J. Interaction of extracellular S100A4 with RAGE prompts prometastatic activation of A375 melanoma cells. J. Cell Mol. Med. 2016, 20, 825–835. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Li, D.; Zhou, Y.M.; Yang, H.; Cheng, D.; Ma, X.X. Effects of receptor for advanced glycation endproducts on microvessel formation in endometrial cancer. BMC Cancer 2016, 16, 93. [Google Scholar] [CrossRef] [Green Version]
- Bao, J.M.; He, M.Y.; Liu, Y.W.; Lu, Y.J.; Hong, Y.Q.; Luo, H.H.; Ren, Z.L.; Zhao, S.C.; Jiang, Y. AGE/RAGE/Akt pathway contributes to prostate cancer cell proliferation by promoting Rb phosphorylation and degradation. Am. J. Cancer Res. 2015, 5, 1741–1750. [Google Scholar] [CrossRef] [Green Version]
- Pusterla, T.; Nemeth, J.; Stein, I.; Wiechert, L.; Knigin, D.; Marhenke, S.; Longerich, T.; Kumar, V.; Arnold, B.; Vogel, A.; et al. Receptor for advanced glycation endproducts (RAGE) is a key regulator of oval cell activation and inflammation-associated liver carcinogenesis in mice. Hepatology 2013, 58, 363–373. [Google Scholar] [CrossRef]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [Green Version]
- Konner, A.C.; Bruning, J.C. Toll-like receptors: Linking inflammation to metabolism. Trends Endocrinol. Metab. 2011, 22, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Jounai, N.; Kobiyama, K.; Takeshita, F.; Ishii, K.J. Recognition of damage-associated molecular patterns related to nucleic acids during inflammation and vaccination. Front. Cell Infect. Microbiol. 2012, 2, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannucci, A.; Caneparo, V.; Raviola, S.; Debernardi, I.; Colangelo, D.; Miggiano, R.; Griffante, G.; Landolfo, S.; Gariglio, M.; De Andrea, M. Toll-like receptor 4-mediated inflammation triggered by extracellular IFI16 is enhanced by lipopolysaccharide binding. PLoS Pathog. 2020, 16, e1008811. [Google Scholar] [CrossRef] [PubMed]
- Arkhypov, I.; Ozbay Kurt, F.G.; Bitsch, R.; Novak, D.; Petrova, V.; Lasser, S.; Hielscher, T.; Groth, C.; Lepper, A.; Hu, X.; et al. HSP90alpha induces immunosuppressive myeloid cells in melanoma via TLR4 signaling. J. Immunother Cancer 2022, 10, e005551. [Google Scholar] [CrossRef]
- Pucci, M.; Raimondo, S.; Urzi, O.; Moschetti, M.; Di Bella, M.A.; Conigliaro, A.; Caccamo, N.; La Manna, M.P.; Fontana, S.; Alessandro, R. Tumor-Derived Small Extracellular Vesicles Induce Pro-Inflammatory Cytokine Expression and PD-L1 Regulation in M0 Macrophages via IL-6/STAT3 and TLR4 Signaling Pathways. Int. J. Mol. Sci. 2021, 22, 12118. [Google Scholar] [CrossRef]
- Hiratsuka, S.; Watanabe, A.; Sakurai, Y.; Akashi-Takamura, S.; Ishibashi, S.; Miyake, K.; Shibuya, M.; Akira, S.; Aburatani, H.; Maru, Y. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat. Cell Biol. 2008, 10, 1349–1355. [Google Scholar] [CrossRef]
- Voelcker, V.; Gebhardt, C.; Averbeck, M.; Saalbach, A.; Wolf, V.; Weih, F.; Sleeman, J.; Anderegg, U.; Simon, J. Hyaluronan fragments induce cytokine and metalloprotease upregulation in human melanoma cells in part by signalling via TLR4. Exp. Dermatol. 2008, 17, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Dang, S.; Peng, Y.; Ye, L.; Wang, Y.; Qian, Z.; Chen, Y.; Wang, X.; Lin, Y.; Zhang, X.; Sun, X.; et al. Stimulation of TLR4 by LMW-HA induces metastasis in human papillary thyroid carcinoma through CXCR7. Clin. Dev. Immunol. 2013, 2013, 712561. [Google Scholar] [CrossRef] [Green Version]
