
Breaking the Gingival Barrier in Periodontitis
 ,
,  , , , , ,
, , , , ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
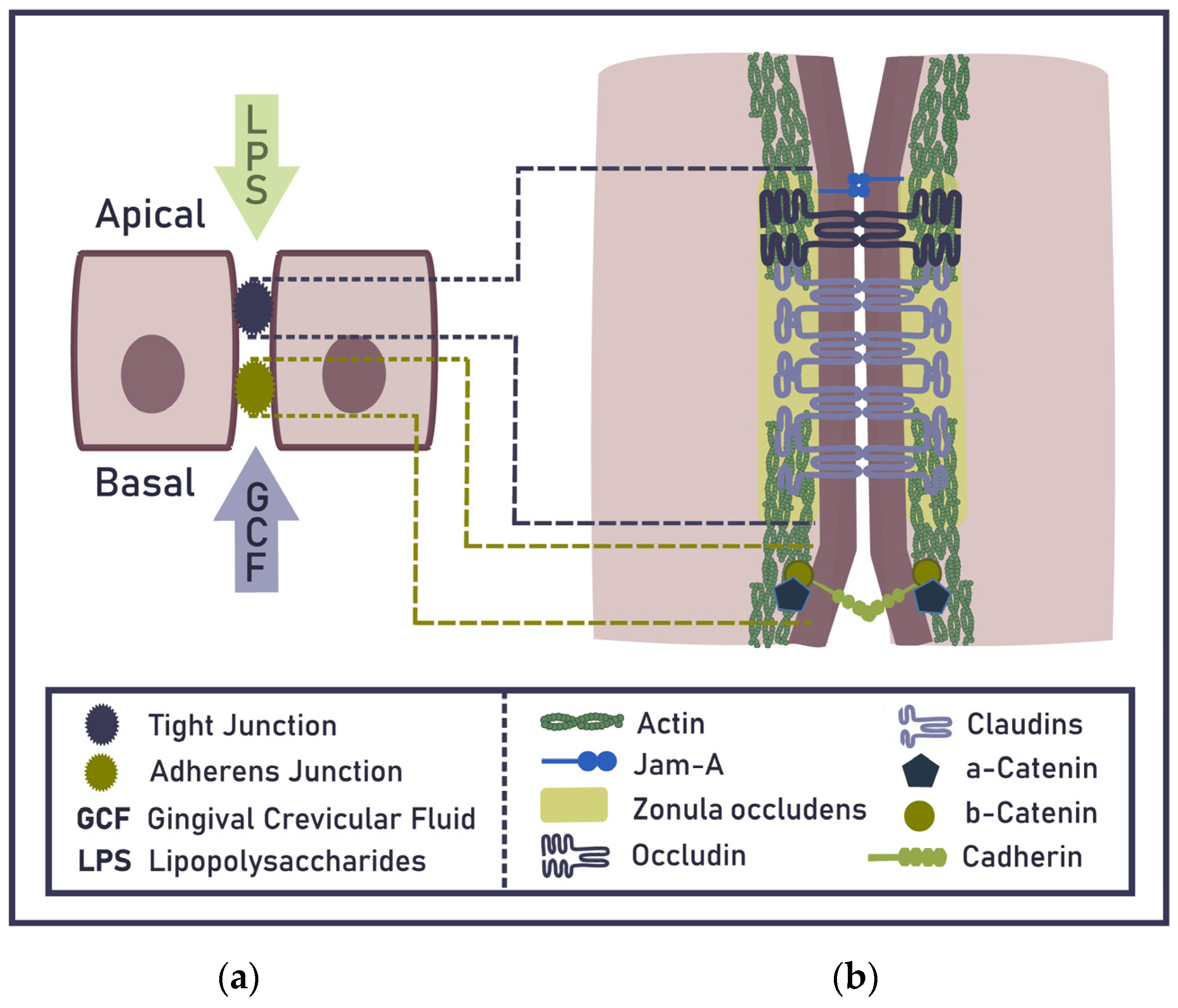
2. Epithelial Barrier of the Gingiva
3. TJ Compromising via a Surplus of LPS and by Oral Pathogens
4. Ageing-Related Impairment of the Epithelial Barrier
5. TJ Compromising via Oncostatin M (OSM) and Neutrophil Proteases
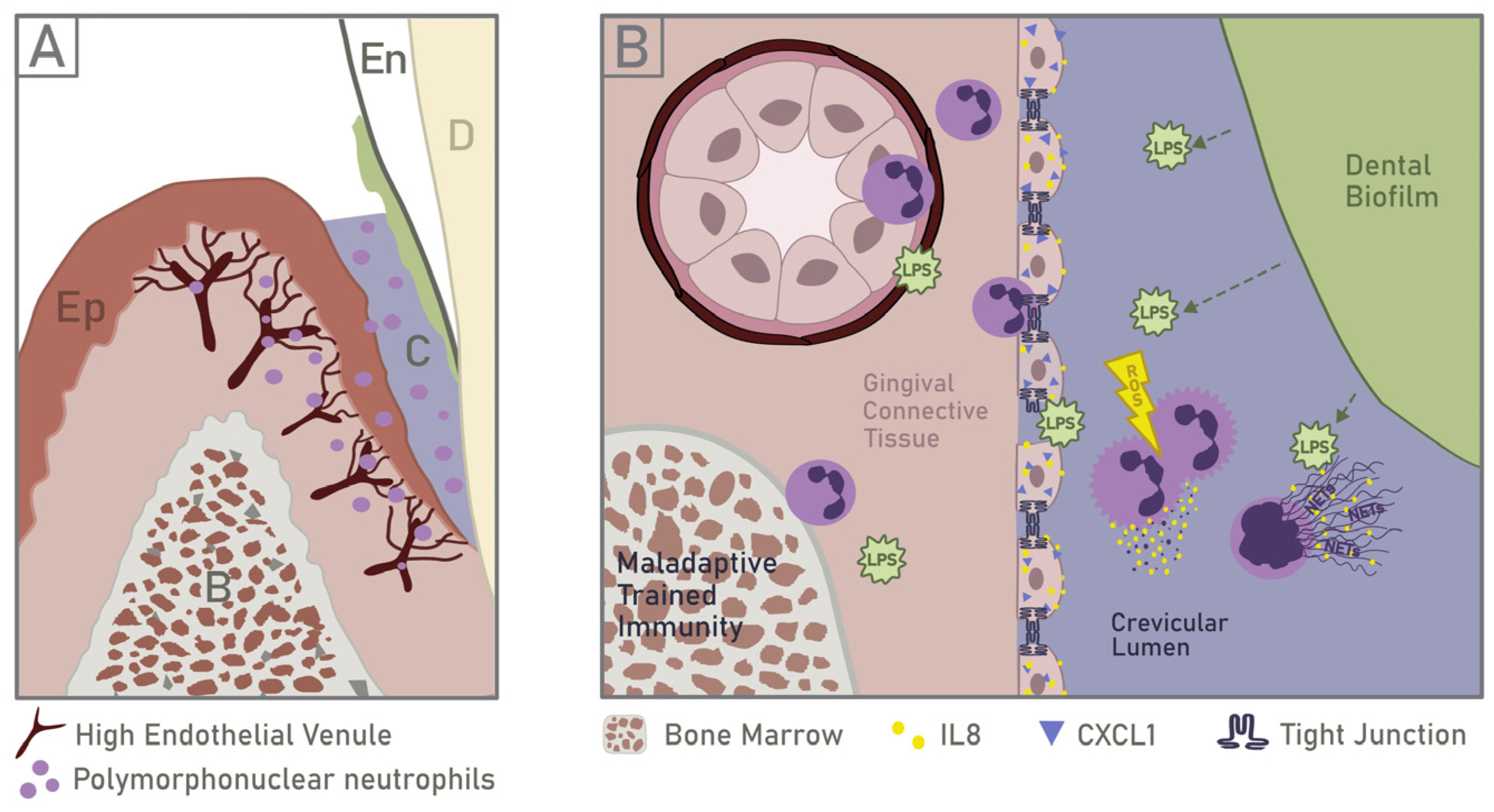
5.1. Neutrophil Recruitment and Response to PAMPs
5.2. Implications of Neutrophil Infiltration on Epithelial Barrier
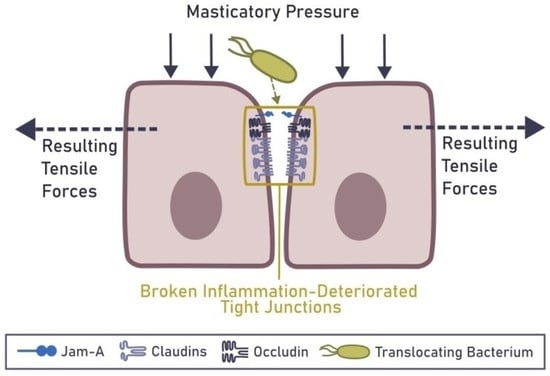
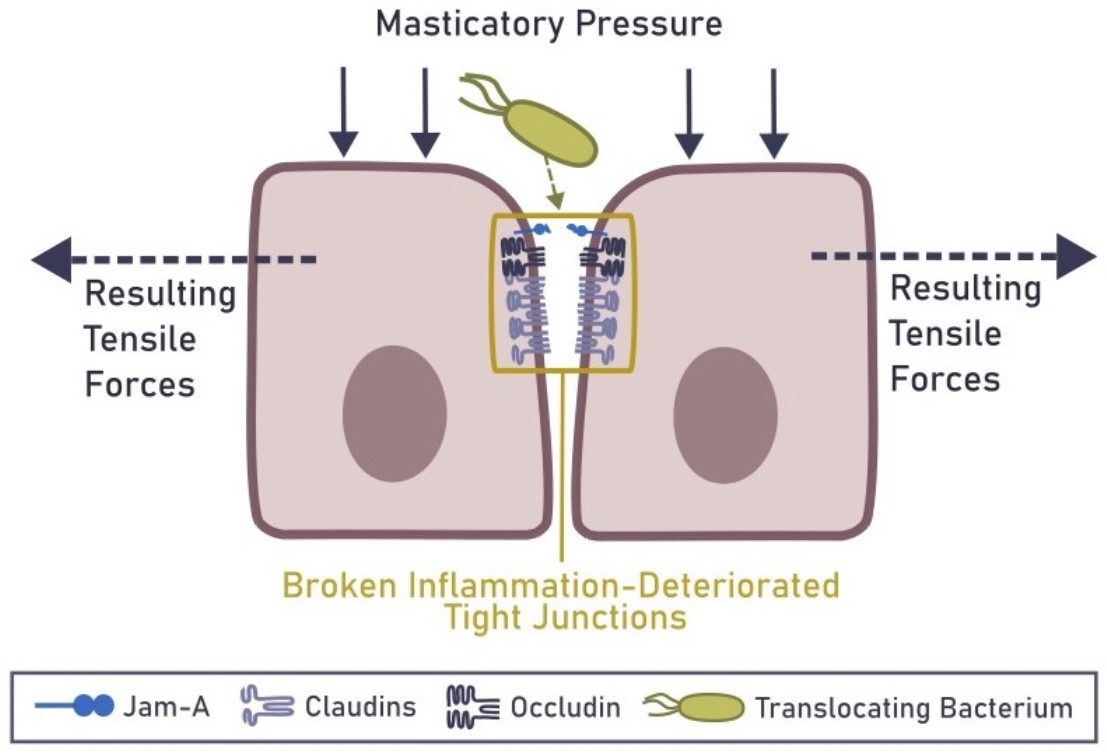
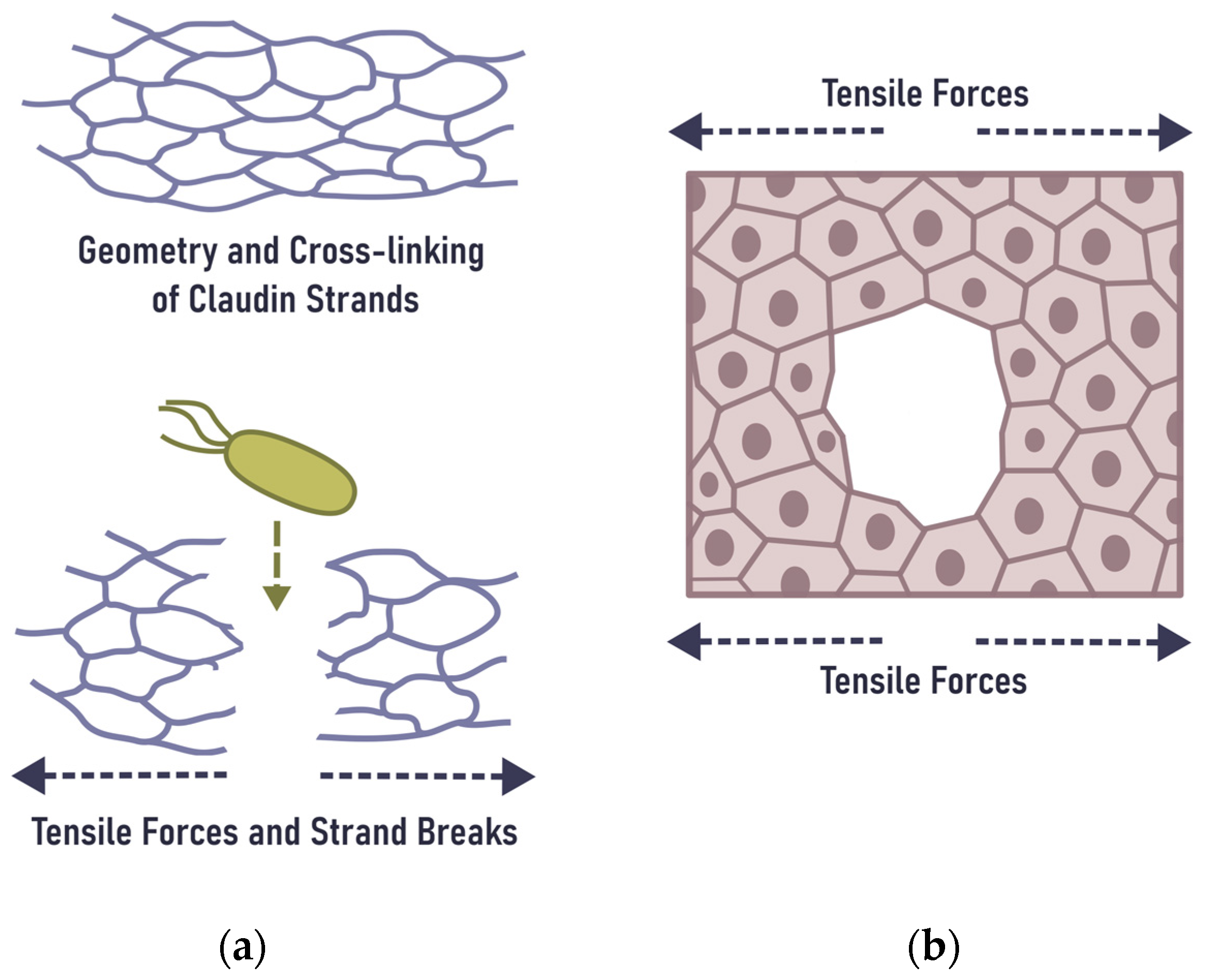
6. Mechanical Rupture of the Epithelial Barrier in the Gingiva
7. State of Knowledge and Perspectives
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DOAJ | directory of open access journals |
| GCF | gingival crevicular fluid |
| IL | interleukin |
| JAM-1 | junctional adhesion molecule-1 |
| LGI | low-grade inflammation |
| LPS | lipopolysaccharide |
| MDPI | multidisciplinary Digital Publishing Institute |
| MTI | maladaptive trained immunity |
| NETs | neutrophil extracellular traps |
| OMS | Oncostatin M |
| OMVs | outer membrane vesicles |
| PAMPs | pathogen associated molecular patterns |
| PAR2 | protease activated receptor 2 |
| ROS | reactive oxygen species |
| TLR | toll-like receptor |
| TNFα | tumor necrosis factor α |
| TJ | tight junction |
| ZO-1 | zonula occludens-1 |
| BOP | Bleeding on Probing |
| FITC | Fluorescein-5-isothiocyanate (FITC) |
References
- Ghosh, S.S.; Wang, J.; Yannie, P.J.; Ghosh, S. Intestinal Barrier Dysfunction, LPS Translocation, and Disease Development. J. Endocr. Soc. 2020, 4, bvz039. [Google Scholar] [CrossRef] [Green Version]
- Vitkov, L.; Klappacher, M.; Hannig, M.; Krautgartner, W.D. Extracellular neutrophil traps in periodontitis. J. Periodontal Res. 2009, 44, 664–672. [Google Scholar] [CrossRef]
- Vitkov, L.; Klappacher, M.; Hannig, M.; Krautgartner, W.D. Neutrophil fate in gingival crevicular fluid. Ultrastruct. Pathol. 2010, 34, 25–30. [Google Scholar] [CrossRef]
- Vitkov, L.; Minnich, B.; Knopf, J.; Schauer, C.; Hannig, M.; Herrmann, M. NETs Are Double-Edged Swords with the Potential to Aggravate or Resolve Periodontal Inflammation. Cells 2020, 9, 2614. [Google Scholar] [CrossRef]
- Vitkov, L.; Munoz, L.E.; Schoen, J.; Knopf, J.; Schauer, C.; Minnich, B.; Herrmann, M.; Hannig, M. Neutrophils Orchestrate the Periodontal Pocket. Front. Immunol. 2021, 12, 788766. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, M.; Komatsu, N.; Nagashima, K.; Nitta, T.; Pluemsakunthai, W.; Shukunami, C.; Iwakura, Y.; Nakashima, T.; Okamoto, K.; Takayanagi, H. Host defense against oral microbiota by bone-damaging T cells. Nat. Commun. 2018, 9, 701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lockhart, P.B.; Brennan, M.T.; Sasser, H.C.; Fox, P.C.; Paster, B.J.; Bahrani-Mougeot, F.K. Bacteremia associated with toothbrushing and dental extraction. Circulation 2008, 117, 3118–3125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosio, N.; Marin, M.J.; Laguna, E.; Herrera, D.; Sanz, M.; Figuero, E. Detection and quantification of Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans in bacteremia induced by interdental brushing in periodontally healthy and periodontitis patients. Arch. Oral. Biol. 2019, 98, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Garcia de Aquino, S.; Manzolli Leite, F.R.; Stach-Machado, D.R.; Francisco da Silva, J.A.; Spolidorio, L.C.; Rossa, C., Jr. Signaling pathways associated with the expression of inflammatory mediators activated during the course of two models of experimental periodontitis. Life Sci. 2009, 84, 745–754. [Google Scholar] [CrossRef] [PubMed]
- de Almeida Brandao, D.; Spolidorio, L.C.; Johnson, F.; Golub, L.M.; Guimaraes-Stabili, M.R.; Rossa, C., Jr. Dose-response assessment of chemically modified curcumin in experimental periodontitis. J. Periodontol. 2019, 90, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Hajishengallis, G. Optimization of the ligature-induced periodontitis model in mice. J. Immunol. Methods 2013, 394, 49–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, S.; Mealey, B.L.; Mariotti, A.; Chapple, I.L.C. Dental plaque–induced gingival conditions. J. Periodontol. 2018, 89, S17–S27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baty, J.J.; Stoner, S.N.; Scoffield, J.A. Oral Commensal Streptococci: Gatekeepers of the Oral Cavity. J. Bacteriol. 2022, 204, e0025722. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Chavakis, T. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat. Rev. Immunol. 2021, 21, 426–440. [Google Scholar] [CrossRef]
- Li, X.; Wang, H.; Yu, X.; Saha, G.; Kalafati, L.; Ioannidis, C.; Mitroulis, I.; Netea, M.G.; Chavakis, T.; Hajishengallis, G. Maladaptive innate immune training of myelopoiesis links inflammatory comorbidities. Cell 2022, 185, 1709–1727.e18. [Google Scholar] [CrossRef]
- Irwandi, R.; Chiesa, S.; Hajishengallis, G.; Papayannopoulos, V.; Deanfield, J.; D’Aiuto, F. The Roles of Neutrophils Linking Periodontitis and Atherosclerotic Cardiovascular Diseases. Front. Immunol. 2022, 13, 915081. [Google Scholar] [CrossRef]
- Vitkov, L.; Knopf, J.; Krunić, J.; Schauer, C.; Schoen, J.; Minnich, B.; Hannig, M.; Herrmann, M. Periodontitis-Derived Dark-NETs in Severe COVID-19. Front. Immunol. 2022, 13, 872695. [Google Scholar] [CrossRef]
- Vitkov, L.; Muñoz, L.E.; Knopf, J.; Schauer, C.; Oberthaler, H.; Minnich, B.; Hannig, M.; Herrmann, M. Connection between Periodontitis-Induced Low-Grade Endotoxemia and Systemic Diseases: Neutrophils as Protagonists and Targets. Int. J. Mol. Sci. 2021, 22, 4647. [Google Scholar] [CrossRef]
- Marchiando, A.M.; Graham, W.V.; Turner, J.R. Epithelial Barriers in Homeostasis and Disease. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 119–144. [Google Scholar] [CrossRef]
- Ko, Y.K.; Hong, S.; Kim, H.M.; Liu, M.; Moon, E.; Kim, P.; Choi, Y. Characterization of junctional structures in the gingival epithelium as barriers against bacterial invasion. J. Periodontal Res. 2022, 57, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Vitkov, L.; Krautgartner, W.D.; Hannig, M.; Fuchs, K. Fimbria-mediated bacterial adhesion to human oral epithelium. FEMS Microbiol. Lett. 2001, 202, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.M.; Van Itallie, C.M. Physiology and function of the tight junction. Cold Spring Harb. Perspect. Biol. 2009, 1, a002584. [Google Scholar] [CrossRef]