- Makkar, S.; Riehl, T.E.; Chen, B.; Yan, Y.; Alvarado, D.M.; Ciorba, M.A.; Stenson, W.F. Hyaluronic Acid Binding to TLR4 Promotes Proliferation and Blocks Apoptosis in Colon Cancer. Mol. Cancer Ther. 2019, 18, 2446–2456. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.; Duan, L.; Cui, F.; Cao, J.; Xiang, Y.; Tang, Y.; Zhou, L. S100A9 promotes human hepatocellular carcinoma cell growth and invasion through RAGE-mediated ERK1/2 and p38 MAPK pathways. Exp. Cell Res. 2015, 334, 228–238. [Google Scholar] [CrossRef]
- Duan, L.; Wu, R.; Ye, L.; Wang, H.; Yang, X.; Zhang, Y.; Chen, X.; Zuo, G.; Zhang, Y.; Weng, Y.; et al. S100A8 and S100A9 are associated with colorectal carcinoma progression and contribute to colorectal carcinoma cell survival and migration via Wnt/beta-catenin pathway. PLoS ONE 2013, 8, e62092. [Google Scholar] [CrossRef]
- Wu, P.; Quan, H.; Kang, J.; He, J.; Luo, S.; Xie, C.; Xu, J.; Tang, Y.; Zhao, S. Downregulation of Calcium Binding Protein S100A9 Inhibits Hypopharyngeal Cancer Cell Proliferation and Invasion Ability Through Inactivation of NFkappaB Signaling. Oncol. Res. 2017, 25, 1479–1488. [Google Scholar] [CrossRef]
- Zhu, W.; Xue, Y.; Liang, C.; Zhang, R.; Zhang, Z.; Li, H.; Su, D.; Liang, X.; Zhang, Y.; Huang, Q.; et al. S100A16 promotes cell proliferation and metastasis via AKT and ERK cell signaling pathways in human prostate cancer. Tumour Biol. 2016, 37, 12241–12250. [Google Scholar] [CrossRef]
- Hua, T.; Liu, S.; Xin, X.; Cai, L.; Shi, R.; Chi, S.; Feng, D.; Wang, H. S100A4 promotes endometrial cancer progress through epithelial-mesenchymal transition regulation. Oncol. Rep. 2016, 35, 3419–3426. [Google Scholar] [CrossRef] [Green Version]
- Grottke, A.; Ewald, F.; Lange, T.; Norz, D.; Herzberger, C.; Bach, J.; Grabinski, N.; Graser, L.; Hoppner, F.; Nashan, B.; et al. Downregulation of AKT3 Increases Migration and Metastasis in Triple Negative Breast Cancer Cells by Upregulating S100A4. PLoS ONE 2016, 11, e0146370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, T.; Zhang, Z.; Yang, X.F.; Luo, J.S. S100A4 expression is closely linked to genesis and progression of glioma by regulating proliferation, apoptosis, migration and invasion. Asian Pac. J. Cancer Prev. 2015, 16, 2883–2887. [Google Scholar] [CrossRef] [Green Version]
- Medapati, M.R.; Dahlmann, M.; Ghavami, S.; Pathak, K.A.; Lucman, L.; Klonisch, T.; Hoang-Vu, C.; Stein, U.; Hombach-Klonisch, S. RAGE Mediates the Pro-Migratory Response of Extracellular S100A4 in Human Thyroid Cancer Cells. Thyroid 2015, 25, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Kuper, C.; Beck, F.X.; Neuhofer, W. NFAT5-mediated expression of S100A4 contributes to proliferation and migration of renal carcinoma cells. Front. Physiol. 2014, 5, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Sato, D.; Saiki, Y.; Sunamura, M.; Fukushige, S.; Horii, A. S100A4 is frequently overexpressed in lung cancer cells and promotes cell growth and cell motility. Biochem. Biophys. Res. Commun. 2014, 447, 459–464. [Google Scholar] [CrossRef]