- Shen, L.; Weber, C.R.; Raleigh, D.R.; Yu, D.; Turner, J.R. Tight junction pore and leak pathways: A dynamic duo. Annu. Rev. Physiol. 2011, 73, 283–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, J.R.; Buschmann, M.M.; Romero-Calvo, I.; Sailer, A.; Shen, L. The role of molecular remodeling in differential regulation of tight junction permeability. Semin. Cell Dev. Biol. 2014, 36, 204–212. [Google Scholar] [CrossRef] [Green Version]
- Van Itallie, C.M.; Tietgens, A.J.; Krystofiak, E.; Kachar, B.; Anderson, J.M. A complex of ZO-1 and the BAR-domain protein TOCA-1 regulates actin assembly at the tight junction. Mol. Biol. Cell 2015, 26, 2769–2787. [Google Scholar] [CrossRef]
- Lee, G.; Kim, H.J.; Kim, H.M. RhoA-JNK Regulates the E-Cadherin Junctions of Human Gingival Epithelial Cells. J. Dent. Res. 2016, 95, 284–291. [Google Scholar] [CrossRef]
- Hollander, D.; Kaunitz, J.D. The “Leaky Gut”: Tight Junctions but Loose Associations? Dig. Dis. Sci. 2020, 65, 1277–1287. [Google Scholar] [CrossRef] [Green Version]
- Guerville, M.; Boudry, G. Gastrointestinal and hepatic mechanisms limiting entry and dissemination of lipopolysaccharide into the systemic circulation. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G1–G15. [Google Scholar] [CrossRef] [Green Version]
- He, D.; Su, Y.; Usatyuk, P.V.; Spannhake, E.W.; Kogut, P.; Solway, J.; Natarajan, V.; Zhao, Y. Lysophosphatidic acid enhances pulmonary epithelial barrier integrity and protects endotoxin-induced epithelial barrier disruption and lung injury. J. Biol. Chem. 2009, 284, 24123–24132. [Google Scholar] [CrossRef] [Green Version]
- Han, F.; Lu, Z.; Liu, Y.; Xia, X.; Zhang, H.; Wang, X.; Wang, Y. Cathelicidin-BF ameliorates lipopolysaccharide-induced intestinal epithelial barrier disruption in rat. Life Sci. 2016, 152, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Lee, S.W.; Kim, J.; Shin, Y.; Chang, F.; Kim, J.M.; Cong, X.; Yu, G.Y.; Park, K. LPS-induced epithelial barrier disruption via hyperactivation of CACC and ENaC. Am. J. Physiol. Cell Physiol. 2021, 320, C448–C461. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Sasaki, N.; Yamaga, S.; Kuboniwa, M.; Matsusaki, M.; Amano, A. Porphyromonas gingivalis induces penetration of lipopolysaccharide and peptidoglycan through the gingival epithelium via degradation of junctional adhesion molecule 1. PLoS Pathog. 2019, 15, e1008124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, H.; Nakamura, E.; Yamaga, S.; Amano, A. Porphyromonas gingivalis Infection Induces Lipopolysaccharide and Peptidoglycan Penetration Through Gingival Epithelium. Front. Oral. Health 2022, 3, 845002. [Google Scholar] [CrossRef]
- Takahashi, N.; Sulijaya, B.; Yamada-Hara, M.; Tsuzuno, T.; Tabeta, K.; Yamazaki, K. Gingival epithelial barrier: Regulation by beneficial and harmful microbes. Tissue Barriers 2019, 7, e1651158. [Google Scholar] [CrossRef]
- Marty, M.; Lemaitre, M.; KÉMoun, P.; Morrier, J.-J.; Monsarrat, P. Trichomonas tenax and periodontal diseases: A concise review. Parasitology 2017, 144, 1417–1425. [Google Scholar] [CrossRef]
- Vitkov, L.; Krautgartner, W.D.; Hannig, M.; Weitgasser, R.; Stoiber, W. Candida attachment to oral epithelium. Oral. Microbiol. Immunol. 2002, 17, 60–64. [Google Scholar] [CrossRef]
- Hassona, Y.; Scully, C.; Delgado-Azanero, W.; de Almeida, O.P. Oral helminthic infestations. J. Investig. Clin. Dent. 2015, 6, 99–107. [Google Scholar] [CrossRef]
- Powell, D.N.; Swimm, A.; Sonowal, R.; Bretin, A.; Gewirtz, A.T.; Jones, R.M.; Kalman, D. Indoles from the commensal microbiota act via the AHR and IL-10 to tune the cellular composition of the colonic epithelium during aging. Proc. Natl. Acad. Sci. USA 2020, 117, 21519–21526. [Google Scholar] [CrossRef]
- Persson, G.R. Dental geriatrics and periodontitis. Periodontol. 2000 2017, 74, 102–115. [Google Scholar] [CrossRef]
- Ebersole, J.L.; Al-Sabbagh, M.; Gonzalez, O.A.; Dawson, D.R., 3rd. Ageing effects on humoral immune responses in chronic periodontitis. J. Clin. Periodontol. 2018, 45, 680–692. [Google Scholar] [CrossRef]
- Billings, M.; Holtfreter, B.; Papapanou, P.N.; Mitnik, G.L.; Kocher, T.; Dye, B.A. Age-dependent distribution of periodontitis in two countries: Findings from NHANES 2009 to 2014 and SHIP-TREND 2008 to 2012. J. Periodontol. 2018, 89 (Suppl. S1), S140–S158. [Google Scholar] [CrossRef]
- Gladyshev, V.N.; Kritchevsky, S.B.; Clarke, S.G.; Cuervo, A.M.; Fiehn, O.; de Magalhães, J.P.; Mau, T.; Maes, M.; Moritz, R.L.; Niedernhofer, L.J.; et al. Molecular damage in aging. Nat. Aging 2021, 1, 1096–1106. [Google Scholar] [CrossRef]
- An, J.Y.; Kerns, K.A.; Ouellette, A.; Robinson, L.; Morris, H.D.; Kaczorowski, C.; Park, S.I.; Mekvanich, T.; Kang, A.; McLean, J.S.; et al. Rapamycin rejuvenates oral health in aging mice. Elife 2020, 9, e54318. [Google Scholar] [CrossRef]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Barros, S.P.; Hefni, E.; Fahimipour, F.; Kim, S.; Arora, P. Maintaining barrier function of infected gingival epithelial cells by inhibition of DNA methylation. J. Periodontol. 2020, 91 (Suppl. S1), S68–S78. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.; Stinson, F.L.; Parker, R.B. The Passage of Tritiated Bacterial Endotoxin across Intact Gingival Crevicular Epithelium. J. Periodontol. 1972, 43, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Gabarin, R.S.; Li, M.; Zimmel, P.A.; Marshall, J.C.; Li, Y.; Zhang, H. Intracellular and Extracellular Lipopolysaccharide Signaling in Sepsis: Avenues for Novel Therapeutic Strategies. J. Innate Immun. 2021, 13, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Zhang, Y.; Feng, S.; Liu, X.; Lü, S.; Long, M. Dynamic contributions of P- and E-selectins to β2-integrin-induced neutrophil transmigration. FASEB J. 2017, 31, 212–223. [Google Scholar] [CrossRef] [Green Version]
- Groeger, S.; Meyle, J. Oral Mucosal Epithelial Cells. Front. Immunol. 2019, 10, 208. [Google Scholar] [CrossRef] [Green Version]