- Tsukamoto, N.; Egawa, S.; Akada, M.; Abe, K.; Saiki, Y.; Kaneko, N.; Yokoyama, S.; Shima, K.; Yamamura, A.; Motoi, F.; et al. The expression of S100A4 in human pancreatic cancer is associated with invasion. Pancreas 2013, 42, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, H.; Chen, X.; Dong, F.; Zhang, Z.; Zhou, Z.; Ma, Z.; Huang, S.; Chen, B.; Zhang, C.; Hou, B. Prognostic values and immune suppression of the S100A family in pancreatic cancer. J. Cell Mol. Med. 2021, 25, 3006–3018. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.Y.; Chen, Y.W.; Hsiao, J.R.; Liu, C.S.; Kuo, Y.Z.; Wang, Y.C.; Chang, K.C.; Tsai, S.T.; Chang, M.Z.; Lin, S.H.; et al. Elevated S100A9 expression in tumor stroma functions as an early recurrence marker for early-stage oral cancer patients through increased tumor cell invasion, angiogenesis, macrophage recruitment and interleukin-6 production. Oncotarget 2015, 6, 28401–28424. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.Y.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Muschel, R.J. Tumor-infiltrating monocytes/macrophages promote tumor invasion and migration by upregulating S100A8 and S100A9 expression in cancer cells. Oncogene 2016, 35, 5735–5745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tidehag, V.; Hammarsten, P.; Egevad, L.; Granfors, T.; Stattin, P.; Leanderson, T.; Wikstrom, P.; Josefsson, A.; Hagglof, C.; Bergh, A. High density of S100A9 positive inflammatory cells in prostate cancer stroma is associated with poor outcome. Eur. J. Cancer 2014, 50, 1829–1835. [Google Scholar] [CrossRef]
- Nasser, M.W.; Wani, N.A.; Ahirwar, D.K.; Powell, C.A.; Ravi, J.; Elbaz, M.; Zhao, H.; Padilla, L.; Zhang, X.; Shilo, K.; et al. RAGE mediates S100A7-induced breast cancer growth and metastasis by modulating the tumor microenvironment. Cancer Res. 2015, 75, 974–985. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.H.; Heo, W.H.; Son, H.Y.; Quan, M.; Hong, B.S.; Kim, J.H.; Lee, H.B.; Han, W.; Park, Y.; Lee, D.S.; et al. S100A8/A9 mediate the reprograming of normal mammary epithelial cells induced by dynamic cell-cell interactions with adjacent breast cancer cells. Sci. Rep. 2021, 11, 1337. [Google Scholar] [CrossRef]
- Shen, Z.; Deng, H.; Fang, Y.; Zhu, X.; Ye, G.T.; Yan, L.; Liu, H.; Li, G. Identification of the interplay between SOX9 and S100P in the metastasis and invasion of colon carcinoma. Oncotarget 2015, 6, 20672–20684. [Google Scholar] [CrossRef] [Green Version]
- Bettum, I.J.; Gorad, S.S.; Barkovskaya, A.; Pettersen, S.; Moestue, S.A.; Vasiliauskaite, K.; Tenstad, E.; Oyjord, T.; Risa, O.; Nygaard, V.; et al. Metabolic reprogramming supports the invasive phenotype in malignant melanoma. Cancer Lett. 2015, 366, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, L.E.; Dingemans, A.C. Heat shock protein antagonists in early stage clinical trials for NSCLC. Expert Opin. Investig. Drugs 2017, 26, 541–550. [Google Scholar] [CrossRef]
- Bagatell, R.; Whitesell, L. Altered Hsp90 function in cancer: A unique therapeutic opportunity. Mol. Cancer Ther. 2004, 3, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Wen, J.; Li, W.; Xue, J.; Zhang, Y.; Liu, H.; Han, J.; Ning, T.; Lu, Z. HSP90 promotes radioresistance of cervical cancer cells via reducing FBXO6-mediated CD147 polyubiquitination. Cancer Sci. 2022, 113, 1463–1474. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Bishop, J.L.; Nip, K.M.; Zardan, A.; Takeuchi, A.; Cordonnier, T.; Beraldi, E.; Bazov, J.; Fazli, L.; Chi, K.; et al. Hsp27 regulates epithelial mesenchymal transition, metastasis, and circulating tumor cells in prostate cancer. Cancer Res. 2013, 73, 3109–3119. [Google Scholar] [CrossRef] [Green Version]