- Hirschfeld, J.; White, P.C.; Milward, M.R.; Cooper, P.R.; Chapple, I.L.C. Modulation of Neutrophil Extracellular Trap and Reactive Oxygen Species Release by Periodontal Bacteria. Infect. Immun. 2017, 85, e00297-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanaja, S.K.; Russo, A.J.; Behl, B.; Banerjee, I.; Yankova, M.; Deshmukh, S.D.; Rathinam, V.A.K. Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell 2016, 165, 1106–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef] [Green Version]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszynski, A.; et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.B.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical inflammasome signaling elicits gasdermin D–dependent neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6676. [Google Scholar] [CrossRef] [Green Version]
- Goren, I.; Kämpfer, H.; Müller, E.; Schiefelbein, D.; Pfeilschifter, J.; Frank, S. Oncostatin M expression is functionally connected to neutrophils in the early inflammatory phase of skin repair: Implications for normal and diabetes-impaired wounds. J. Investig. Dermatol. 2006, 126, 628–637. [Google Scholar] [CrossRef] [Green Version]
- Pothoven, K.L.; Norton, J.E.; Suh, L.A.; Carter, R.G.; Harris, K.E.; Biyasheva, A.; Welch, K.; Shintani-Smith, S.; Conley, D.B.; Liu, M.C.; et al. Neutrophils are a major source of the epithelial barrier disrupting cytokine oncostatin M in patients with mucosal airways disease. J. Allergy Clin. Immunol. 2017, 139, 1966–1978.e9. [Google Scholar] [CrossRef]
- Pothoven, K.L.; Schleimer, R.P. The barrier hypothesis and Oncostatin M: Restoration of epithelial barrier function as a novel therapeutic strategy for the treatment of type 2 inflammatory disease. Tissue Barriers 2017, 5, e1341367. [Google Scholar] [CrossRef] [Green Version]
- Manojkumar, S.T.; Pradeep, A.R.; Garg, G.; Raju, A. Gingival crevicular fluid levels of oncostatin M in periodontal conditions. Cytokine 2010, 50, 248–252. [Google Scholar] [CrossRef]
- Headland, S.E.; Dengler, H.S.; Xu, D.; Teng, G.; Everett, C.; Ratsimandresy, R.A.; Yan, D.; Kang, J.; Ganeshan, K.; Nazarova, E.V.; et al. Oncostatin M expression induced by bacterial triggers drives airway inflammatory and mucus secretion in severe asthma. Sci. Transl. Med. 2022, 14, eabf8188. [Google Scholar] [CrossRef] [PubMed]
- West, N.R.; Hegazy, A.N.; Owens, B.M.J.; Bullers, S.J.; Linggi, B.; Buonocore, S.; Coccia, M.; Görtz, D.; This, S.; Stockenhuber, K.; et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat. Med. 2017, 23, 579–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.M.; Vanyo, S.T.; Ibraheem, W.; Maddi, A.; Visser, M.B. Treponema denticola stimulates Oncostatin M cytokine release and de novo synthesis in neutrophils and macrophages. J. Leukoc. Biol. 2020, 108, 1527–1541. [Google Scholar] [CrossRef]
- de Oliveira, S.; Rosowski, E.E.; Huttenlocher, A. Neutrophil migration in infection and wound repair: Going forward in reverse. Nat. Rev. Immunol. 2016, 16, 378–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mäntylä, P.; Stenman, M.; Kinane, D.F.; Tikanoja, S.; Luoto, H.; Salo, T.; Sorsa, T. Gingival crevicular fluid collagenase-2 (MMP-8) test stick for chair-side monitoring of periodontitis: MMP-8 test in monitoring periodontitis. J. Periodontal Res. 2003, 38, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Figueredo, C.M.S.; Fischer, R.G.; Gustafsson, A. Aberrant Neutrophil Reactions in Periodontitis. J. Periodontol. 2005, 76, 951–955. [Google Scholar] [CrossRef] [PubMed]
- Sorsa, T.; Gursoy, U.K.; Nwhator, S.; Hernandez, M.; Tervahartiala, T.; Leppilahti, J.; Gursoy, M.; Könönen, E.; Emingil, G.; Pussinen, P.J.; et al. Analysis of matrix metalloproteinases, especially MMP-8, in gingival crevicular fluid, mouthrinse and saliva for monitoring periodontal diseases. Periodontol. 2000 2016, 70, 142–163. [Google Scholar] [CrossRef] [PubMed]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil Extracellular Traps Directly Induce Epithelial and Endothelial Cell Death: A Predominant Role of Histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef] [PubMed]
- Allam, R.; Scherbaum, C.R.; Darisipudi, M.N.; Mulay, S.R.; Hägele, H.; Lichtnekert, J.; Hagemann, J.H.; Rupanagudi, K.V.; Ryu, M.; Schwarzenberger, C.; et al. Histones from Dying Renal Cells Aggravate Kidney Injury via TLR2 and TLR4. J. Am. Soc. Nephrol. 2012, 23, 1375–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Wang, J.; Wang, Y.; Wang, C.; Liu, X.; Han, Z.; Fu, Y.; Yang, Z. Effects of Neutrophil Extracellular Traps on Bovine Mammary Epithelial Cells in vitro. Front. Immunol. 2019, 10, 1003. [Google Scholar] [CrossRef] [Green Version]
- Knopf, J.; Leppkes, M.; Schett, G.; Herrmann, M.; Muñoz, L.E. Aggregated NETs Sequester and Detoxify Extracellular Histones. Front. Immunol. 2019, 10, 2176. [Google Scholar] [CrossRef] [Green Version]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 2019, 133, 2178–2185. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Rivera, C.; Carlucci, P.M.; Goel, R.R.; James, E.; Brooks, S.R.; Rims, C.; Hoffmann, V.; Fox, D.A.; Buckner, J.H.; Kaplan, M.J. Neutrophil extracellular traps mediate articular cartilage damage and enhance cartilage component immunogenicity in rheumatoid arthritis. JCI Insight 2020, 5, e139388. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonello, S.; Rizzi, M.; Migliario, M.; Rocchetti, V.; Renò, F. Low concentrations of neutrophil extracellular traps induce proliferation in human keratinocytes via NF-kB activation. J. Dermatol. Sci. 2017, 88, 110–116. [Google Scholar] [CrossRef] [Green Version]
- Hiyoshi, T.; Domon, H.; Maekawa, T.; Tamura, H.; Isono, T.; Hirayama, S.; Sasagawa, K.; Takizawa, F.; Tabeta, K.; Terao, Y. Neutrophil elastase aggravates periodontitis by disrupting gingival epithelial barrier via cleaving cell adhesion molecules. Sci. Rep. 2022, 12, 8159. [Google Scholar] [CrossRef]
- Olsen, I. Update on bacteraemia related to dental procedures. Transfus. Apher. Sci. 2008, 39, 173–178. [Google Scholar] [CrossRef]
- Varadarajan, S.; Stephenson, R.E.; Miller, A.L. Multiscale dynamics of tight junction remodeling. J. Cell Sci. 2019, 132, jcs229286. [Google Scholar] [CrossRef]
- Paradis, T.; Bègue, H.; Basmaciyan, L.; Dalle, F.; Bon, F. Tight Junctions as a Key for Pathogens Invasion in Intestinal Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 2506. [Google Scholar] [CrossRef]
- Higashi, T.; Saito, A.C.; Fukazawa, Y.; Furuse, M.; Higashi, A.Y.; Ono, M.; Chiba, H. EpCAM proteolysis and release of complexed claudin-7 repair and maintain the tight junction barrier. J. Cell Biol. 2022, 222, e202204079. [Google Scholar] [CrossRef] [PubMed]