- Cordonnier, T.; Bishop, J.L.; Shiota, M.; Nip, K.M.; Thaper, D.; Vahid, S.; Heroux, D.; Gleave, M.; Zoubeidi, A. Hsp27 regulates EGF/beta-catenin mediated epithelial to mesenchymal transition in prostate cancer. Int. J. Cancer 2015, 136, E496–E507. [Google Scholar] [CrossRef]
- Ren, X.; Li, T.; Zhang, W.; Yang, X. Targeting Heat-Shock Protein 90 in Cancer: An Update on Combination Therapy. Cells 2022, 11, 2556. [Google Scholar] [CrossRef]
- Xue, N.; Du, T.; Lai, F.; Jin, J.; Ji, M.; Chen, X. Secreted HSP90alpha-LRP1 Signaling Promotes Tumor Metastasis and Chemoresistance in Pancreatic Cancer. Int. J. Mol. Sci. 2022, 23, 5532. [Google Scholar] [CrossRef]
- Feng, H.; Guo, Z.; Chen, X.; Liu, K.; Li, H.; Jia, W.; Wang, C.; Luo, F.; Ji, X.; Zhang, T.; et al. Excessive HSP70/TLR2 activation leads to remodeling of the tumor immune microenvironment to resist chemotherapy sensitivity of mFOLFOX in colorectal cancer. Clin. Immunol. 2022, 245, 109157. [Google Scholar] [CrossRef]
- Su, K.; Liu, Y.; Wang, P.; He, K.; Wang, F.; Chi, H.; Rao, M.; Li, X.; Wen, L.; Song, Y.; et al. Heat-shock protein 90alpha is a potential prognostic and predictive biomarker in hepatocellular carcinoma: A large-scale and multicenter study. Hepatol. Int. 2022, 16, 1208–1219. [Google Scholar] [CrossRef]
- Pahwa, R.; Dubhashi, J.; Singh, A.; Jailwala, P.; Lobanov, A.; Thomas, C.J.; Ceribelli, M.; Wilson, K.; Ricketts, C.J.; Vocke, C.D.; et al. Inhibition of HSP 90 is associated with potent anti-tumor activity in Papillary Renal Cell Carcinoma. J. Exp. Clin. Cancer Res. 2022, 41, 208. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Cho, T.M.; Park, J.M.; Park, S.; Park, M.; Nam, K.D.; Ko, D.; Seo, J.; Kim, S.; Jung, E.; et al. A novel HSP90 inhibitor SL-145 suppresses metastatic triple-negative breast cancer without triggering the heat shock response. Oncogene 2022, 41, 3289–3297. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kang, R.; Kroemer, G.; Tang, D. Targeting HSP90 sensitizes pancreas carcinoma to PD-1 blockade. Oncoimmunology 2022, 11, 2068488. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.A.; Kumar, V.; Tailor, D.; Garcia-Marques, F.J.; Hsu, E.C.; Liu, S.; Bermudez, A.; Kanchustambham, V.; Shankar, V. SU086, an inhibitor of HSP90, impairs glycolysis and represents a treatment strategy for advanced prostate cancer. Cell Rep. Med. 2022, 3, 100502. [Google Scholar] [CrossRef]
- Wei, S.; Yin, D.; Yu, S.; Lin, X.; Savani, M.R.; Du, K.; Ku, Y.; Wu, D.; Li, S.; Liu, H.; et al. Antitumor Activity of a Mitochondrial-Targeted HSP90 Inhibitor in Gliomas. Clin. Cancer Res. 2022, 28, 2180–2195. [Google Scholar] [CrossRef]
- Killock, D. HSP90 inhibition improves GIST survival. Nat. Rev. Clin. Oncol. 2022, 19, 568. [Google Scholar] [CrossRef]