- Deppermann, C.; Peiseler, M.; Zindel, J.; Zbytnuik, L.; Lee, W.Y.; Pasini, E.; Baciu, C.; Matelski, J.; Lee, Y.; Kumar, D.; et al. Tacrolimus Impairs Kupffer Cell Capacity to Control Bacteremia: Why Transplant Recipients Are Susceptible to Infection. Hepatology 2021, 73, 1967–1984. [Google Scholar] [CrossRef]
- Bonfanti, A.; Duque, J.; Kabla, A.; Charras, G. Fracture in living tissues. Trends Cell Biol. 2022, 32, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Javed, T.A.; Yimlamai, D.; Mukherjee, A.; Xiao, X.; Husain, S.Z. Transient High Pressure in Pancreatic Ducts Promotes Inflammation and Alters Tight Junctions via Calcineurin Signaling in Mice. Gastroenterology 2018, 155, 1250–1263.e5. [Google Scholar] [CrossRef]
- Zhou, J.; Zhou, X.D.; Xu, R.; Du, X.Z.; Li, Q.; Li, B.; Zhang, G.Y.; Chen, L.X.; Perelman, J.M.; Kolosov, V.P. The Degradation of Airway Epithelial Tight Junctions in Asthma Under High Airway Pressure Is Probably Mediated by Piezo-1. Front. Physiol. 2021, 12, 637790. [Google Scholar] [CrossRef] [PubMed]
- Douglas, P. Re-thinking benign inflammation of the lactating breast: A mechanobiological model. Womens Health 2022, 18, 17455065221075907. [Google Scholar] [CrossRef]
- Sato, T.; Hara, T.; Mori, S.; Shirai, H.; Minagi, S. Threshold for bone resorption induced by continuous and intermittent pressure in the rat hard palate. J. Dent. Res. 1998, 77, 387–392. [Google Scholar] [CrossRef]
- Madea, B. Asphyxiation, Suffocation, and Neck Pressure Deaths; CRC Press: Boca Raton, FL, USA, 2020. [Google Scholar]
- Dutzan, N.; Abusleme, L.; Bridgeman, H.; Greenwell-Wild, T.; Zangerle-Murray, T.; Fife, M.E.; Bouladoux, N.; Linley, H.; Brenchley, L.; Wemyss, K.; et al. On-going Mechanical Damage from Mastication Drives Homeostatic Th17 Cell Responses at the Oral Barrier. Immunity 2017, 46, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Konkel, J.E.; Moutsopoulos, N.M. Unique Tailoring of Th17 at the Gingival Oral Mucosal Barrier. J. Dent. Res. 2018, 97, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Geerts, S.O.; Nys, M.; Mol, P.D.; Charpentier, J.; Albert, A.; Legrand, V.; Rompen, E.H. Systemic Release of Endotoxins Induced by Gentle Mastication: Association With Periodontitis Severity. J. Periodontol. 2002, 73, 73–78. [Google Scholar] [CrossRef]
- Crasta, K.; Daly, C.G.; Mitchell, D.; Curtis, B.; Stewart, D.; Heitz-Mayfield, L.J.A. Bacteraemia due to dental flossing. J. Clin. Periodontol. 2009, 36, 323–332. [Google Scholar] [CrossRef]
- Tomás, I.; Diz, P.; Tobías, A.; Scully, C.; Donos, N. Periodontal health status and bacteraemia from daily oral activities: Systematic review/meta-analysis. J. Clin. Periodontol. 2012, 39, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Bosshardt, D.D. The periodontal pocket: Pathogenesis, histopathology and consequences. Periodontol. 2000 2018, 76, 43–50. [Google Scholar] [CrossRef]
- Aguirre, J.I.; Akhter, M.P.; Neuville, K.G.; Trcalek, C.R.; Leeper, A.M.; Williams, A.A.; Rivera, M.; Kesavalu, L.; Ke, H.Z.; Liu, M.; et al. Age-related periodontitis and alveolar bone loss in rice rats. Arch. Oral Biol. 2017, 73, 193–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira, P.A.; de Pizzol-Júnior, J.P.; Longhini, R.; Sasso-Cerri, E.; Cerri, P.S. Cimetidine Reduces Interleukin-6, Matrix Metalloproteinases-1 and -9 Immunoexpression in the Gingival Mucosa of Rat Molars With Induced Periodontal Disease. J. Periodontol. 2017, 88, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Hujoel, P.P.; White, B.A.; García, R.I.; Listgarten, M.A. The dentogingival epithelial surface area revisited. J. Periodontal Res. 2001, 36, 48–55. [Google Scholar] [CrossRef]