- Sasame, J.; Ikegaya, N.; Kawazu, M.; Natsumeda, M.; Hayashi, T.; Isoda, M.; Satomi, K.; Tomiyama, A.; Oshima, A.; Honma, H.; et al. HSP90 Inhibition Overcomes Resistance to Molecular Targeted Therapy in BRAFV600E-mutant High-grade Glioma. Clin. Cancer Res. 2022, 28, 2425–2439. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ren, X.; Wu, J.; Gao, X.; Cen, X.; Wang, S.; Sheng, S.; Chen, Q.; Tang, Y.J.; Liang, X.H.; et al. HSP27 associates with epithelial-mesenchymal transition, stemness and radioresistance of salivary adenoid cystic carcinoma. J. Cell Mol. Med. 2018, 22, 2283–2298. [Google Scholar] [CrossRef] [Green Version]
- Yao, K.; He, L.; Gan, Y.; Liu, J.; Tang, J.; Long, Z.; Tan, J. HMGN5 promotes IL-6-induced epithelial-mesenchymal transition of bladder cancer by interacting with Hsp27. Aging 2020, 12, 7282–7298. [Google Scholar] [CrossRef]
- Fang, Z.; Liang, W.; Luo, L. HSP27 promotes epithelial-mesenchymal transition through activation of the beta-catenin/MMP3 pathway in pancreatic ductal adenocarcinoma cells. Transl. Cancer Res. 2019, 8, 1268–1278. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Jiang, Y.; Han, D.; Tan, W. Hsp27 regulates epithelial mesenchymal transition, metastasis and proliferation in colorectal carcinoma. Oncol. Lett. 2018, 16, 5309–5316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.K.; Kam, H.; Kim, K.Y.; Park, S.I.; Lee, Y.S. Targeting Heat Shock Protein 27 in Cancer: A Druggable Target for Cancer Treatment? Cancers 2019, 11, 1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katakam, S.; Anand, S.; Martin, P.; Riggi, N.; Stamenkovic, I. Necrotic debris and STING exert therapeutically relevant effects on tumor cholesterol homeostasis. Life Sci. Alliance 2022, 5, e202101256. [Google Scholar] [CrossRef]
- Mehrabi, M.; Amini, F.; Mehrabi, S. Active Role of the Necrotic Zone in Desensitization of Hypoxic Macrophages and Regulation of CSC-Fate: A hypothesis. Front. Oncol. 2018, 8, 235. [Google Scholar] [CrossRef] [Green Version]
- ElShamy, W.M.; Sinha, A.; Said, N. Aggressiveness Niche: Can It Be the Foster Ground for Cancer Metastasis Precursors? Stem Cells Int. 2016, 2016, 4829106. [Google Scholar] [CrossRef] [Green Version]
- Cormier, S.A.; Taranova, A.G.; Bedient, C.; Nguyen, T.; Protheroe, C.; Pero, R.; Dimina, D.; Ochkur, S.I.; O’Neill, K.; Colbert, D.; et al. Pivotal Advance: Eosinophil infiltration of solid tumors is an early and persistent inflammatory host response. J. Leukoc. Biol. 2006, 79, 1131–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotfi, R.; Kaltenmeier, C.; Lotze, M.T.; Bergmann, C. Until Death Do Us Part: Necrosis and Oxidation Promote the Tumor Microenvironment. Transfus Med. Hemother 2016, 43, 120–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yee, P.P.; Li, W. Tumor necrosis: A synergistic consequence of metabolic stress and inflammation. Bioessays 2021, 43, e2100029. [Google Scholar] [CrossRef]
- Ocana, A.; Nieto-Jimenez, C.; Pandiella, A.; Templeton, A.J. Neutrophils in cancer: Prognostic role and therapeutic strategies. Mol. Cancer 2017, 16, 137. [Google Scholar] [CrossRef] [Green Version]
- Di Virgilio, F.; Sarti, A.C.; Coutinho-Silva, R. Purinergic signaling, DAMPs, and inflammation. Am. J. Physiol. Cell Physiol. 2020, 318, C832–C835. [Google Scholar] [CrossRef] [Green Version]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Hasko, G. Immunity, inflammation and cancer: A leading role for adenosine. Nat. Rev. Cancer 2013, 13, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.R.; Deng, W.W.; Liu, J.F.; Mao, L.; Yu, G.T.; Bu, L.L.; Kulkarni, A.B.; Zhang, W.F.; Sun, Z.J. Blockade of adenosine A2A receptor enhances CD8(+) T cells response and decreases regulatory T cells in head and neck squamous cell carcinoma. Mol. Cancer 2017, 16, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, G.; Vuerich, M.; Pellegatti, P.; Marimpietri, D.; Emionite, L.; Marigo, I.; Bronte, V.; Di Virgilio, F.; Pistoia, V.; Raffaghello, L. ATP/P2X7 axis modulates myeloid-derived suppressor cell functions in neuroblastoma microenvironment. Cell Death Dis. 2014, 5, e1135. [Google Scholar] [CrossRef] [Green Version]
- Baghdadi, M.; Yoneda, A.; Yamashina, T.; Nagao, H.; Komohara, Y.; Nagai, S.; Akiba, H.; Foretz, M.; Yoshiyama, H.; Kinoshita, I.; et al. TIM-4 glycoprotein-mediated degradation of dying tumor cells by autophagy leads to reduced antigen presentation and increased immune tolerance. Immunity 2013, 39, 1070–1081. [Google Scholar] [CrossRef] [Green Version]
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.D.; et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 2012, 13, 832–842. [Google Scholar] [CrossRef] [Green Version]
- Wild, C.A.; Bergmann, C.; Fritz, G.; Schuler, P.; Hoffmann, T.K.; Lotfi, R.; Westendorf, A.; Brandau, S.; Lang, S. HMGB1 conveys immunosuppressive characteristics on regulatory and conventional T cells. Int. Immunol. 2012, 24, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Demoulin, S.; Herfs, M.; Somja, J.; Roncarati, P.; Delvenne, P.; Hubert, P. HMGB1 secretion during cervical carcinogenesis promotes the acquisition of a tolerogenic functionality by plasmacytoid dendritic cells. Int. J. Cancer 2015, 137, 345–358. [Google Scholar] [CrossRef] [Green Version]
- Shiau, D.J.; Kuo, W.T.; Davuluri, G.V.N.; Shieh, C.C.; Tsai, P.J.; Chen, C.C.; Lin, Y.S.; Wu, Y.Z.; Hsiao, Y.P.; Chang, C.P. Hepatocellular carcinoma-derived high mobility group box 1 triggers M2 macrophage polarization via a TLR2/NOX2/autophagy axis. Sci. Rep. 2020, 10, 13582. [Google Scholar] [CrossRef]
- Ye, L.; Zhang, Q.; Cheng, Y.; Chen, X.; Wang, G.; Shi, M.; Zhang, T.; Cao, Y.; Pan, H.; Zhang, L.; et al. Tumor-derived exosomal HMGB1 fosters hepatocellular carcinoma immune evasion by promoting TIM-1(+) regulatory B cell expansion. J. Immunother Cancer 2018, 6, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Chapman, N.M.; Zhang, B.; Li, M.; Fan, M.; Laribee, R.N.; Zaidi, M.R.; Pfeffer, L.M.; Chi, H.; Wu, Z.H. Upregulation of PD-L1 via HMGB1-Activated IRF3 and NF-kappaB Contributes to UV Radiation-Induced Immune Suppression. Cancer Res. 2019, 79, 2909–2922. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Okoro, C.; Foell, D.; Freeze, H.H.; Ostrand-Rosenberg, S.; Srikrishna, G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J. Immunol. 2008, 181, 4666–4675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, P.; Corzo, C.A.; Luetteke, N.; Yu, B.; Nagaraj, S.; Bui, M.M.; Ortiz, M.; Nacken, W.; Sorg, C.; Vogl, T.; et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J. Exp. Med. 2008, 205, 2235–2249. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Xu, H.; Li, N.; Wang, H.; Ma, L.; Chen, S.; Liu, J.; Zheng, Y.; Zhang, Y. Renal cancer-derived exosomes induce tumor immune tolerance by MDSCs-mediated antigen-specific immunosuppression. Cell Commun. Signal 2020, 18, 106. [Google Scholar] [CrossRef]