- Müller-Glauser, W.; Schroeder, H.E. The pocket epithelium: A light- and electronmicroscopic study. J. Periodontol. 1982, 53, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Zoellner, H.; Chapple, C.C.; Hunter, N. Microvasculature in gingivitis and chronic periodontitis: Disruption of vascular networks with protracted inflammation. Microsc. Res. Tech. 2002, 56, 15–31. [Google Scholar] [CrossRef]
- Iglesias-Bartolome, R.; Uchiyama, A.; Molinolo, A.A.; Abusleme, L.; Brooks, S.R.; Callejas-Valera, J.L.; Edwards, D.; Doci, C.; Asselin-Labat, M.L.; Onaitis, M.W.; et al. Transcriptional signature primes human oral mucosa for rapid wound healing. Sci. Transl. Med. 2018, 10, eaap8798. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Chen, J.; Grauer, J.A.; Xu, Q.; Van Brunt, L.A.; Helms, J.A. The Junctional Epithelium Is Maintained by a Stem Cell Population. J. Dent. Res. 2021, 100, 209–216. [Google Scholar] [CrossRef]
- Wang, X.; Yong, C.C.; Oh, S. Metabolites of Latilactobacillus curvatus BYB3 and Indole Activate Aryl Hydrocarbon Receptor to Attenuate Lipopolysaccharide-Induced Intestinal Barrier Dysfunction. Food Sci. Anim. Resour. 2022, 42, 1046–1060. [Google Scholar] [CrossRef]
- Shan, Q.; Liu, N.; Wang, X.; Zhu, Y.; Yin, J.; Wang, J. Lactobacillus rhamnosus GR-1 attenuates foodborne Bacillus cereus-induced NLRP3 inflammasome activity in bovine mammary epithelial cells by protecting intercellular tight junctions. J. Anim. Sci. Biotechnol. 2022, 13, 101. [Google Scholar] [CrossRef] [PubMed]
- Elbadawi, M.; Ammar, R.M.; Aziz-Kalbhenn, H.; Rabini, S.; Klauck, S.M.; Dawood, M.; Saeed, M.E.M.; Kampf, C.J.; Efferth, T. Anti-inflammatory and tight junction protective activity of the herbal preparation STW 5-II on mouse intestinal organoids. Phytomedicine 2021, 88, 153589. [Google Scholar] [CrossRef] [PubMed]
- Colamonici, M.A.; Epshtein, Y.; Chen, W.; Jacobson, J.R. Haloperidol Attenuates Lung Endothelial Cell Permeability In Vitro and In Vivo. Cells 2021, 10, 2186. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitkov, L.; Singh, J.; Schauer, C.; Minnich, B.; Krunić, J.; Oberthaler, H.; Gamsjaeger, S.; Herrmann, M.; Knopf, J.; Hannig, M. Breaking the Gingival Barrier in Periodontitis. Int. J. Mol. Sci. 2023, 24, 4544. https://doi.org/10.3390/ijms24054544
Vitkov L, Singh J, Schauer C, Minnich B, Krunić J, Oberthaler H, Gamsjaeger S, Herrmann M, Knopf J, Hannig M. Breaking the Gingival Barrier in Periodontitis. International Journal of Molecular Sciences. 2023; 24(5):4544. https://doi.org/10.3390/ijms24054544
Chicago/Turabian StyleVitkov, Ljubomir, Jeeshan Singh, Christine Schauer, Bernd Minnich, Jelena Krunić, Hannah Oberthaler, Sonja Gamsjaeger, Martin Herrmann, Jasmin Knopf, and Matthias Hannig. 2023. "Breaking the Gingival Barrier in Periodontitis" International Journal of Molecular Sciences 24, no. 5: 4544. https://doi.org/10.3390/ijms24054544