- Block, H.; Rossaint, J.; Zarbock, A. The Fatal Circle of NETs and NET-Associated DAMPs Contributing to Organ Dysfunction. Cells 2022, 11, 1919. [Google Scholar] [CrossRef] [PubMed]
- Munir, H.; Jones, J.O.; Janowitz, T.; Hoffmann, M.; Euler, M.; Martins, C.P.; Welsh, S.J.; Shields, J.D. Stromal-driven and Amyloid beta-dependent induction of neutrophil extracellular traps modulates tumor growth. Nat. Commun. 2021, 12, 683. [Google Scholar] [CrossRef] [PubMed]
- Leijte, G.P.; Custers, H.; Gerretsen, J.; Heijne, A.; Roth, J.; Vogl, T.; Scheffer, G.J.; Pickkers, P.; Kox, M. Increased Plasma Levels of Danger-Associated Molecular Patterns Are Associated With Immune Suppression and Postoperative Infections in Patients Undergoing Cytoreductive Surgery and Hyperthermic Intraperitoneal Chemotherapy. Front. Immunol. 2018, 9, 663. [Google Scholar] [CrossRef]
- Timmermans, K.; Kox, M.; Vaneker, M.; van den Berg, M.; John, A.; van Laarhoven, A.; van der Hoeven, H.; Scheffer, G.J.; Pickkers, P. Plasma levels of danger-associated molecular patterns are associated with immune suppression in trauma patients. Intensive Care Med. 2016, 42, 551–561. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Tait, S.W.G. Targeting immunogenic cell death in cancer. Mol. Oncol. 2020, 14, 2994–3006. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Type of DAMP | DAMPs | Receptors |
|---|---|---|
| Proteins | HMGB1 | TLR2, TLR4, RAGE |
| Histone | TLR2, TLR4 | |
| S100 | TLR2, TLR4, RAGE | |
| HSPs | TLR2, TLR4, CD91, RAGE | |
| Annexin A1 | FPR1 | |
| Versican | TLR2, TLR6, CD14 | |
| Fibronectin (EDA domain) | TLR4 | |
| Fibrinogen | TLR4 | |
| Tenascin C | TLR4 | |
| F-actin | DNGR-1 | |
| Cyclophilin A | CD147 | |
| Aβ | TLR2, NLRP1, NLRP3, CD36, RAGE | |
| IL1α | IL-1R | |
| IL33 | ST2 | |
| Formyl peptide | FPR1 | |
| Calreticulin | CD91 | |
| Defensins | TLR4 | |
| Cathelicidin (LL37) | P2X7, FPR2 | |
| Granulysin | TLR4 | |
| Lipids and carbohydrates | LMW hyaluronan | TLR2, TLR4, NLRP3 |
| SAA | TLR2, TLR4 | |
| Heparan sulfate | TLR4 | |
| Metabolite-related DAMPs | ATP | P2X7, P2Y2 |
| Uric acid | NLRP3, P2X7 | |
| Nucleic acids | DNA | TLR9, AIM2 |
| RNA | TLR3, TLR7/8, RIG-I, MDA5 | |
| mtDNA | TLR9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zapletal, E.; Vasiljevic, T.; Busson, P.; Matijevic Glavan, T. Dialog beyond the Grave: Necrosis in the Tumor Microenvironment and Its Contribution to Tumor Growth. Int. J. Mol. Sci. 2023, 24, 5278. https://doi.org/10.3390/ijms24065278
Zapletal E, Vasiljevic T, Busson P, Matijevic Glavan T. Dialog beyond the Grave: Necrosis in the Tumor Microenvironment and Its Contribution to Tumor Growth. International Journal of Molecular Sciences. 2023; 24(6):5278. https://doi.org/10.3390/ijms24065278
Chicago/Turabian StyleZapletal, Emilija, Tea Vasiljevic, Pierre Busson, and Tanja Matijevic Glavan. 2023. "Dialog beyond the Grave: Necrosis in the Tumor Microenvironment and Its Contribution to Tumor Growth" International Journal of Molecular Sciences 24, no. 6: 5278. https://doi.org/10.3390/ijms24065278